
Revolutionizing Cancer Treatments through Stem Cell-Derived CAR T Cells for Immunotherapy: Opening New Horizons for the Future of Oncology



Abstract
1. Introduction
2. Generation of Allogeneic CAR T Cells
3. Cord Blood-Derived CAR T Cells
4. iPSC-Derived CAR T Cells
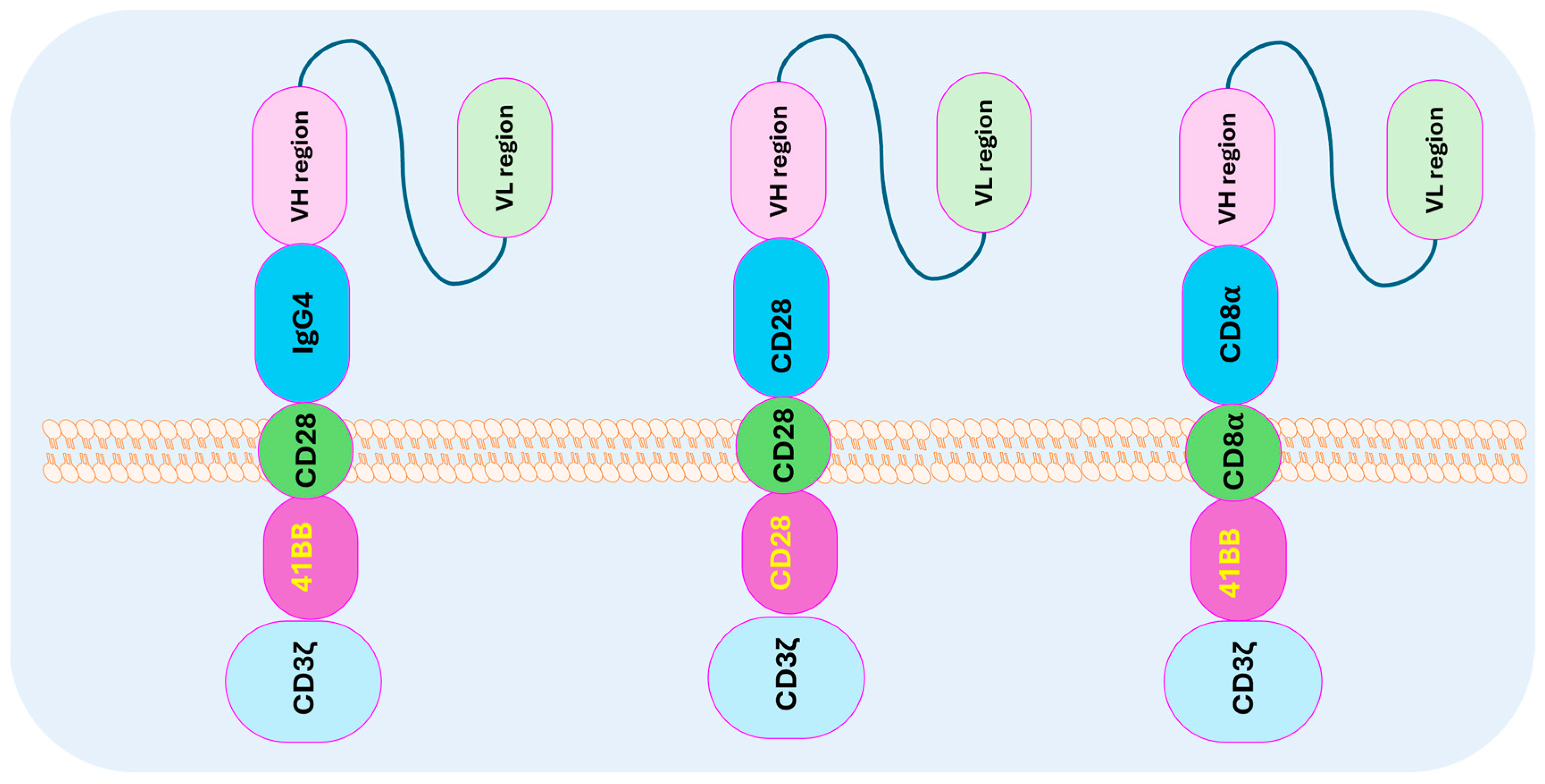
5. CAR T-Cell Structure and Design
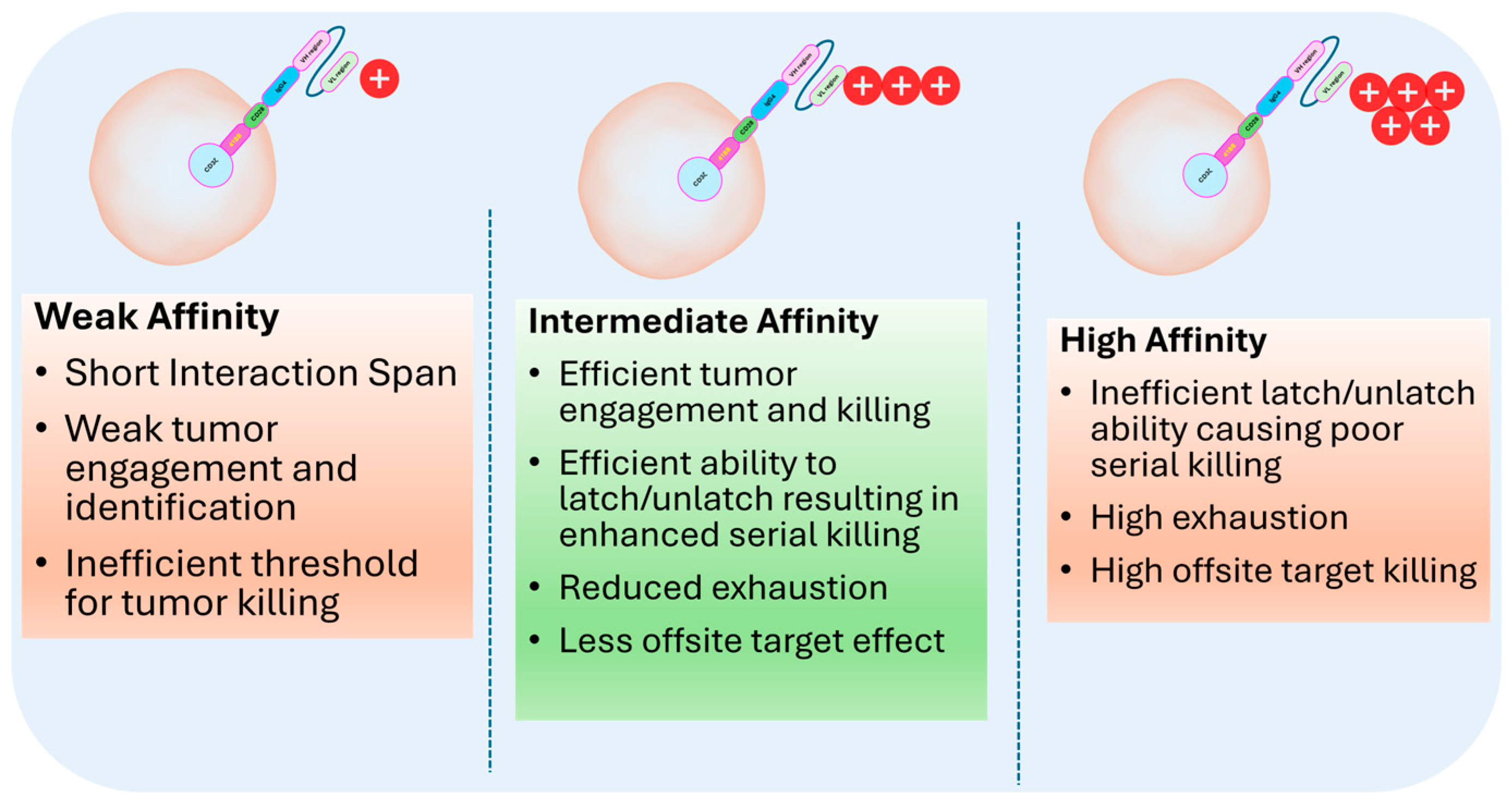
6. Selection of Optimized scFv
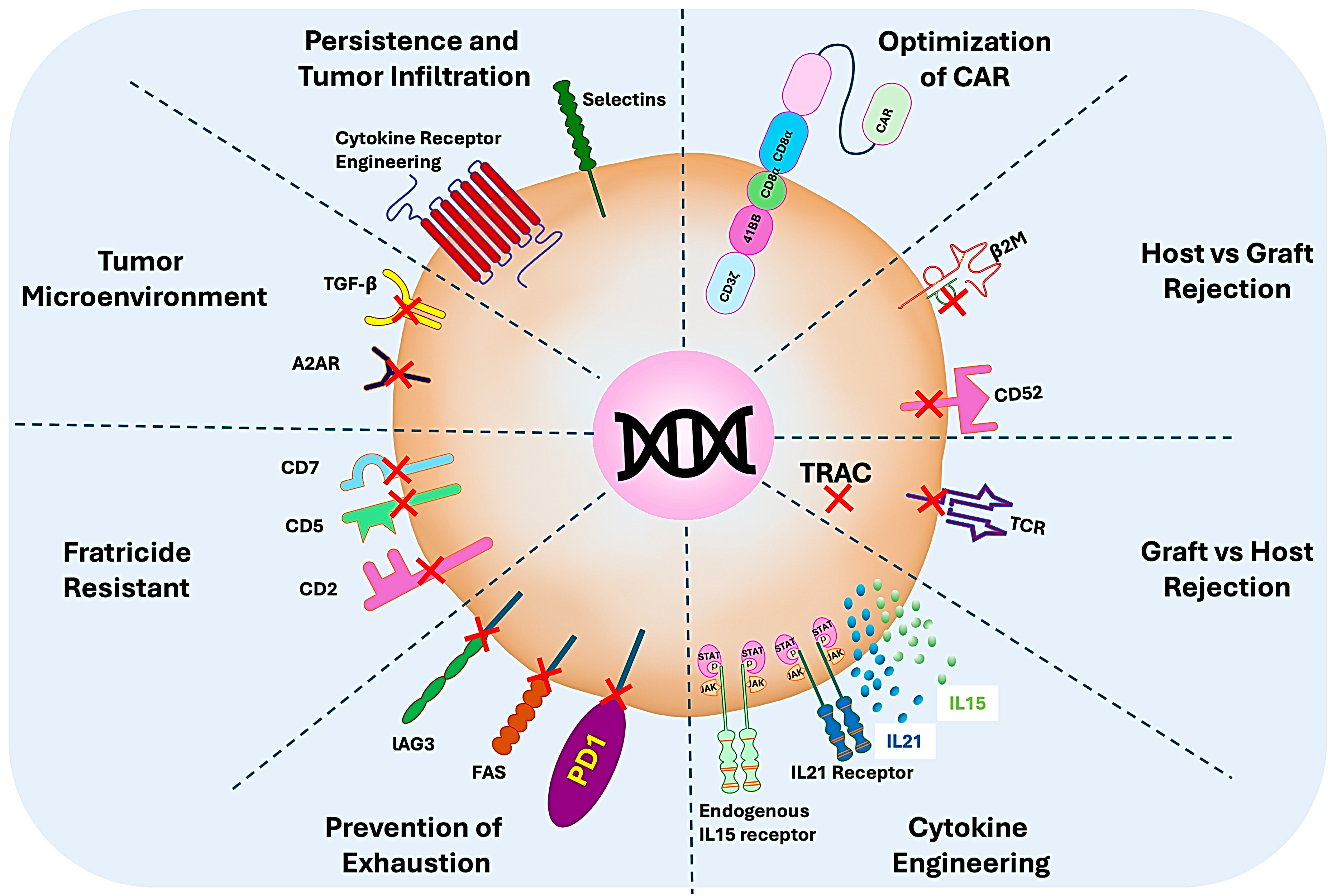
7. Enhancing the Allogenicity and Efficacy of CAR T Cells
8. Future Direction and Scope
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cerneckis, J.; Cai, H.; Shi, Y. Induced pluripotent stem cells (iPSCs): Molecular mechanisms of induction and applications. Signal Transduct. Target Ther. 2024, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef]
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef]
- Gajra, A.; Zalenski, A.; Sannareddy, A.; Jeune-Smith, Y.; Kapinos, K.; Kansagra, A. Barriers to Chimeric Antigen Receptor T-Cell (CAR-T) Therapies in Clinical Practice. Pharm. Med. 2022, 36, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Kandra, P.; Nandigama, R.; Eul, B.; Huber, M.; Kobold, S.; Seeger, W.; Grimminger, F.; Savai, R. Utility and Drawbacks of Chimeric Antigen Receptor T Cell (CAR-T) Therapy in Lung Cancer. Front. Immunol. 2022, 13, 903562. [Google Scholar] [CrossRef]
- Ilic, D.; Ogilvie, C. Pluripotent Stem Cells in Clinical Setting-New Developments and Overview of Current Status. Stem Cells 2022, 40, 791–801. [Google Scholar] [CrossRef]
- Ueda, T.; Shiina, S.; Iriguchi, S.; Terakura, S.; Kawai, Y.; Kabai, R.; Sakamoto, S.; Watanabe, A.; Ohara, K.; Wang, B.; et al. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat. Biomed. Eng. 2023, 7, 24–37. [Google Scholar] [CrossRef]
- Srour, S.A.; Akin, S. Chimeric Antigen Receptor T-Cell Therapy for Solid Tumors: The Past and the Future. J. Immunother. Precis. Oncol. 2023, 6, 19–30. [Google Scholar] [CrossRef]
- Pessach, I.; Nagler, A. Leukapheresis for CAR-T cell production and therapy. Transfus. Apher. Sci. 2023, 62, 103828. [Google Scholar] [CrossRef]
- Tyagarajan, S.; Schmitt, D.; Acker, C.; Rutjens, E. Autologous cryopreserved leukapheresis cellular material for chimeric antigen receptor-T cell manufacture. Cytotherapy 2019, 21, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Qayed, M.; McGuirk, J.P.; Myers, G.D.; Parameswaran, V.; Waller, E.K.; Holman, P.; Rodrigues, M.; Clough, L.F.; Willert, J. Leukapheresis guidance and best practices for optimal chimeric antigen receptor T-cell manufacturing. Cytotherapy 2022, 24, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Schlenk, R.F.; Steiner, M.; Kluter, H.; Wuchter, P. Predicting Successful Hematopoietic Stem Cell Collection in Healthy Allogeneic Donors. Transfus. Med. Hemother. 2023, 50, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Martinez Bedoya, D.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef]
- Liu, D.D.; Hong, W.C.; Qiu, K.Y.; Li, X.Y.; Liu, Y.; Zhu, L.W.; Lai, W.X.; Chen, H.; Yang, H.Q.; Xu, L.H.; et al. Umbilical cord blood: A promising source for allogeneic CAR-T cells. Front. Oncol. 2022, 12, 944248. [Google Scholar] [CrossRef]
- Tang, T.C.Y.; Xu, N.; Nordon, R.; Haber, M.; Micklethwaite, K.; Dolnikov, A. Donor T cells for CAR T cell therapy. Biomark. Res. 2022, 10, 14. [Google Scholar] [CrossRef]
- Sanchez-Petitto, G.; Rezvani, K.; Daher, M.; Rafei, H.; Kebriaei, P.; Shpall, E.J.; Olson, A. Umbilical Cord Blood Transplantation: Connecting Its Origin to Its Future. Stem Cells Transl. Med. 2023, 12, 55–71. [Google Scholar] [CrossRef]
- Cael, B.; Galaine, J.; Bardey, I.; Marton, C.; Fredon, M.; Biichle, S.; Poussard, M.; Godet, Y.; Angelot-Delettre, F.; Barisien, C.; et al. Umbilical Cord Blood as a Source of Less Differentiated T Cells to Produce CD123 CAR-T Cells. Cancers 2022, 14, 3168. [Google Scholar] [CrossRef] [PubMed]
- Marra, J.D.; Galli, E.; Giammarco, S.; Metafuni, E.; Minnella, G.; Fosso, F.; Marietti, S.; Sica, S.; Sora, F.; Chiusolo, P. CAR-T from cord blood in a patient with Ph+ acute lymphoblastic leukemia relapsing after hematopoietic stem cell transplantation. Eur. J. Haematol. 2024, 113, 127–129. [Google Scholar] [CrossRef]
- Yu, T.; Luo, C.; Zhang, H.; Tan, Y.; Yu, L. Cord blood-derived CD19-specific chimeric antigen receptor T cells: An off-the-shelf promising therapeutic option for treatment of diffuse large B-cell lymphoma. Front. Immunol. 2023, 14, 1139482. [Google Scholar] [CrossRef]
- Sharma, A.; Mucke, M.; Seidman, C.E. Human Induced Pluripotent Stem Cell Production and Expansion from Blood using a Non-Integrating Viral Reprogramming Vector. Curr. Protoc. Mol. Biol. 2018, 122, e58. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Scarfo, I.; Najia, M.A.; Lummertz da Rocha, E.; Han, A.; Sanborn, M.; Bingham, T.; Kubaczka, C.; Jha, D.K.; Falchetti, M.; et al. EZH1 repression generates mature iPSC-derived CAR T cells with enhanced antitumor activity. Cell Stem Cell 2022, 29, 1181–1196.e6. [Google Scholar] [CrossRef]
- Qin, V.M.; D’Souza, C.; Neeson, P.J.; Zhu, J.J. Chimeric Antigen Receptor beyond CAR-T Cells. Cancers 2021, 13, 404. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wang, C.; Cao, C.; Huang, J.; Holzhauer, S.; Desilva, H.; Wesley, E.M.; Evans, D.B.; Benci, J.; Wichroski, M.; et al. DGKzeta exerts greater control than DGKalpha over CD8(+) T cell activity and tumor inhibition. Oncoimmunology 2021, 10, 1941566. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Luo, F.; Chu, Y. Strategies for overcoming bottlenecks in allogeneic CAR-T cell therapy. Front. Immunol. 2023, 14, 1199145. [Google Scholar] [CrossRef]
- Guzman, G.; Reed, M.R.; Bielamowicz, K.; Koss, B.; Rodriguez, A. CAR-T Therapies in Solid Tumors: Opportunities and Challenges. Curr. Oncol. Rep. 2023, 25, 479–489. [Google Scholar] [CrossRef]
- Patel, S.J.; Yamauchi, T.; Ito, F. Induced Pluripotent Stem Cell-Derived T Cells for Cancer Immunotherapy. Surg. Oncol. Clin. N. Am. 2019, 28, 489–504. [Google Scholar] [CrossRef]
- Sadeqi Nezhad, M.; Abdollahpour-Alitappeh, M.; Rezaei, B.; Yazdanifar, M.; Seifalian, A.M. Induced Pluripotent Stem Cells (iPSCs) Provide a Potentially Unlimited T Cell Source for CAR-T Cell Development and Off-the-Shelf Products. Pharm. Res. 2021, 38, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; van der Stegen, S.J.C.; Miller, J.S. Engineered and banked iPSCs for advanced NK- and T-cell immunotherapies. Blood 2023, 141, 846–855. [Google Scholar] [CrossRef]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-organoid culture supports differentiation of human CAR+ iPSCs into highly functional CAR T cells. Cell Stem Cell 2022, 29, 515–527.e8. [Google Scholar] [CrossRef]
- Lahimchi, M.R.; Maroufi, F.; Maali, A. Induced Pluripotent Stem Cell-Derived Chimeric Antigen Receptor T Cells: The Intersection of Stem Cells and Immunotherapy. Cell Reprogram. 2023, 25, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; Xu, H.; Cao, W.; Cheng, C.; Tan, S.; Zhao, T. Optimizing in vitro T cell differentiation by using induced pluripotent stem cells with GFP-RUNX1 and mCherry-TCF7 labelling. Cell Prolif. 2024, e13661. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug. FDA Approval of KYMRIAH. FDA News, 3 October 2017. [Google Scholar]
- U.S. Food and Drug. FDA Approves CAR-T Cell Therapy to Treat Adults with Certain Types of Large B-Cell Lymphoma. FDA News, 18 October 2017. [Google Scholar]
- U.S. Food and Drug. FDA Grants Accelerated Approval to Axicabtagene Ciloleucel for Relapsed or Refractory Follicular Lymphoma. FDA News, 5 March 2021. [Google Scholar]
- U.S. Food and Drug. Accelerated Approval of TECARTUS. FDA News, 23 July 2020. [Google Scholar]
- U.S. Food and Drug. FDA Approval of BREYANZI (Lisocabtagene Maraleucel). FDA News, 1 February 2021. [Google Scholar]
- U.S. Food and Drug. FDA Approval of ABECMA (Idecabtagene Vicleucel). FDA News, 26 March 2021. [Google Scholar]
- U.S. Food and Drug. FDA Approval of CARVYKTI Ciltacabtagene Autoleucel. FDA News, 28 February 2022. [Google Scholar]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Benjamin, R.; Jain, N.; Maus, M.V.; Boissel, N.; Graham, C.; Jozwik, A.; Yallop, D.; Konopleva, M.; Frigault, M.J.; Teshima, T.; et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): A phase 1, dose-escalation trial. Lancet Haematol. 2022, 9, e833–e843. [Google Scholar] [CrossRef]
- Pan, J.; Tang, K.; Luo, Y.; Seery, S.; Tan, Y.; Deng, B.; Liu, F.; Xu, X.; Ling, Z.; Song, W.; et al. Sequential CD19 and CD22 chimeric antigen receptor T-cell therapy for childhood refractory or relapsed B-cell acute lymphocytic leukaemia: A single-arm, phase 2 study. Lancet Oncol. 2023, 24, 1229–1241. [Google Scholar] [CrossRef]
- Schultz, L.M.; Jeyakumar, N.; Kramer, A.M.; Sahaf, B.; Srinagesh, H.; Shiraz, P.; Agarwal, N.; Hamilton, M.; Erickson, C.; Jacobs, A.; et al. CD22 CAR T cells demonstrate high response rates and safety in pediatric and adult B-ALL: Phase 1b results. Leukemia 2024, 38, 963–968. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, B.; Wu, Z.; Bo, J.; Tong, C.; Chen, D.; Wang, J.; Wang, H.; Wang, Y.; Han, W. Mutant B2M-HLA-E and B2M-HLA-G fusion proteins protects universal chimeric antigen receptor-modified T cells from allogeneic NK cell-mediated lysis. Eur. J. Immunol. 2021, 51, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Nai, Y.; Shen, M.; Li, T.; Huang, J.; Han, X.; Wang, W.; Pang, D.; Jin, A. IL-21 Optimizes the CAR-T Cell Preparation Through Improving Lentivirus Mediated Transfection Efficiency of T Cells and Enhancing CAR-T Cell Cytotoxic Activities. Front. Mol. Biosci. 2021, 8, 675179. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Song, Y.; Hu, K.; Zu, W.; Zhang, F.; Chen, L.; Ma, Q.; Zhou, Z.; Jiao, S. CXCL10 and IL15 co-expressing chimeric antigen receptor T cells enhance anti-tumor effects in gastric cancer by increasing cytotoxic effector cell accumulation and survival. Oncoimmunology 2024, 13, 2358590. [Google Scholar] [CrossRef]
- Lau, E.; Kwong, G.; Fowler, T.W.; Sun, B.C.; Donohoue, P.D.; Davis, R.T.; Bryan, M.; McCawley, S.; Clarke, S.C.; Williams, C.; et al. Allogeneic chimeric antigen receptor-T cells with CRISPR-disrupted programmed death-1 checkpoint exhibit enhanced functional fitness. Cytotherapy 2023, 25, 750–762. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Montalvo, M.J.; Bandey, I.N.; Rezvan, A.; Wu, K.L.; Saeedi, A.; Kulkarni, R.; Li, Y.; An, X.; Sefat, K.; Varadarajan, N. Decoding the mechanisms of chimeric antigen receptor (CAR) T cell-mediated killing of tumors: Insights from granzyme and Fas inhibition. Cell Death Dis. 2024, 15, 109. [Google Scholar] [CrossRef]
- Xiang, J.; Devenport, J.M.; Carter, A.J.; Staser, K.W.; Kim, M.Y.; Neal, J.O.; Ritchey, J.K.; Rettig, M.P.; Gao, F.; Rettig, G.; et al. An “off-the-shelf” CD2 universal CAR-T therapy for T-cell malignancies. Leukemia 2023, 37, 2448–2456. [Google Scholar] [CrossRef]
- Ma, R.; Woods, M.; Burkhardt, P.; Crooks, N.; van Leeuwen, D.G.; Shmidt, D.; Couturier, J.; Chaumette, A.; Popat, D.; Hill, L.C.; et al. Chimeric antigen receptor-induced antigen loss protects CD5.CART cells from fratricide without compromising on-target cytotoxicity. Cell Rep. Med. 2024, 5, 101628. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, D.; Fu, X.; Meng, H.; Nan, F.; Sun, Z.; Yu, H.; Zhang, L.; Li, L.; Li, X.; et al. Autologous Nanobody-Derived Fratricide-Resistant CD7-CAR T-cell Therapy for Patients with Relapsed and Refractory T-cell Acute Lymphoblastic Leukemia/Lymphoma. Clin. Cancer Res. 2022, 28, 2830–2843. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.R.F.; Corveloni, A.C.; Caruso, S.R.; Macedo, N.A.; Brussolo, N.M.; Haddad, F.; Fernandes, T.R.; de Andrade, P.V.; Orellana, M.D.; Guerino-Cunha, R.L. Cytokines as an important player in the context of CAR-T cell therapy for cancer: Their role in tumor immunomodulation, manufacture, and clinical implications. Front. Immunol. 2022, 13, 947648. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Si, X.; Shao, M.; Teng, X.; Xiao, G.; Huang, H. Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells. J. Hematol. Oncol. 2022, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shao, M.; Teng, X.; Si, X.; Wu, L.; Jiang, P.; Liu, L.; Cai, B.; Wang, X.; Han, Y.; et al. Inhibition of CD38 enzymatic activity enhances CAR-T cell immune-therapeutic efficacy by repressing glycolytic metabolism. Cell Rep. Med. 2024, 5, 101400. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sal, M.; Schmueck-Henneresse, M. CAR-T overdrive: Harnessing inosine for metabolic rewiring and stemness induction. Signal Transduct. Target. Ther. 2024, 9, 120. [Google Scholar] [CrossRef]
- Klysz, D.D.; Fowler, C.; Malipatlolla, M.; Stuani, L.; Freitas, K.A.; Chen, Y.; Meier, S.; Daniel, B.; Sandor, K.; Xu, P.; et al. Inosine induces stemness features in CAR-T cells and enhances potency. Cancer Cell 2024, 42, 266–282.e8. [Google Scholar] [CrossRef]
- U.S. Food and Drug. Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products; FDA-2021-D-0404; U.S. Food and Drug: Silver Spring, MD, USA, 2024.
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef]
- Kawai, Y.; Kawana-Tachikawa, A.; Kitayama, S.; Ueda, T.; Miki, S.; Watanabe, A.; Kaneko, S. Generation of highly proliferative, rejuvenated cytotoxic T cell clones through pluripotency reprogramming for adoptive immunotherapy. Mol. Ther. 2021, 29, 3027–3041. [Google Scholar] [CrossRef]
- Itoh, M.; Kawagoe, S.; Nakagawa, H.; Asahina, A.; Okano, H.J. Generation of induced pluripotent stem cell (iPSC) from NY-ESO-I-specific cytotoxic T cells isolated from the melanoma patient with minor HLAs: The practical pilot study for the adoptive immunotherapy for melanoma using iPSC technology. Exp. Dermatol. 2023, 32, 126–134. [Google Scholar] [CrossRef]
- Harada, S.; Ando, M.; Ando, J.; Ishii, M.; Yamaguchi, T.; Yamazaki, S.; Toyota, T.; Ohara, K.; Ohtaka, M.; Nakanishi, M.; et al. Dual-antigen targeted iPSC-derived chimeric antigen receptor-T cell therapy for refractory lymphoma. Mol. Ther. 2022, 30, 534–549. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAR T-Cell Therapy Name | Target Antigen | Indication | Manufacturer | Reference |
|---|---|---|---|---|
| Kymriah (tisagenlecleucel) | CD19 | B-cell acute lymphoblastic leukemia (ALL) | Novartis | FDA approval (2017) [33,34] |
| Yescarta (axicabtagene ciloleucel) | CD19 | Large B-cell lymphoma | Kite Pharma/Gilead | FDA approval (2017) [35] |
| Kymriah (tisagenlecleucel) | CD19 | Large B-cell lymphoma | Novartis | FDA approval (2018) [34] |
| Yescarta (axicabtagene ciloleucel) | CD19 | Follicular lymphoma | Kite Pharma/Gilead | FDA approval (2021) [36] |
| Tecartus (brexucabtagene autoleucel) | CD19 | Mantle cell lymphoma | Kite Pharma/Gilead | FDA approval (2020) [37] |
| Breyanzi (lisocabtagene maraleucel) | CD19 | Large B-cell lymphoma | Bristol-Myers Squibb | FDA approval (2021) [38] |
| Abecma (idecabtagene vicleucel) | BCMA | Multiple myeloma | Bristol-Myers Squibb | FDA approval (2021) [39] |
| Carvykti (ciltacabtagene autoleucel) | BCMA | Relapsed or refractory multiple myeloma | Johnson & Johnson/Legend Biotech | FDA approval (2022) [40] |
| Domains | Protein Region |
|---|---|
| Intracellular | CD3ζ, CD28, and 41BB |
| Transmembrane | CD3ζ, CD28, 41BB, CD8α, FcγRIIIA, TCRαβ, and DAP12 |
| Hinge domain | CD8α, CD28, IgG4, IgG2, IgG1, and DAP12 |
| Extracellular | VH-VL, VL-VH, and VH |
| Criteria | Description |
|---|---|
| Eligibility of donor | Donors must be screened for transmissible diseases as per FDA regulations (21 CFR 1271). |
| Transduction efficiency | Efficiency of gene transduction should be monitored to ensure correct expression of the CAR construct in T cells. |
| Vector copy number (VCN) | VCN per cell must be monitored and controlled, generally targeting 1–5 copies per cell to avoid toxicity and insertional mutagenesis. |
| T-cell expansion | Post-transduction, T cells are expanded in a controlled environment, ensuring high viability and potency. |
| Purity check | The final product must meet specific purity (e.g., CD3+ cells) and identity (e.g., CAR expression) standards. |
| Potency assays | Potency assays must be performed to ensure CAR T cells are capable of recognizing and killing target cancer cells. |
| Safety check | Sterility, endotoxin levels, and mycoplasma testing must be performed to ensure no contamination during production. |
| Viability of CAR T cells | The FDA mandates the minimum viability (usually > 70%) of CAR T cells at the time of infusion. |
| Cryopreservation | Cells are typically cryopreserved for shipment and storage; freezing methods must ensure cell stability post-thaw. The concentration of DMSO must be ≤10% v/v; however, ≤5% v/v is often preferred to further reduce DMSO-related cytotoxicity. |
| Release criteria | The final product must meet pre-defined release criteria, including CAR expression, viability, identity, purity, and vector copy number (VCN). |
| Adventitious agent or other contaminants | Testing is required to confirm that the product is free from unintended viruses or contaminants. |
| Preclinical studies | Preclinical models must demonstrate safety and efficacy before clinical trials can commence. |
| Clinical manufacturing | Must adhere to Good Manufacturing Practice (GMP) guidelines (21 CFR Parts 210 and 211), including controlled and reproducible processes. |
| Labeling | CAR T-cell products must have clear labeling for identity, dose, and instructions as per FDA labeling regulations (21 CFR Part 610). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, H.K.; Kalyuzhny, A. Revolutionizing Cancer Treatments through Stem Cell-Derived CAR T Cells for Immunotherapy: Opening New Horizons for the Future of Oncology. Cells 2024, 13, 1516. https://doi.org/10.3390/cells13181516
Mishra HK, Kalyuzhny A. Revolutionizing Cancer Treatments through Stem Cell-Derived CAR T Cells for Immunotherapy: Opening New Horizons for the Future of Oncology. Cells. 2024; 13(18):1516. https://doi.org/10.3390/cells13181516
Chicago/Turabian StyleMishra, Hemant K., and Alex Kalyuzhny. 2024. "Revolutionizing Cancer Treatments through Stem Cell-Derived CAR T Cells for Immunotherapy: Opening New Horizons for the Future of Oncology" Cells 13, no. 18: 1516. https://doi.org/10.3390/cells13181516
APA StyleMishra, H. K., & Kalyuzhny, A. (2024). Revolutionizing Cancer Treatments through Stem Cell-Derived CAR T Cells for Immunotherapy: Opening New Horizons for the Future of Oncology. Cells, 13(18), 1516. https://doi.org/10.3390/cells13181516