
C1orf106 (INAVA) Is a SMAD3-Dependent TGF-β Target Gene That Promotes Clonogenicity and Correlates with Poor Prognosis in Breast Cancer

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Plasmids, siRNAs and Generation of Stable Cell Lines
2.3. Measurement of Gene and Protein Expression
2.4. Migration and Invasion Assays
2.5. Colony Formation and Clonogenicity Assays
2.6. Mammary Fat Pad Tumour Initiation
2.7. Survival Analysis of Clinical Datasets
2.8. Statistical Analysis
3. Results
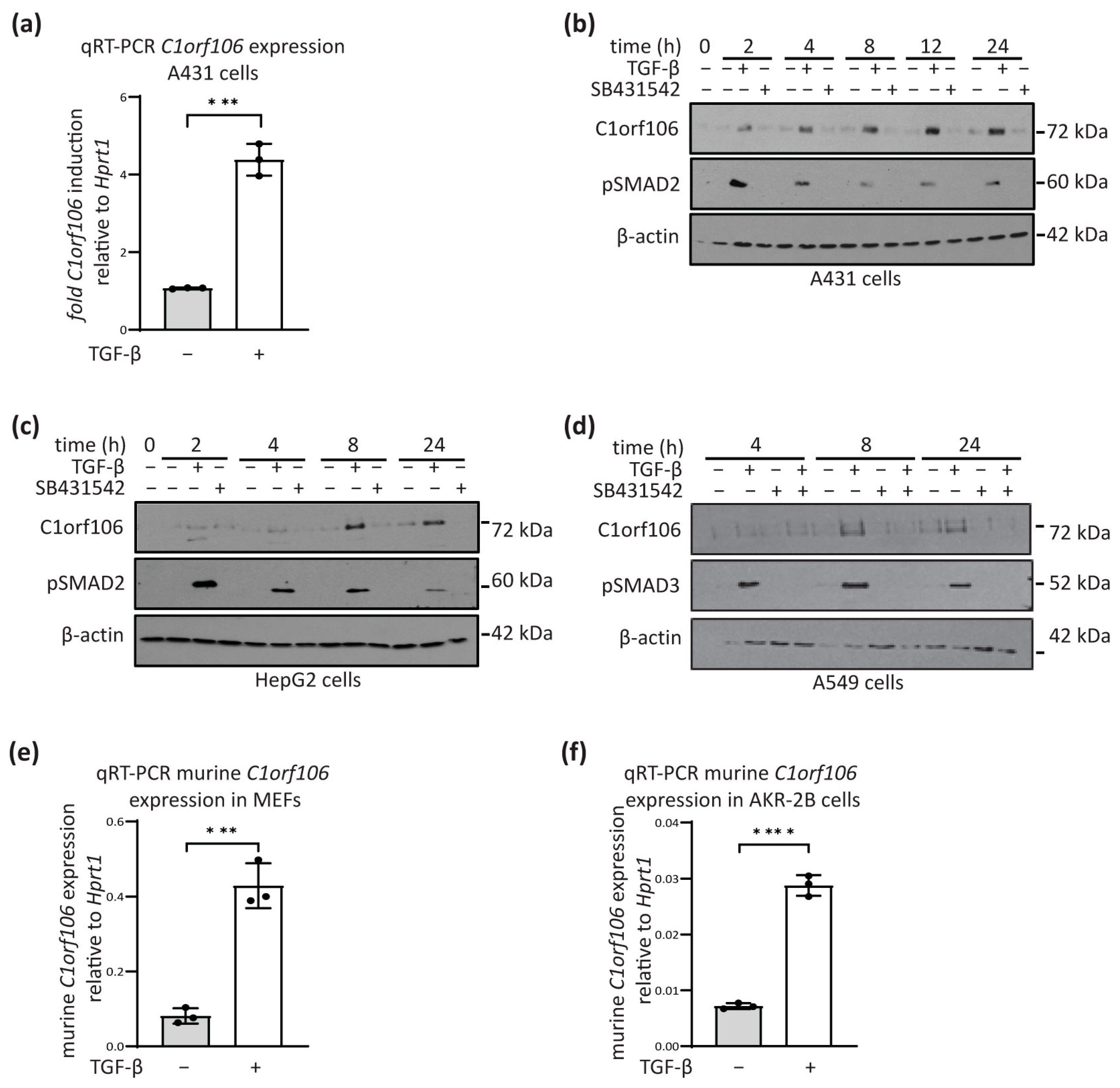
3.1. C1orf106 Is a TGF-β Target Gene in Human and Mouse Cells
3.2. C1orf106 Induction by TGF-β Is SMAD3-Dependent
3.3. C1orf106 Expression Increases with Tumourigenic or Metastatic Potential in Breast Cancer Cell Line Models and Is a Mediator of Pro-Migratory and Pro-Invasive TGF-β Signalling
3.4. C1orf106 Enhances Clonogenicity In Vitro and Tumour Initiation Frequency In Vivo
3.5. High C1orf106 Expression Is Associated with Poor Clinical Outcome in Breast Cancer Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, A.; Smith, P.; Kalna, G.; Lo Nigro, C.; Orange, C.; O’Brien, D.I.; Shah, R.; Syed, N.; Spender, L.C.; Herrera, B.; et al. Epigenetic downregulation of human disabled homolog 2 switches TGF-beta from a tumor suppressor to a tumor promoter. J. Clin. Investig. 2010, 120, 2842–2857. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Sheppard, D. TGF-beta signaling in health and disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Nicolas, F.J.; Hill, C.S. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol. Cell 2002, 10, 283–294. [Google Scholar] [CrossRef]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Inman, G.J. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr. Opin. Genet. Dev. 2011, 21, 93–99. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Bottinger, E.P.; Jakubczak, J.L.; Haines, D.C.; Bagnall, K.; Wakefield, L.M. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor beta receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 1997, 57, 5564–5570. [Google Scholar]
- Gorska, A.E.; Jensen, R.A.; Shyr, Y.; Aakre, M.E.; Bhowmick, N.A.; Moses, H.L. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-beta receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am. J. Pathol. 2003, 163, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, R.S.; Koh, Y.; Roebuck, L.R.; Sanders, M.E.; Brantley-Sieders, D.; Gorska, A.E.; Moses, H.L.; Arteaga, C.L. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol. Cell Biol. 2003, 23, 8691–8703. [Google Scholar] [CrossRef] [PubMed]
- Pierce, D.F., Jr.; Gorska, A.E.; Chytil, A.; Meise, K.S.; Page, D.L.; Coffey, R.J., Jr.; Moses, H.L. Mammary tumor suppression by transforming growth factor beta 1 transgene expression. Proc. Natl. Acad. Sci. USA 1995, 92, 4254–4258. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massague, J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massague, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massague, J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef]
- Tang, B.; Vu, M.; Booker, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef]
- Dalal, B.I.; Keown, P.A.; Greenberg, A.H. Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am. J. Pathol. 1993, 143, 381–389. [Google Scholar]
- Gorsch, S.M.; Memoli, V.A.; Stukel, T.A.; Gold, L.I.; Arrick, B.A. Immunohistochemical staining for transforming growth factor beta 1 associates with disease progression in human breast cancer. Cancer Res. 1992, 52, 6949–6952. [Google Scholar]
- Liu, Y.; Xu, H.; Chen, S.; Chen, X.; Zhang, Z.; Zhu, Z.; Qin, X.; Hu, L.; Zhu, J.; Zhao, G.P.; et al. Genome-wide interaction-based association analysis identified multiple new susceptibility Loci for common diseases. PLoS Genet. 2011, 7, e1001338. [Google Scholar] [CrossRef]
- Rivas, M.A.; Beaudoin, M.; Gardet, A.; Stevens, C.; Sharma, Y.; Zhang, C.K.; Boucher, G.; Ripke, S.; Ellinghaus, D.; Burtt, N.; et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011, 43, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hedl, M.; Abraham, C. An inflammatory bowel disease-risk variant in INAVA decreases pattern recognition receptor-induced outcomes. J. Clin. Investig. 2017, 127, 2192–2205. [Google Scholar] [CrossRef] [PubMed]
- Manzanillo, P.; Mouchess, M.; Ota, N.; Dai, B.; Ichikawa, R.; Wuster, A.; Haley, B.; Alvarado, G.; Kwon, Y.; Caothien, R.; et al. Inflammatory Bowel Disease Susceptibility Gene C1ORF106 Regulates Intestinal Epithelial Permeability. Immunohorizons 2018, 2, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, V.; Nakata, T.; Desch, A.N.; Levesque, C.; Boroughs, A.; Guzman, G.; Cao, Z.; Creasey, E.; Yao, J.; Boucher, G.; et al. C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science 2018, 359, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, C.; Yang, D.; Song, J.; Zhang, J.; Wang, T.; Wang, M.; Xu, W.; Li, X.; Ding, S.; et al. C1orf106, an innate immunity activator, is amplified in breast cancer and is required for basal-like/luminal progenitor fate decision. Sci. China Life Sci. 2019, 62, 1229–1242. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef]
- Jezequel, P.; Campone, M.; Gouraud, W.; Guerin-Charbonnel, C.; Leux, C.; Ricolleau, G.; Campion, L. bc-GenExMiner: An easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res. Treat. 2012, 131, 765–775. [Google Scholar] [CrossRef]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar]
- Zhao, M.; Yang, X.; Fu, Y.; Wang, H.; Ning, Y.; Yan, J.; Chen, Y.G.; Wang, G. Mediator MED15 modulates transforming growth factor beta (TGFbeta)/Smad signaling and breast cancer cell metastasis. J. Mol. Cell Biol. 2013, 5, 57–60. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Smad4 dependency defines two classes of transforming growth factor beta (TGF-beta) target genes and distinguishes TGF-beta-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Isogaya, K.; Koinuma, D.; Tsutsumi, S.; Saito, R.A.; Miyazawa, K.; Aburatani, H.; Miyazono, K. A Smad3 and TTF-1/NKX2-1 complex regulates Smad4-independent gene expression. Cell Res. 2014, 24, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Morikawa, M.; Ren, J.; Vasilaki, E.; Kawasaki, N.; Kobayashi, M.; Koinuma, D.; Aburatani, H.; Miyazono, K.; Heldin, C.H.; et al. JUNB governs a feed-forward network of TGFbeta signaling that aggravates breast cancer invasion. Nucleic Acids Res. 2018, 46, 1180–1195. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, C.N.; Smith, Y.E.; Cao, Y.; Burrows, A.D.; Cross, R.S.; Ling, X.; Redvers, R.P.; Doherty, J.P.; Eckhardt, B.L.; Natoli, A.L.; et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Model. Mech. 2015, 8, 237–251. [Google Scholar] [CrossRef]
- Guan, H.; Guo, Y.; Liu, L.; Ye, R.; Liang, W.; Li, H.; Xiao, H.; Li, Y. INAVA promotes aggressiveness of papillary thyroid cancer by upregulating MMP9 expression. Cell Biosci. 2018, 8, 26. [Google Scholar] [CrossRef]
- Wiercinska, E.; Naber, H.P.; Pardali, E.; van der Pluijm, G.; van Dam, H.; ten Dijke, P. The TGF-beta/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res. Treat. 2011, 128, 657–666. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Umeda, M. FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc. Natl. Acad. Sci. USA 2010, 107, 748–753. [Google Scholar] [CrossRef]
- Huang, N.; Xia, Y.; Zhang, D.; Wang, S.; Bao, Y.; He, R.; Teng, J.; Chen, J. Hierarchical assembly of centriole subdistal appendages via centrosome binding proteins CCDC120 and CCDC68. Nat. Commun. 2017, 8, 15057. [Google Scholar] [CrossRef]
- Luong, P.; Hedl, M.; Yan, J.; Zuo, T.; Fu, T.M.; Jiang, X.; Thiagarajah, J.R.; Hansen, S.H.; Lesser, C.F.; Wu, H.; et al. INAVA-ARNO complexes bridge mucosal barrier function with inflammatory signaling. eLife 2018, 7, e38539. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strathearn, L.S.; Spender, L.C.; Schoenherr, C.; Mason, S.; Edwards, R.; Blyth, K.; Inman, G.J. C1orf106 (INAVA) Is a SMAD3-Dependent TGF-β Target Gene That Promotes Clonogenicity and Correlates with Poor Prognosis in Breast Cancer. Cells 2024, 13, 1530. https://doi.org/10.3390/cells13181530
Strathearn LS, Spender LC, Schoenherr C, Mason S, Edwards R, Blyth K, Inman GJ. C1orf106 (INAVA) Is a SMAD3-Dependent TGF-β Target Gene That Promotes Clonogenicity and Correlates with Poor Prognosis in Breast Cancer. Cells. 2024; 13(18):1530. https://doi.org/10.3390/cells13181530
Chicago/Turabian StyleStrathearn, Lauren S., Lindsay C. Spender, Christina Schoenherr, Susan Mason, Ruaridh Edwards, Karen Blyth, and Gareth J. Inman. 2024. "C1orf106 (INAVA) Is a SMAD3-Dependent TGF-β Target Gene That Promotes Clonogenicity and Correlates with Poor Prognosis in Breast Cancer" Cells 13, no. 18: 1530. https://doi.org/10.3390/cells13181530