

Pravastatin Protects Cytotrophoblasts from Hyperglycemia-Induced Preeclampsia Phenotype

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Cell Treatments
2.3. Primers and Reagents
2.4. Real-Time PCR
2.5. Migration Assay
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Statistical Analysis
3. Results
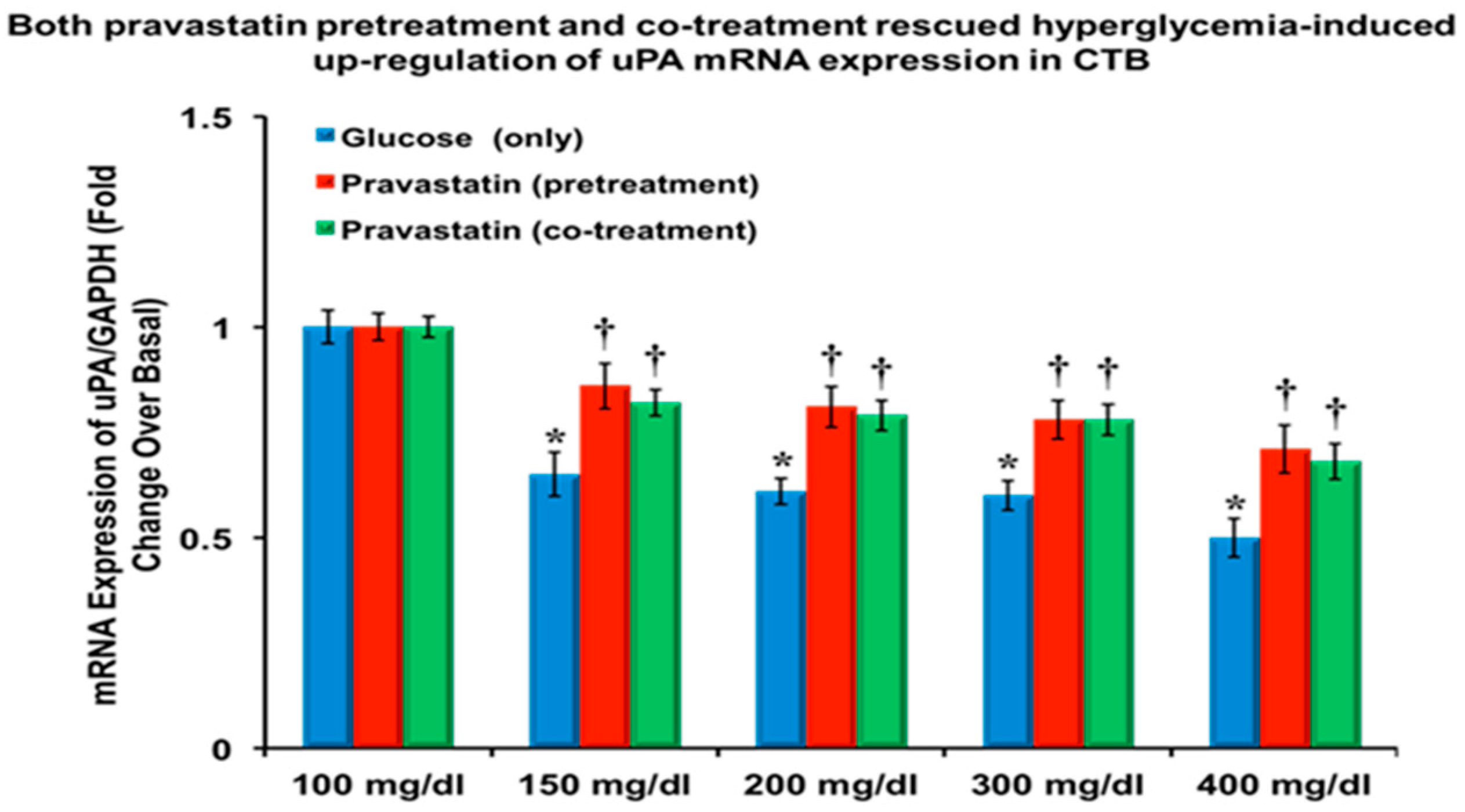
3.1. Pravastatin Attenuated the Hyperglycemia-Induced Downregulation of uPA and PAI-1, and mRNA Expression
3.2. Pravastatin Attenuated the Hyperglycemia-Induced Reduction in VEGF and PlGF
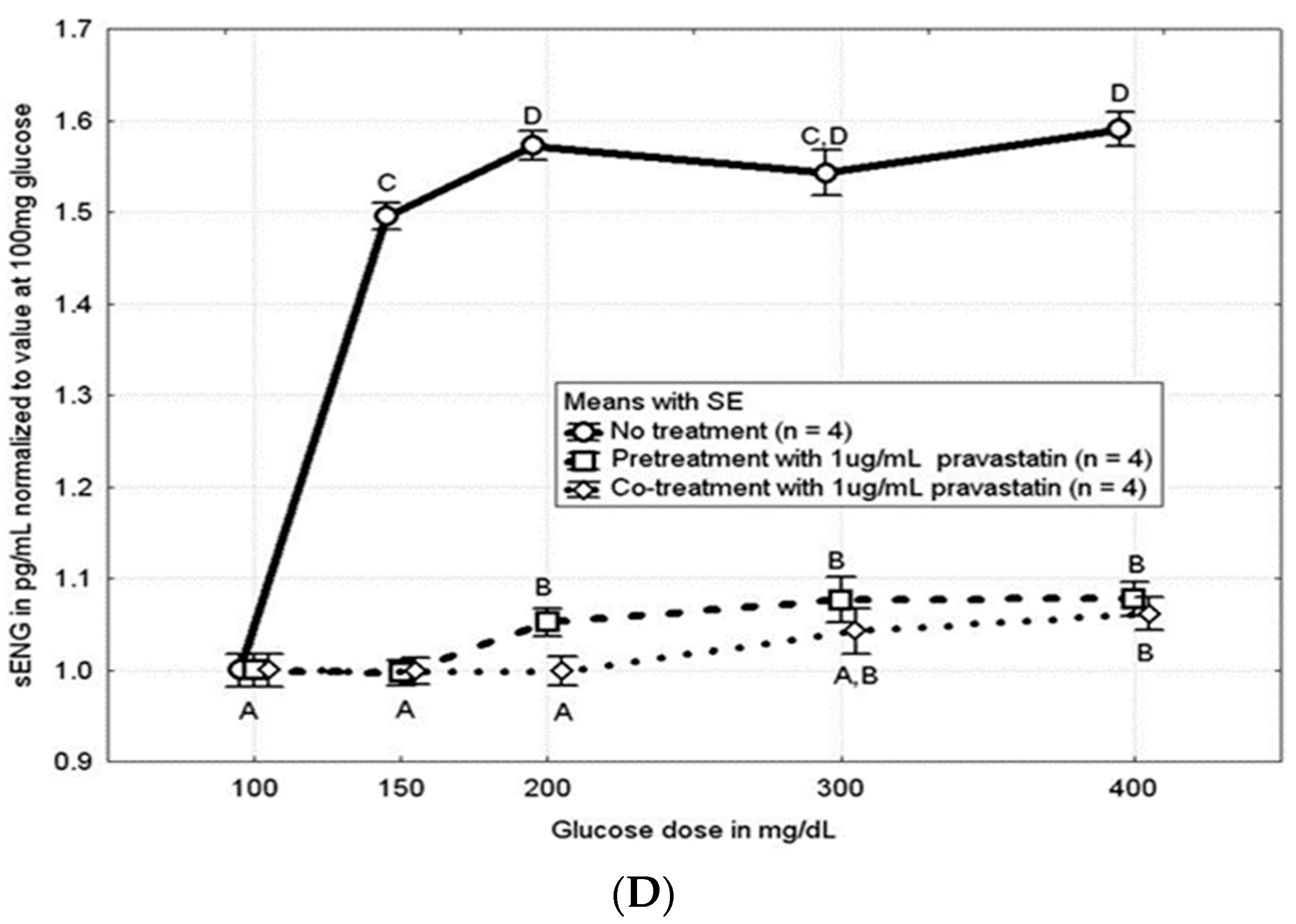
3.3. Pravastatin Attenuated the Hyperglycemia-Induced Increase in sFLT and sENG
3.4. Pravastatin Attenuated the Hyperglycemia-Induced Loss of CTB Cell Migration
3.5. Effects of Pravastatin on Pathways of Abnormal Placentation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, U.D.; Olsson, M.G.; Kristensen, K.H.; Åkerström, B.; Hansson, S.R. Review: Biochemical markers to predict preeclampsia. Placenta 2012, 33, S42–S47. [Google Scholar] [CrossRef] [PubMed]
- Pridjian, G.; Puschett, J.B. Preeclampsia. Part I: Clinical and pathophysiological considerations. Obstet. Gynecol. Surv. 2002, 57, 598–618. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Allen, S.; Jones, R.; Zawieja, D.; Kuehl, T. Pathogenesis of preeclampsia: Marinobufagenin and angiogenic imbalance as biomarkers of the syndrome. Transl. Res. 2012, 160, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jenkins, A.J.; Nankervis, A.J.; Hanssen, K.F.; Scholz, H.; Henriksen, T.; Lorentzen, B.; Clausen, T.; Garg, S.K.; Menard, M.K.; et al. Anti-angiogenic factors and pre-eclampsia in type 1 diabetic women. Diabetologia 2009, 52, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Beeram, M.R.; Kuehl, T.J. Diabetes Mellitus and Preeclampsia. Med. J. Obstet. Gynecol. 2013, 1, 1016. [Google Scholar]
- Giannubilo, S.R.; Marzioni, D.; Tossetta, G.; Montironi, R.; Meccariello, M.L.; Ciavattini, A. The “Bad Father”: Paternal Role in Biology of Pregnancy and in Birth Outcome. Biology 2024, 13, 165. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gesuita, R.; Licini, C.; Picchiassi, E.; Tarquini, F.; Coata, G.; Fantone, S.; Tossetta, G.; Ciavattini, A.; Castellucci, M.; Di Renzo, G.C.; et al. Association between first trimester plasma htra1 level and subsequent preeclampsia: A possible early marker? Pregnancy Hypertens. 2019, 18, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Damsky, C.H.; Chiu, K.; Roberts, J.M.; Fisher, S.J. Preeclampsia is associated with abnormal expression of adhesion molecules by invasive cytotrophoblasts. J. Clin. Investig. 1993, 91, 950–960. [Google Scholar] [CrossRef]
- Zhou, Y.; Fisher, S.J.; Janatpour, M.; Genbacev, O.; Dejana, E.; Wheelock, M.; Damsky, C.H. Human cytotrophoblasts adopt a vascular phenotype as they differentiate: A strategy for successful endovascular invasion? J. Clin. Investig. 1997, 99, 2139–2151. [Google Scholar] [CrossRef]
- Harris, L.K.; Keogh, R.J.; Wareing, M.; Baker, P.N.; Cartwright, J.E.; Aplin, J.D.; Whitley, G.S.J. Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am. J. Pathol. 2006, 169, 1863–1874. [Google Scholar] [CrossRef]
- Brosens, I.A.; Robertson, W.B.; Dixon, H.G. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet. Gynecol. Annu. 1972, 1, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Cawyer, C.R.; Horvat, D.; Leonard, D.; Allen, S.R.; Jones, R.O.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Hyperglycemia impairs cytotrophoblast function via stress signaling. Am. J. Obstet. Gynecol. 2014, 211, 541.e1–541.e8. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.G.; Kenneth, J.L.; Bloom, S.L.; Hauth, J.C.; Rouse, D.J.; Spong, C.Y. Implantation, Embryogenesis, and Placental Development. In Williams Obstetrics, 23rd ed.; McGraw-Hill: New York, NY, USA, 2010; pp. 36–77. [Google Scholar]
- Chapman, H.A. Plasminogen activators, integrins and the coordinated regulation of cell adhesion and migration. Curr. Opin. Cell Biol. 1997, 9, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Waltz, D.A.; Natkin, L.R.; Fujita, R.M.; Wei, Y.; Chapman, H.A. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J. Clin. Investig. 1997, 100, 58–67. [Google Scholar] [CrossRef]
- Feng, Q.; Liu, Y.; Liu, K.; Byrne, S.; Liu, G.; Wang, X.; Li, Z.; Ockleford, C. Expression of the urokinase, plasminogen activator inhibitors and urokinase receptor in pregnant rhesus monkey uterus during early placentation. Placenta 2000, 21, 184–193. [Google Scholar] [CrossRef]
- Vayalil, P.K.; Iles, K.E.; Choi, J.; Yi, A.K.; Postlethwait, E.M.; Liu, R.M. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1281–L1292. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signaling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Garrington, T.P.; Johnson, G.L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 1999, 11, 211–218. [Google Scholar] [CrossRef]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Nebreda, A.R.; Porras, A. p38 MAP kinases: Beyond the stress response. Trends Biochem. Sci. 2000, 25, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.N.; Horvat, D.; Glaser, S.S.; Mitchell, B.M.; Puschett, J.B. Examination of the cellular mechanisms by which marinobufagenin inhibits cytotrophoblast function. J. Biol. Chem. 2008, 283, 17946–17953. [Google Scholar] [CrossRef] [PubMed]
- Unek, G.; Ozmen, A.; Mendilcioglu, I.; Simsek, M.; Korgun, E. The expression of cell cycle related proteins PCNA, Ki67, p27 and p57 in normal and preeclamptic human placentas. Tissue Cell 2014, 46, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Costantine, M.M.; Cleary, K. Pravastatin for the Prevention of Preeclampsia in High-Risk Pregnant Women. Obstet. Gynecol. 2013, 121, 349–353. [Google Scholar] [CrossRef]
- Cleary, K.L.; Roney, K.; Costatine, M. Challenges of studying drugs in pregnancy for off-label indications: Pravastatin for preeclampsia prevention. Semin. Perinatol. 2014, 38, 523–527. [Google Scholar] [CrossRef]
- Lefkou, E.; Mamopoulos, A.; Fragakis, N. Clinical Improvement and Successful Pregnancy in a Preeclamptic Patient with Antiphospholipid Syndrome Treated with Pravastatin. Hypertension 2014, 63, e118–e119. [Google Scholar] [CrossRef]
- Gangooly, S.; Muttukrishna, S.; Jauniaux, E. In-vitro Study of the effect of anti-hypertensive drugs on placental hormones and angiogenic proteins synythesis in pre-eclampsia. PLoS ONE 2014, 9, e107644. [Google Scholar] [CrossRef]
- Kumasawa, K.; Ikawa, M.; Kidoya, H.; Hasuwa, H.; Saito-Fujita, T.; Morioka, Y.; Takakura, N.; Kimura, T.; Okabe, M. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc. Natl. Acad. Sci. USA 2011, 108, 1451–1455. [Google Scholar] [CrossRef]
- Forbes, K.; Shah, V.; Siddals, K.; Gibson, J.M.; Aplin, J.; Westwood, M. Statins inhibit insulin-like growth factor action in first trimester placenta by altering insulin-like growth factor I receptor glycosylation. Mol. Hum. Reprod. 2015, 21, 105–114. [Google Scholar] [CrossRef]
- Hatanaka, T. Clinical pharmacokinetics of pravastatin: Mechanisms of pharmacokinetic events. Clin. Pharmacokinet. 2000, 39, 397–412. [Google Scholar] [CrossRef]
- Nanovskaya, T.N.; Patrikeeva, S.L.; Paul, J.; Costantine, M.M.; Hankins, G.D.; Ahmed, M.S. Transplacental transfer and distribution of pravastatin. Am. J. Obstet. Gynecol. 2013, 209, 373.e1–373.e5. [Google Scholar] [CrossRef] [PubMed]
- Zarek, J.; DeGorter, M.; Lubetsky, A.; Kim, R.; Laskin, C.; Berger, H.; Koren, G. The transfer of pravastatin in the dually perfused human placenta. Placenta 2013, 34, 719–721. [Google Scholar] [CrossRef] [PubMed]
- FDA. Fetal risk summary. Fed. Regist. 1980, 44, 37434–37467. [Google Scholar]
- Petersen, E.E.; Mitchell, A.A.; Carey, J.C.; Werler, M.M.; Louik, C.; Rasmussen, S.A. Maternal exposure to statins and risk for birth defects: A case-series approach. Am. J. Med. Genet. A 2008, 146A, 2701–2705. [Google Scholar] [CrossRef]
- Edison, R.; Muenke, M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am. J. Med. Gen. 2004, 131A, 287. [Google Scholar] [CrossRef]
- Kazmin, A.; Garcia-Bournissen, F.; Koren, G. Risk of statin use during pregnancy: A systematic review. J. Obstet. Gynecol. Can. 2007, 29, 906. [Google Scholar] [CrossRef]
- Fisher, S.J. The placental problem: Linking abnormal cytotrophoblast differentiation to the maternal symptoms of preeclampsia. Reprod. Biol. Endocrinol. 2004, 2, 53. [Google Scholar] [CrossRef]
- Myatt, L. Role of placenta in preeclampsia. Endocrine 2002, 19, 103–111. [Google Scholar] [CrossRef]
- Zhou, L.; Xue, H.; Wang, Z.; Ni, J.; Yao, T.; Huang, Y.; Yu, C.; Lu, L. Angiotensin-(1–7) attenuates high glucose-induced proximal tubular epithelial-to-mesenchymal transition via inhibiting ERK1/2 and p38 phosphorylation. Life Sci. 2012, 90, 454–462. [Google Scholar] [CrossRef]
- Desoye, G.; Mouzon, S.H.; Shafrir, E. The placenta in diabetic pregnancy. In Textbook of Diabetes and Pregnancy; Hod, M., Jovanovic, L., Di Renzo, G.C., Leiva, A., Langer, O., Eds.; Martin Dunitz: London, UK, 2003; pp. 126–143. [Google Scholar]
- Smith, D.D.; Costantine, M.M. The role of statins in the prevention of preeclampsia. Am. J. Obstet. Gynecol. 2022, 226 (Suppl. S2), S1171–S1181. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mészáros, B.; Veres, D.S.; Nagyistók, L.; Somogyi, A.; Rosta, K.; Herold, Z.; Kukor, Z.; Valent, S. Pravastatin in preeclampsia: A meta-analysis and systematic review. Front. Med. 2023, 9, 1076372. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantho, A.F.; Mohamed, S.; Govande, J.V.; Rane, R.; Vora, N.; Kelso, K.R.; Kuehl, T.J.; Lindheim, S.R.; Uddin, M.N. Pravastatin Protects Cytotrophoblasts from Hyperglycemia-Induced Preeclampsia Phenotype. Cells 2024, 13, 1534. https://doi.org/10.3390/cells13181534
Pantho AF, Mohamed S, Govande JV, Rane R, Vora N, Kelso KR, Kuehl TJ, Lindheim SR, Uddin MN. Pravastatin Protects Cytotrophoblasts from Hyperglycemia-Induced Preeclampsia Phenotype. Cells. 2024; 13(18):1534. https://doi.org/10.3390/cells13181534
Chicago/Turabian StylePantho, Ahmed F., Sara Mohamed, Janhavi V. Govande, Riddhi Rane, Niraj Vora, Kelsey R. Kelso, Thomas J. Kuehl, Steven R. Lindheim, and Mohammad N. Uddin. 2024. "Pravastatin Protects Cytotrophoblasts from Hyperglycemia-Induced Preeclampsia Phenotype" Cells 13, no. 18: 1534. https://doi.org/10.3390/cells13181534