
The Interactions of T Cells with Myeloid-Derived Suppressor Cells in Peripheral Blood Stem Cell Grafts

 and
and
Abstract
:1. Introduction
2. Methods
2.1. Mobilization and Collection of PBSCs
2.2. Graft Analysis
2.3. T Cell Proliferation and Cytokine Secretion from Graft Cells
2.4. Effects of MDSC Subsets on the Proliferation and Expansion of T Cells
2.5. Statistical Analysis
3. Results
3.1. Variation between Individual Grafts and between Different Graft Sources
3.2. Characterization of T Cells in the Grafts
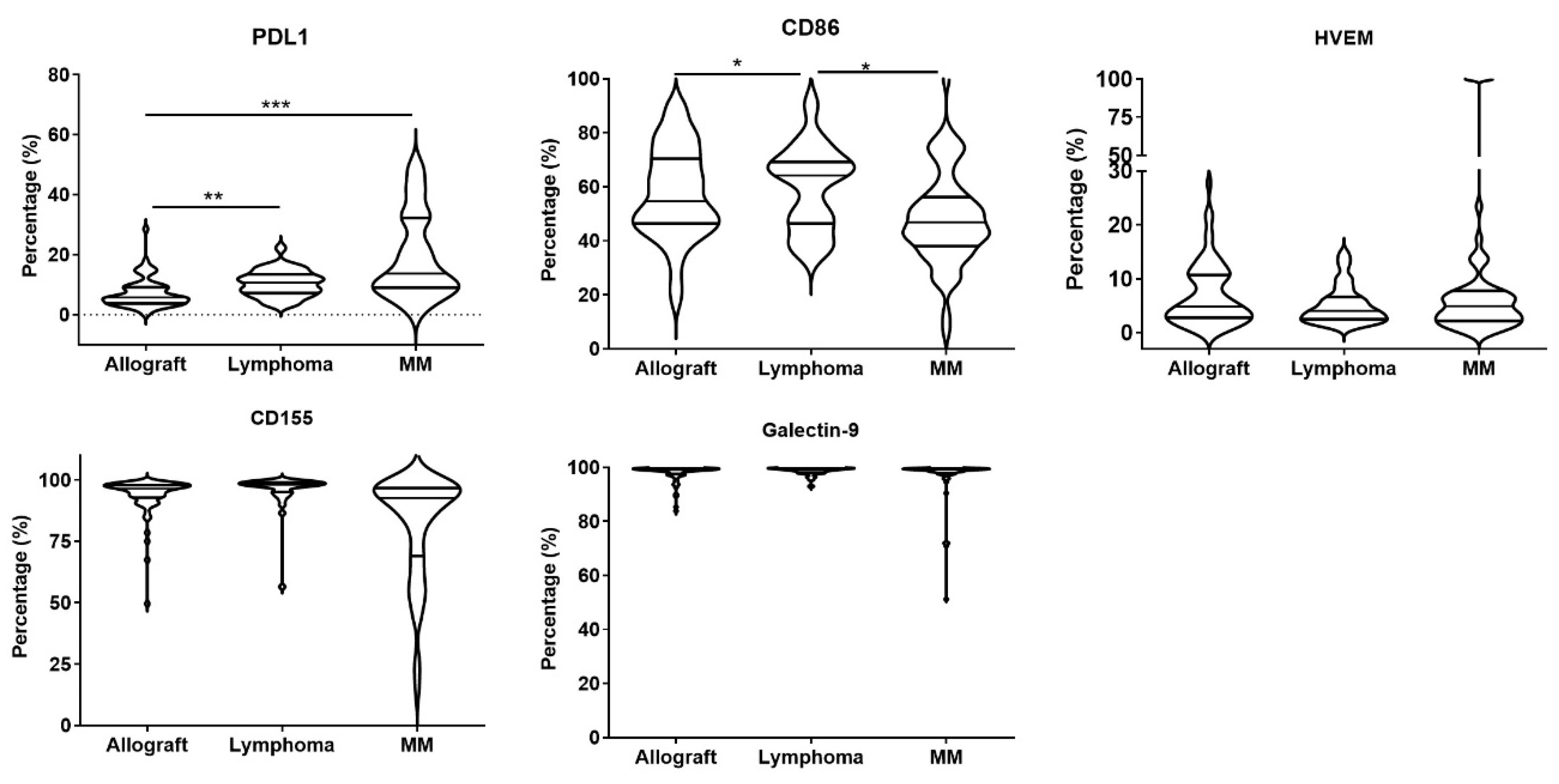
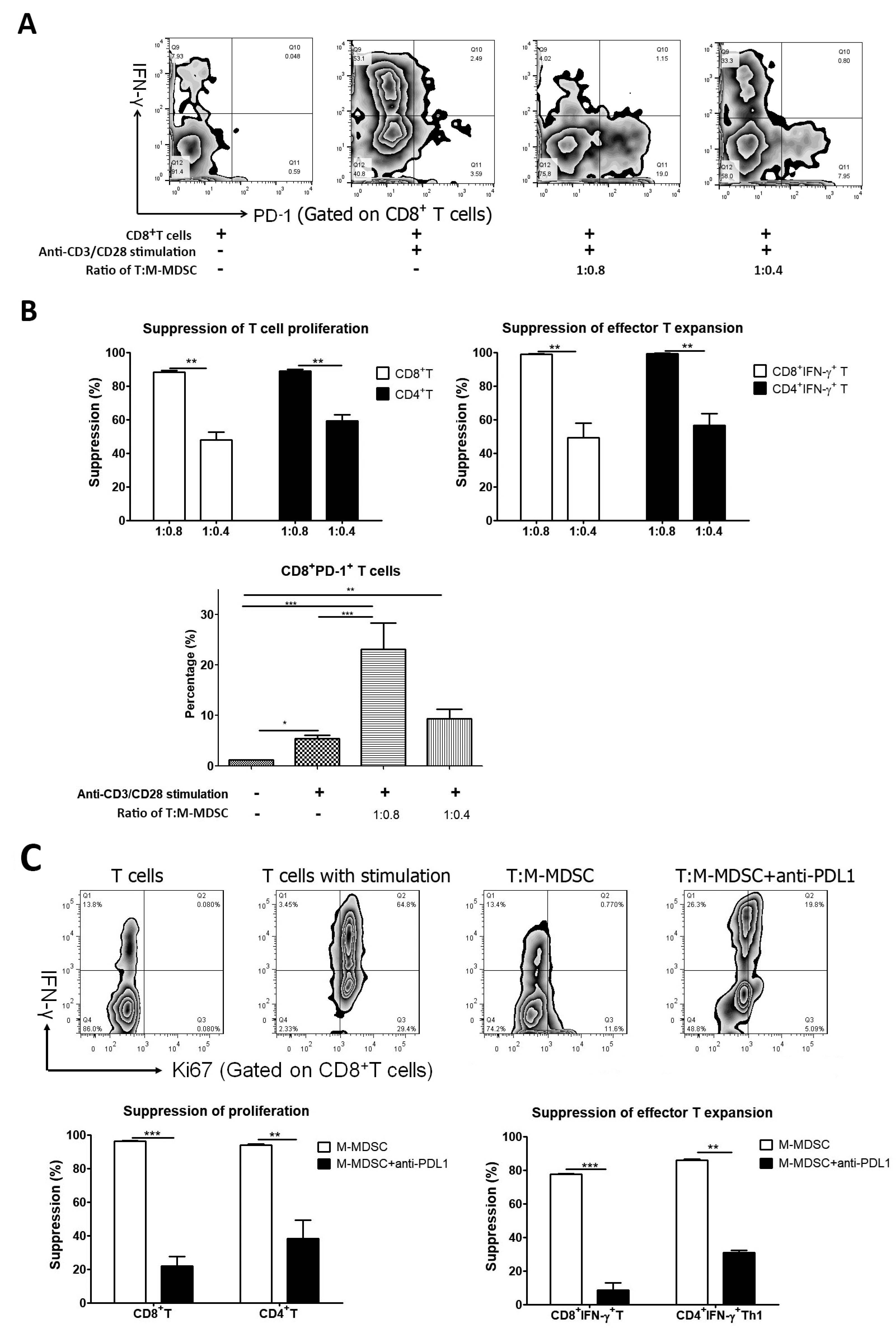
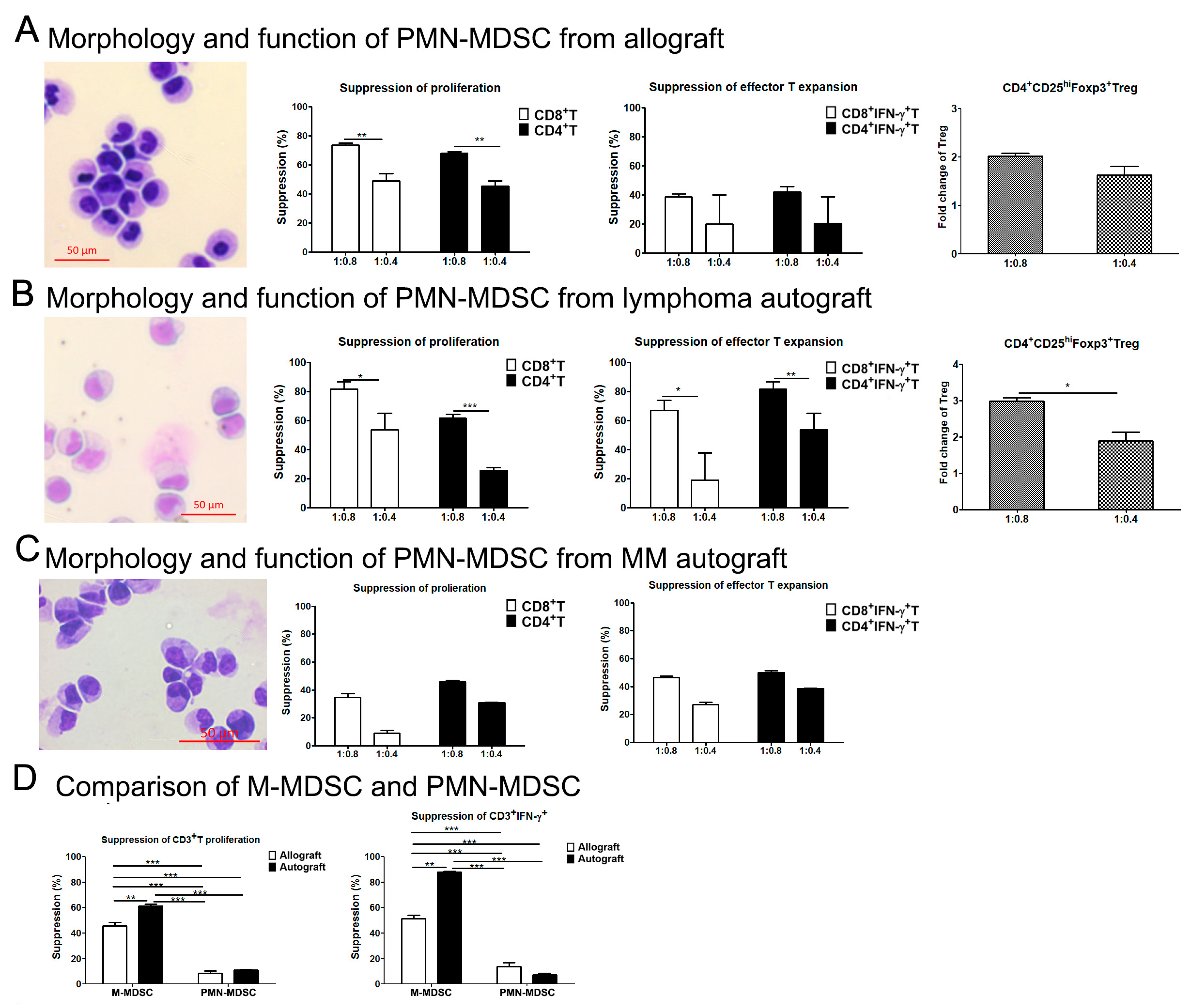
3.3. M-MDSCs Expressed Multiple Ligands of Inhibitory Receptors at Variable Levels and Had Immune Regulatory Function
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hopman, R.K.; DiPersio, J.F. Advances in stem cell mobilization. Blood Rev. 2014, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Balint, M.T.; Lemajic, N.; Jurisic, V.; Pantelic, S.; Stanisavljevic, D.; Kurtovic, N.K.; Balint, B. An evidence-based and risk-adapted GSF versus GSF plus plerixafor mobilization strategy to obtain a sufficient CD34(+) cell yield in the harvest for autologous stem cell transplants. Transl. Oncol. 2024, 39, 101811. [Google Scholar] [CrossRef] [PubMed]
- Saraceni, F.; Shem-Tov, N.; Olivieri, A.; Nagler, A. Mobilized peripheral blood grafts include more than hematopoietic stem cells: The immunological perspective. Bone Marrow Transplant. 2015, 50, 886–891. [Google Scholar] [CrossRef]
- Guan, Q.; Blankstein, A.R.; Anjos, K.; Synova, O.; Tulloch, M.; Giftakis, A.; Yang, B.; Lambert, P.; Peng, Z.; Cuvelier, G.D.; et al. Functional Myeloid-Derived Suppressor Cell Subsets Recover Rapidly after Allogeneic Hematopoietic Stem/Progenitor Cell Transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1205–1214. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Guan, Q.; Yang, B.; Warrington, R.J.; Mink, S.; Kalicinsky, C.; Becker, A.B.; Simons, E.; Peng, Z. Myeloid-derived suppressor cells: Roles and relations with Th2, Th17, and Treg cells in asthma. Allergy 2019, 74, 2233–2237. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.A.; DiPersio, J.F. Mouse models of graft-versus-host disease: Advances and limitations. Dis. Model. Mech. 2011, 4, 318–333. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.M.; Ma, G.; Zhou, Z.; Raulet, D.; Rivera, A.L.; Chen, S.H.; Pan, P.Y. The mechanistic study behind suppression of GVHD while retaining GVL activities by myeloid-derived suppressor cells. Leukemia 2019, 33, 2078–2089. [Google Scholar] [CrossRef]
- Vendramin, A.; Gimondi, S.; Bermema, A.; Longoni, P.; Rizzitano, S.; Corradini, P.; Carniti, C. Graft monocytic myeloid-derived suppressor cell content predicts the risk of acute graft-versus-host disease after allogeneic transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood stem cells. Biol. Blood Marrow Transplant. 2014, 20, 2049–2055. [Google Scholar] [CrossRef]
- Lv, M.; Zhao, X.S.; Hu, Y.; Chang, Y.J.; Zhao, X.Y.; Kong, Y.; Zhang, X.H.; Xu, L.P.; Liu, K.Y.; Huang, X.J. Monocytic and promyelocytic myeloid-derived suppressor cells may contribute to G-CSF-induced immune tolerance in haplo-identical allogeneic hematopoietic stem cell transplantation. Am. J. Hematol. 2015, 90, E9–E16. [Google Scholar] [CrossRef] [PubMed]
- Kansagra, A.; Inwards, D.J.; Ansell, S.M.; Micallef, I.N.; Johnston, P.B.; Hogan, W.J.; Markovic, S.N.; Porrata, L.F. Infusion of autograft natural killer cell/CD14(+)HLA-DR(DIM) cell ratio predicts survival in lymphoma post autologous stem cell transplantation. Bone Marrow Transplant. 2018, 53, 146–154. [Google Scholar] [CrossRef]
- Porrata, L.F.; Inwards, D.J.; Ansell, S.M.; Micallef, I.N.; Johnston, P.B.; Villasboas, J.C.; Markovic, S.N. Autograft immune content and survival in non-Hodgkin’s lymphoma: A post hoc analysis. Leuk. Res. 2019, 81, 1–9. [Google Scholar] [CrossRef]
- Godwin, C.D.; Fromm, J.R.; Othus, M.; Sandmaier, B.M.; Mielcarek, M.B.; Wood, B.L.; Appelbaum, F.R.; Storb, R.; Walter, R.B. Pre-transplant bone marrow monocytic myeloid-derived suppressor cell frequency is not associated with outcome after allogeneic hematopoietic cell transplantation for acute myeloid leukemia in remission. Bone Marrow Transplant. 2019, 54, 1511–1514. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Patil, S.; Rao, R.S.; Majumdar, B. T-cell Exhaustion and Cancer Immunotherapy. J. Int. Oral. Health 2015, 7, i–ii. [Google Scholar] [PubMed]
- Liu, L.; Chang, Y.J.; Xu, L.P.; Zhang, X.H.; Wang, Y.; Liu, K.Y.; Huang, X.J. Reversal of T Cell Exhaustion by the First Donor Lymphocyte Infusion Is Associated with the Persistently Effective Antileukemic Responses in Patients with Relapsed AML after Allo-HSCT. Biol. Blood Marrow Transplant. 2018, 24, 1350–1359. [Google Scholar] [CrossRef]
- Chung, D.J.; Pronschinske, K.B.; Shyer, J.A.; Sharma, S.; Leung, S.; Curran, S.A.; Lesokhin, A.M.; Devlin, S.M.; Giralt, S.A.; Young, J.W. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer Immunol. Res. 2016, 4, 61–71. [Google Scholar] [CrossRef]
- Minnie, S.A.; Kuns, R.D.; Gartlan, K.H.; Zhang, P.; Wilkinson, A.N.; Samson, L.; Guillerey, C.; Engwerda, C.; MacDonald, K.P.A.; Smyth, M.J.; et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018, 132, 1675–1688. [Google Scholar] [CrossRef]
- Porrata, L.F.; Inwards, D.J.; Ansell, S.M.; Micallef, I.N.; Johnston, P.B.; Hogan, W.J.; Markovic, S.N. Infused autograft lymphocyte-to-monocyte ratio and survival in T-cell lymphoma post-autologous peripheral blood hematopoietic stem cell transplantation. J. Hematol. Oncol. 2015, 8, 80. [Google Scholar] [CrossRef]
- Porrata, L.F.; Inwards, D.J.; Ansell, S.M.; Micallef, I.N.; Johnston, P.B.; Hogan, W.J.; Markovic, S.N. Infused autograft lymphocyte to monocyte ratio predicts survival in classical Hodgkin lymphoma. J. Blood Med. 2015, 6, 45–53. [Google Scholar] [CrossRef]
- Sutherland, D.R.; Anderson, L.; Keeney, M.; Nayar, R.; Chin-Yee, I. The ISHAGE guidelines for CD34+ cell determination by flow cytometry. International Society of Hematotherapy and Graft Engineering. J. Hematother 1996, 5, 213–226. [Google Scholar] [CrossRef]
- Mullen, K.M.; Gocke, A.R.; Allie, R.; Ntranos, A.; Grishkan, I.V.; Pardo, C.; Calabresi, P.A. Expression of CCR7 and CD45RA in CD4+ and CD8+ subsets in cerebrospinal fluid of 134 patients with inflammatory and non-inflammatory neurological diseases. J. Neuroimmunol. 2012, 249, 86–92. [Google Scholar] [CrossRef]
- Guan, Q.; Moreno, S.; Qing, G.; Weiss, C.R.; Lu, L.; Bernstein, C.N.; Warrington, R.J.; Ma, Y.; Peng, Z. The role and potential therapeutic application of myeloid-derived suppressor cells in TNBS-induced colitis. J. Leukoc. Biol. 2013, 94, 803–811. [Google Scholar] [CrossRef]
- Guan, Q.; Li, Y.; Shpiruk, T.; Bhagwat, S.; Wall, D.A. Inducible indoleamine 2,3-dioxygenase 1 and programmed death ligand 1 expression as the potency marker for mesenchymal stromal cells. Cytotherapy 2018, 20, 639–649. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Jurisic, V.; Colovic, M. Correlation of sera TNF-alpha with percentage of bone marrow plasma cells, LDH, beta2-microglobulin, and clinical stage in multiple myeloma. Med. Oncol. 2002, 19, 133–139. [Google Scholar] [CrossRef]
- Wang, K.; Lv, M.; Chang, Y.J.; Zhao, X.Y.; Zhao, X.S.; Zhang, Y.Y.; Sun, Y.Q.; Wang, Z.D.; Suo, P.; Zhou, Y.; et al. Early myeloid-derived suppressor cells (HLA-DR(-)/(low)CD33(+)CD16(-)) expanded by granulocyte colony-stimulating factor prevent acute graft-versus-host disease (GVHD) in humanized mouse and might contribute to lower GVHD in patients post allo-HSCT. J. Hematol. Oncol. 2019, 12, 31. [Google Scholar] [CrossRef]
- Kim, T.W.; Park, S.S.; Lim, J.Y.; Min, G.J.; Park, S.; Jeon, Y.W.; Yahng, S.A.; Shin, S.H.; Lee, S.E.; Yoon, J.H.; et al. Predictive Role of Circulating Immune Cell Subtypes Early after Allogeneic Hematopoietic Stem Cell Transplantation in Patients with Acute Leukemia. Int. J. Stem Cells 2018, 12, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Lim, J.Y.; Kim, T.W.; Ryu, D.B.; Park, S.S.; Jeon, Y.W.; Yoon, J.H.; Cho, B.S.; Eom, K.S.; Kim, Y.J.; et al. Different role of circulating myeloid-derived suppressor cells in patients with multiple myeloma undergoing autologous stem cell transplantation. J. Immunother. Cancer 2019, 7, 35. [Google Scholar] [CrossRef]
- Highfill, S.L.; Rodriguez, P.C.; Zhou, Q.; Goetz, C.A.; Koehn, B.H.; Veenstra, R.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Serody, J.S.; Munn, D.H.; et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood 2010, 116, 5738–5747. [Google Scholar] [CrossRef]
- Lim, J.Y.; Ryu, D.B.; Park, M.Y.; Lee, S.E.; Park, G.; Kim, T.G.; Min, C.K. Ex Vivo Generated Human Cord Blood Myeloid-Derived Suppressor Cells Attenuate Murine Chronic Graft-versus-Host Diseases. Biol. Blood Marrow Transplant. 2018, 24, 2381–2396. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Ansell, S.M. Targeting immune checkpoints in lymphoma. Curr. Opin. Hematol. 2015, 22, 337–342. [Google Scholar] [CrossRef]
- Kreamer, K.M. Immune Checkpoint Blockade: A New Paradigm in Treating Advanced Cancer. J. Adv. Pract. Oncol. 2014, 5, 418–431. [Google Scholar]
- Marquez-Rodas, I.; Cerezuela, P.; Soria, A.; Berrocal, A.; Riso, A.; Gonzalez-Cao, M.; Martin-Algarra, S. Immune checkpoint inhibitors: Therapeutic advances in melanoma. Ann. Transl. Med. 2015, 3, 267. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Liang, A.B.; Ansell, S.M. T-cell-mediated antitumor immunity in B-cell non-Hodgkin lymphoma: Activation, suppression and exhaustion. Leuk. Lymphoma 2015, 56, 2498–2504. [Google Scholar] [CrossRef]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef]
- McClanahan, F.; Hanna, B.; Miller, S.; Clear, A.J.; Lichter, P.; Gribben, J.G.; Seiffert, M. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood 2015, 126, 203–211. [Google Scholar] [CrossRef]
- Zhu, H.; Gu, Y.; Xue, Y.; Yuan, M.; Cao, X.; Liu, Q. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 2017, 8, 114554–114567. [Google Scholar] [CrossRef]
- Kong, X.; Sun, R.; Chen, Y.; Wei, H.; Tian, Z. gammadeltaT cells drive myeloid-derived suppressor cell-mediated CD8+ T cell exhaustion in hepatitis B virus-induced immunotolerance. J. Immunol. 2014, 193, 1645–1653. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, B.; Zhang, Z.; Zhang, Y.; Yang, R. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin. Immunol. 2008, 129, 471–481. [Google Scholar] [CrossRef]
- Kotsakis, A.; Harasymczuk, M.; Schilling, B.; Georgoulias, V.; Argiris, A.; Whiteside, T.L. Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. J. Immunol. Methods 2012, 381, 14–22. [Google Scholar] [CrossRef]
- Tumino, N.; Besi, F.; Di Pace, A.L.; Mariotti, F.R.; Merli, P.; Pira, G.L.; Galaverna, F.; Pitisci, A.; Ingegnere, T.; Pelosi, A.; et al. PMN-MDSC are a new target to rescue graft-versus-leukemia activity of NK cells in haplo-HSC transplantation. Leukemia 2020, 34, 932–937. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Donor for Allogeneic Transplant (N = 68) | Patient Donor for Autologous Transplant (N = 94) | |
|---|---|---|
| Age, years, median | 45 (15–67) | Lymphoma: 48 (18–70) Multiple myeloma: 59 (34–71) |
| Sex | 44 Male/24 Female | Lymphoma: 12 Male/22 Female Multiple myeloma: 32 Male/29 Female |
| Disease | N/A | Lymphoma: HL (11), NHL (23) |
| Multiple myeloma: 60 | ||
| Mobilization | G-CSF 10 mcg/kg/d × 5 days | Chemotherapy (disease-specific) and G-CSF 10 mcg/kg/d × 5 days |
| Days of collection | 1 | 1 |
| Total nucleated cells/kg collected (×108/kg) | 6.98 (0.86–18.9) | Lymphoma: 5.37 (1.7–11.7) |
| Multiple myeloma: 2.59 (0.73–12.0) | ||
| CD34 + cells/kg collected (×106/kg) | 5.35 (2.4–12.8) | Lymphoma: 6.75 (1.9–34.6) |
| Multiple myeloma: 5.98 (1.5–18.1) |
| Allograft | Correlation (R Value) | p Value | |
|---|---|---|---|
| PD-L1+M-MDSC | PD-1+CD4+T | −0.192 | 0.126 |
| PD-L1+M-MDSC | PD-1+CD8+T | −0.143 | 0.251 |
| CD86+M-MDSC | CTLA-4+CD4+T | −0.388 | 0.001 |
| CD86+M-MDSC | CTLA-4+CD8+T | −0.271 | <0.05 |
| Galectin-9+M-MDSC | TIM-3+CD4+T | −0.341 | <0.01 |
| Galectin-9+M-MDSC | TIM-3+CD8+T | −0.200 | 0.117 |
| CD155+M-MDSC | TIGIT+CD4+T | −0.45 | <0.0001 |
| CD155+M-MDSC | TIGIT+CD8+T | −0.205 | 0.098 |
| HVEM+M-MDSC | BTLA+CD4+T | −0.358 | <0.01 |
| HVEM+M-MDSC | BTLA+CD8+T | −0.327 | <0.05 |
| Lymphoma autograft | |||
| PD-L1+M-MDSC | PD-1+CD4+T | −0.350 | <0.05 |
| PD-L1+M-MDSC | PD-1+CD8+T | −0.444 | <0.01 |
| CD86+M-MDSC | CTLA-4+CD4+T | −0.379 | <0.05 |
| CD86+M-MDSC | CTLA-4+CD8+T | −0.481 | <0.01 |
| Galectin-9+M-MDSC | TIM-3+CD4+T | −0.733 | <0.001 |
| Galectin-9+M-MDSC | TIM-3+CD8+T | −0.691 | <0.001 |
| CD155+M-MDSC | TIGIT+CD4+T | −0.648 | <0.0001 |
| CD155+M-MDSC | TIGIT+CD8+T | −0.440 | <0.01 |
| HVEM+M-MDSC | BTLA+CD4+T | −0.128 | 0.471 |
| HVEM+M-MDSC | BTLA+CD8+T | −0.211 | 0.23 |
| MM autograft | |||
| PD-L1+M-MDSC | PD-1+CD4+T | −0.547 | <0.0001 |
| PD-L1+M-MDSC | PD-1+CD8+T | −0.531 | <0.0001 |
| CD86+M-MDSC | CTLA-4+CD4+T | −0.497 | <0.0001 |
| CD86+M-MDSC | CTLA-4+CD8+T | −0.295 | <0.05 |
| Galectin-9+M-MDSC | TIM-3+CD4+T | −0.524 | <0.0001 |
| Galectin-9+M-MDSC | TIM-3+CD8+T | −0.354 | <0.01 |
| CD155+M-MDSC | TIGIT+CD4+T | −0.597 | <0.0001 |
| CD155+M-MDSC | TIGIT+CD8+T | −0.528 | <0.0001 |
| HVEM+M-MDSC | BTLA+CD4+T | −0.359 | <0.01 |
| HVEM+M-MDSC | BTLA+CD8+T | −0.271 | <0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, Q.; Gilpin, S.G.; Doerksen, J.; Bath, L.; Lam, T.; Li, Y.; Lambert, P.; Wall, D.A. The Interactions of T Cells with Myeloid-Derived Suppressor Cells in Peripheral Blood Stem Cell Grafts. Cells 2024, 13, 1545. https://doi.org/10.3390/cells13181545
Guan Q, Gilpin SG, Doerksen J, Bath L, Lam T, Li Y, Lambert P, Wall DA. The Interactions of T Cells with Myeloid-Derived Suppressor Cells in Peripheral Blood Stem Cell Grafts. Cells. 2024; 13(18):1545. https://doi.org/10.3390/cells13181545
Chicago/Turabian StyleGuan, Qingdong, Scott G. Gilpin, James Doerksen, Lauren Bath, Tracy Lam, Yun Li, Pascal Lambert, and Donna A. Wall. 2024. "The Interactions of T Cells with Myeloid-Derived Suppressor Cells in Peripheral Blood Stem Cell Grafts" Cells 13, no. 18: 1545. https://doi.org/10.3390/cells13181545
APA StyleGuan, Q., Gilpin, S. G., Doerksen, J., Bath, L., Lam, T., Li, Y., Lambert, P., & Wall, D. A. (2024). The Interactions of T Cells with Myeloid-Derived Suppressor Cells in Peripheral Blood Stem Cell Grafts. Cells, 13(18), 1545. https://doi.org/10.3390/cells13181545