
Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets

 , and
, and
Abstract
1. Introduction
2. Rewired Metabolic Pathways in GBM
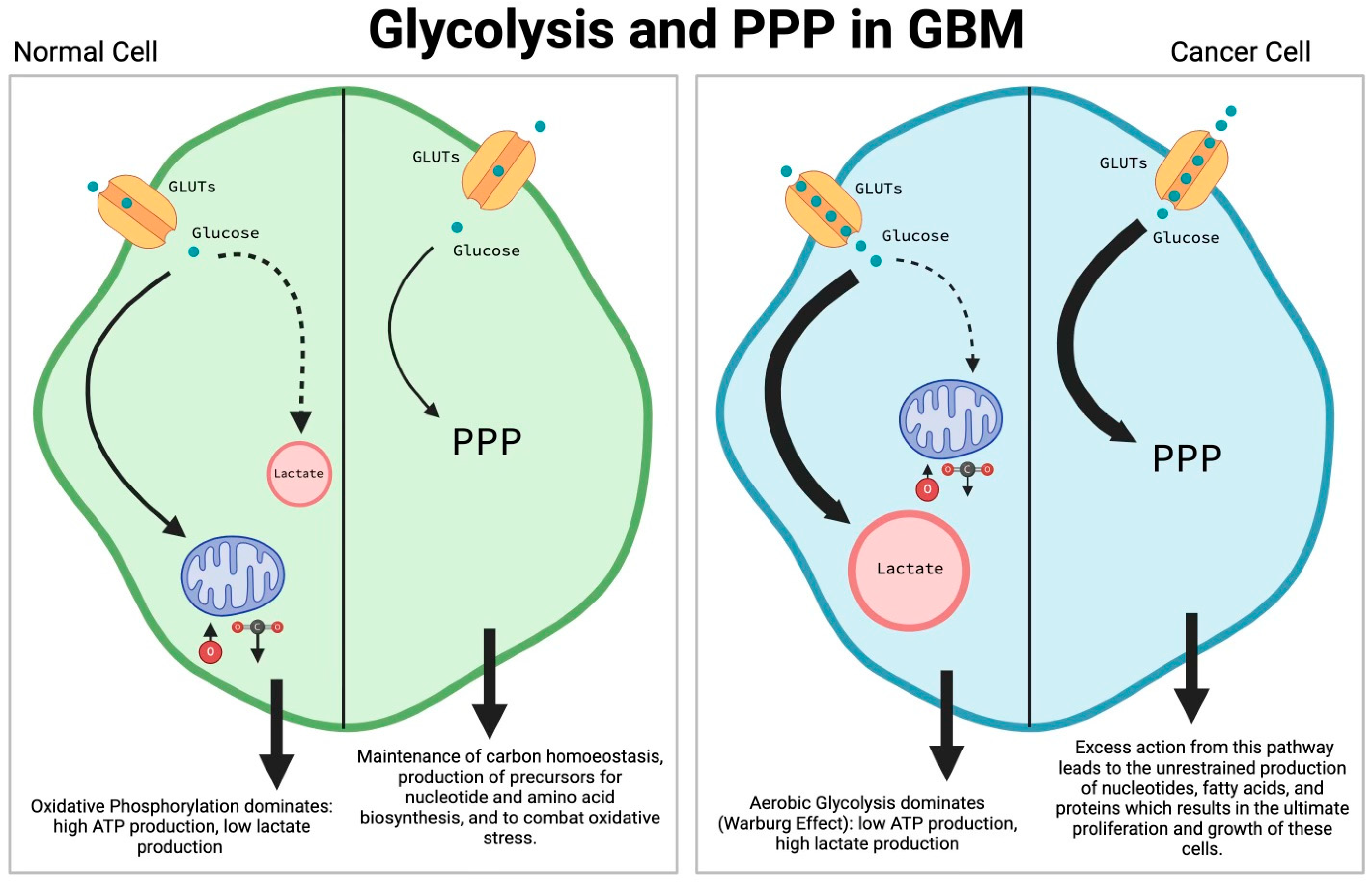
2.1. Glycolysis (Warburg Effect) and Pentose Phosphate Pathway (PPP)
2.2. Amino-Acid Metabolism
2.3. Lipid Metabolism
2.4. Nucleotide Metabolism
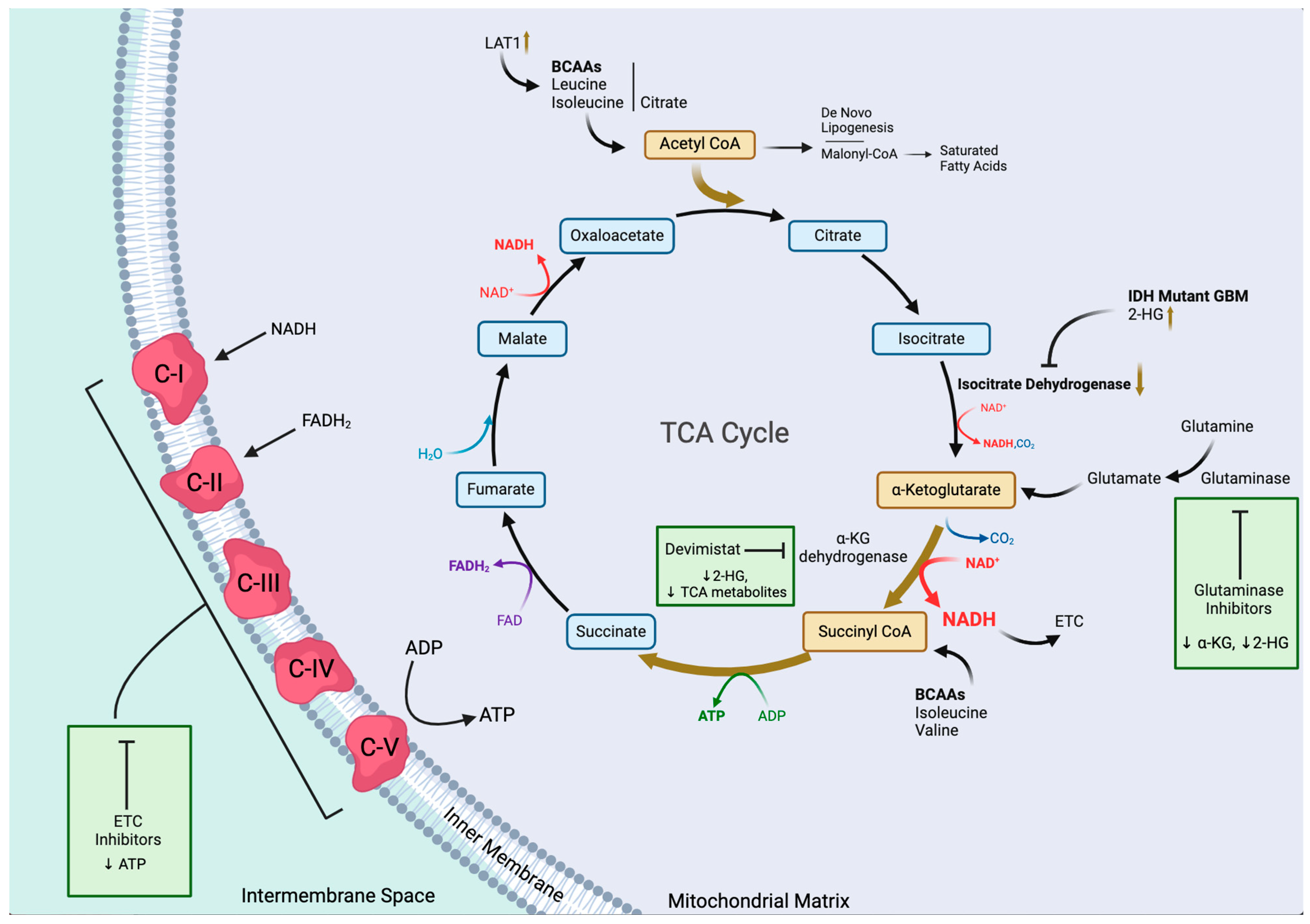
2.5. TCA Cycle and Oxidative Phosphorylation
3. Drivers of Metabolic Reprogramming in GBMs
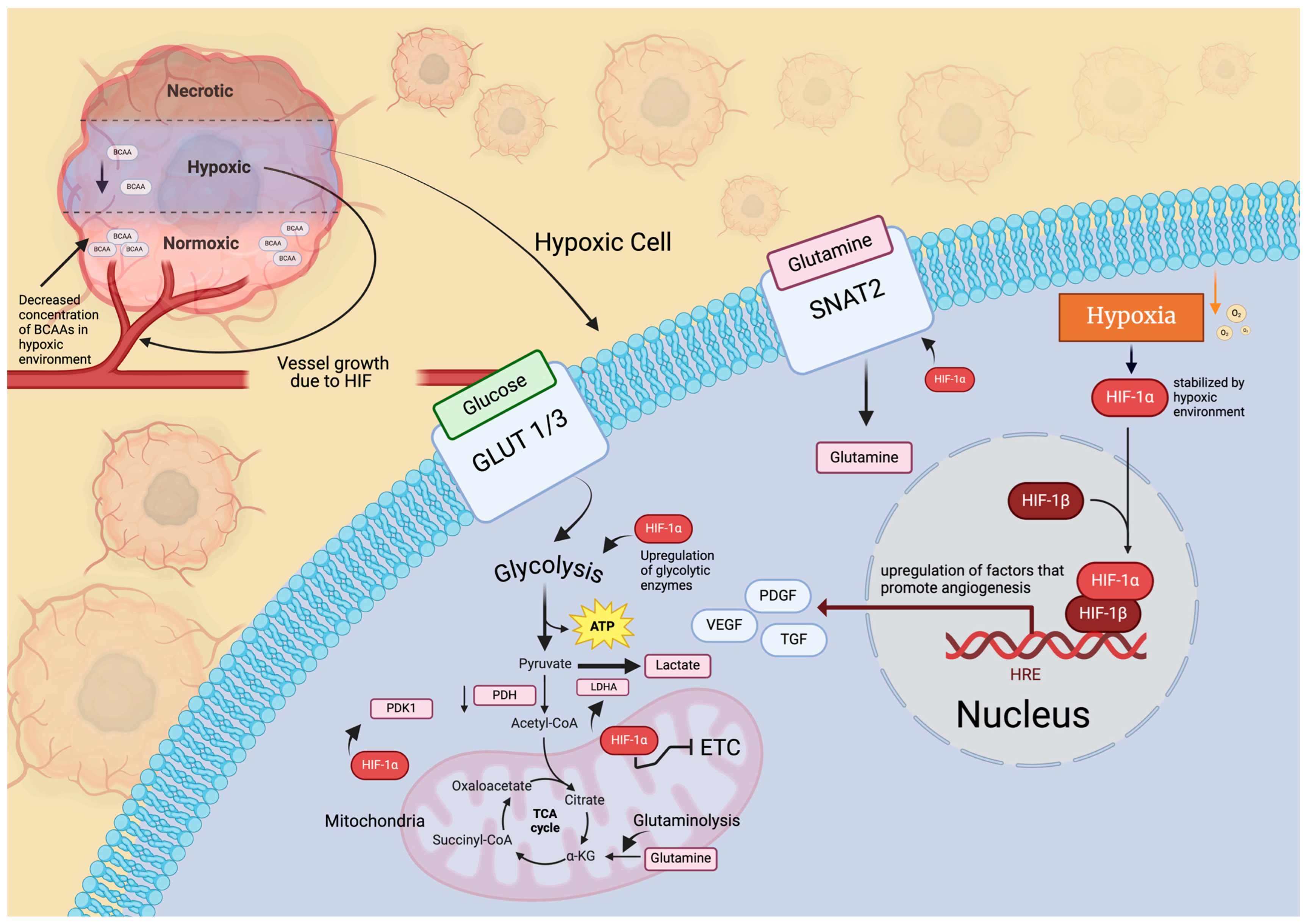
3.1. Hypoxia
3.2. Microenvironment and Extracellular Matrix
3.3. Immune System
3.4. Stem Cells and Nanotubes
3.5. Effects of Therapeutic Drugs
3.6. Epigenetic Factors
4. Targeting Metabolic Pathways and Reprogramming
4.1. Glycolysis (Warburg Effect) and the PPP

{kind=link}
{kind=link}
{kind=link}
| Section | Targets | Treatments | References |
|---|---|---|---|
| Glycolysis (Warburg effect) and pentose phosphate pathway | Glycolytic pathway (carbohydrate intake) | Dietary interventions (ketogenic diet) | [139,140] |
| Praja2 | Praja2 silencers/inhibitors | [142] | |
| Glucose uptake transporters (GLUTs) | Fasentin (GLUT1), ritonavir (GLUT1), idinivar (GLUT1), piperazin, glutor (GLUT1, GLUT2, GLUT3) | [27,145,146] | |
| 2-deoxyglucose (2-DG) | Phosphorylation of 2-DG | [147,148] | |
| Gboxin | Currently under investigation | [149] | |
| PIKE-A | PIKE-A knockdown | [150] | |
| HIF-1α | HIF-1α inhibitors | [13,15,151] | |
| PI3-Akt-mTOR pathway | Currently under investigation | ||
| Both the oxidative and non-oxidative branches of the PPP | DHEA (G6PD inhibitor), genistein, and imatinib mesylate | [22,23,152] | |
| Amino-acid metabolism | Glutaminase (GLS) | GLS inhibitors (e.g., BPTES, GLS1 inhibitor) | [153] |
| GLUD1 | GLUD1 inhibitors (e.g., R162) combined with docetaxel | [154,155] | |
| mTOR | mTOR inhibitors (e.g., AZD8055) | [156] | |
| Relevant signaling proteins (e.g., PI3) | Methionine | [31] | |
| Glutamine | L-asparaginase, further research required to mitigate drug toxicity | [157,158] | |
| Branched-chain amino acids, BCAT1 | BCAT1 inhibitors | [26,159] | |
| Lipid metabolism | ATP-dependent citrate lyase (ACLY) | ACLY inhibitors (e.g., hydroxycitrate, SB-204990, bempedoic acid) | [160] |
| Cholesterol synthesis | Bempedocic acid | [161] | |
| Acetyl-CoA carboxylase (ACC) | Currently under investigation | [162] | |
| Fatty acid synthase (FASN) | FASN inhibitors (e.g., cerulenin, C75, orlistat, C93, TVB-2640 in combination with bevacizumab) combined with chemotherapy and radiation | [163,164] | |
| Nucleotide metabolism | Amidophosphoribosyltransferase (PPAT) (de novo purine synthesis) | PRPP analogs, molecules that target the function of PPAT | [161] |
| Purine synthesis | Compounds that inhibit phosphoribosyl pyrophosphate synthetase | [161] | |
| Carbamoyl phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (de novo pyrimidine synthesis) | CAD inhibitors | [162] | |
| Dihydroorotate dehydrogenase (pyrimidine synthesis) | DHODH inhibitors | [55] | |
| Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) | Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) inhibitors | [163,164] | |
| Tyrosine kinase (TK) | TK inhibitors | [163,164] | |
| Xanthine oxidase (purine and pyrimidine catabolism) | Xanthine oxidase inhibitors | ||
| Dihydropyrimidine dehydrogenase | Dihydropyrimidine dehydrogenase (DPD) inhibitors | [165] | |
| TCA cycle and oxidative phosphorylation | Glutaminase | Glutaminase inhibitors (e.g., CB-839) | [166] |
| α-KG | Currently under investigation | [64] | |
| OXPHOS/ETC | ETC inhibitors that target components of mitochondrial ETC: such as complex I (e.g., metformin), complex II (e.g., sorafenib), and complex III (e.g., antimycin A) | [142,167,168,169,170] | |
| Mitochondrial metabolism | Mitochondrial metabolism inhibitors that target components of mitochondrial function: such as inhibitors of ATP synthase (e.g., oligomycin) or mitochondrial pyruvate carrier (e.g., UK-5099), AMPK inhibitors | [171,172,173,174] | |
| Pyruvate dehydrogenase and α-KG dehydrogenase | Devimistat | [121,175] | |
| TCA cycle and cellular respiration | Imiprodones | [121,176] |
4.2. Amino-Acid Metabolism
4.3. Lipid Metabolism
4.4. Nucleotide Metabolism
4.5. TCA Cycle and Oxidative Phosphorylation
5. Conclusions and Future Directions
6. Limitations
Author Contributions
Funding
Conflicts of Interest
References
- Rock, K.; McArdle, O.; Forde, P.; Dunne, M.; Fitzpatrick, D.; O’Neill, B.; Faul, C. A clinical review of treatment outcomes in glioblastoma multiforme—The validation in a non-trial population of the results of a randomised Phase III clinical trial: Has a more radical approach improved survival? Br. J. Radiol. 2012, 85, e729–e733. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro-Oncology 2023, 25, iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solis, C.; Silva-Adaya, D.; Serrano-García, N.; Magaña-Maldonado, R.; Jimenez-Farfan, D.; Ferreira-Guerrero, E.; Cruz-Salgado, A.; Castillo-Rodriguez, R.A. Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. Int. J. Mol. Sci. 2023, 24, 17633. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Gao, M.; Huang, J.; Jiang, X.; Yuan, Y.; Pang, H.; Luo, S.; Wang, N.; Yao, C.; Lin, Z.; Pu, D.; et al. Regulation of aerobic glycolysis to decelerate tumor proliferation by small molecule inhibitors targeting glucose transporters. Protein Cell 2020, 11, 446–451. [Google Scholar] [CrossRef]
- Bao, Z.; Chen, K.; Krepel, S.; Tang, P.; Gong, W.; Zhang, M.; Liang, W.; Trivett, A.; Zhou, M.; Wang, J.M. High Glucose Promotes Human Glioblastoma Cell Growth by Increasing the Expression and Function of Chemoattractant and Growth Factor Receptors. Transl. Oncol. 2019, 12, 1155–1163. [Google Scholar] [CrossRef]
- Papavassiliou, K.A.; Papavassiliou, A.G. Transcription factors in glioblastoma–Molecular pathogenesis and clinical implications. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1877, 188667. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Mongiardi, M.P.; Savino, M.; Falchetti, M.L.; Illi, B.; Bozzo, F.; Valle, C.; Helmer-Citterich, M.; Ferrè, F.; Nasi, S.; Levi, A. c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget 2016, 7, 33257–33271. [Google Scholar] [CrossRef]
- Dang, C.V. The interplay between MYC and HIF in the Warburg effect. In Oncogenes Meet Metabolism; Ernst Schering Foundation Symposium Proceedings; Springer: Berlin/Heidelberg, Germany, 2007; pp. 35–53. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef]
- Sengupta, T.; Datta, C.; Dasgupta, J.; De, K.; De, S.; Sengupta, D. Role of hexosemonophosphate shunt pathway in glucose metabolism in developing human foetal brain. Indian J. Biochem. Biophys. 1985, 22, 208–210. [Google Scholar]
- Adams, J.D.; Klaidman, L.K.; Chang, M.L.; Yang, J. Brain oxidative stress-analytical chemistry and thermodynamics of glutathione and NADPH. Curr. Top. Med. Chem. 2001, 1, 473–482. [Google Scholar] [CrossRef]
- Bilger, A.; Nehlig, A. Quantitative histochemical changes in enzymes involved in energy metabolism in the rat brain during postnatal development. II. Glucose-6-phosphate dehydrogenase and beta-hydroxybutyrate dehydrogenase. Int. J. Dev. Neurosci. 1992, 10, 143–152. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Panosyan, E.H.; Lin, H.J.; Koster, J.; Lasky, J.L. In search of druggable targets for GBM amino acid metabolism. BMC Cancer 2017, 17, 162. [Google Scholar] [CrossRef]
- Garcia, J.H.; Jain, S.; Aghi, M.K. Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Yuan, H.; Wu, X.; Wu, Q.; Chatoff, A.; Megill, E.; Gao, J.; Huang, T.; Duan, T.; Yang, K.; Jin, C.; et al. Lysine catabolism reprograms tumour immunity through histone crotonylation. Nature 2023, 617, 818–826. [Google Scholar] [CrossRef]
- Mestre-Farrera, A.; Bruch-Oms, M.; Peña, R.; Rodríguez-Morató, J.; Alba-Castellón, L.; Comerma, L.; Quintela-Fandino, M.; Duñach, M.; Baulida, J.; Pozo, Ó.; et al. Glutamine-Directed Migration of Cancer-Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res. 2021, 81, 438–451. [Google Scholar] [CrossRef]
- Hou, X.; Chen, S.; Zhang, P.; Guo, D.; Wang, B. Targeted Arginine Metabolism Therapy: A Dilemma in Glioma Treatment. Front. Oncol. 2022, 12, 938847. [Google Scholar] [CrossRef]
- Palanichamy, K.; Thirumoorthy, K.; Kanji, S.; Gordon, N.; Singh, R.; Jacob, J.R.; Sebastian, N.; Litzenberg, K.T.; Patel, D.; Bassett, E.; et al. Methionine and Kynurenine Activate Oncogenic Kinases in Glioblastoma, and Methionine Deprivation Compromises Proliferation. Clin. Cancer Res. 2016, 22, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, K.; Chakravarti, A. Diagnostic and Prognostic Significance of Methionine Uptake and Methionine Positron Emission Tomography Imaging in Gliomas. Front. Oncol. 2017, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-R.; Wang, J.; Wang, Z.-J.; Xi, M.-J.; Xia, B.-H.; Deng, K.; Yang, J.-L. Lipid metabolic reprogramming in tumor microenvironment: From mechanisms to therapeutics. J. Hematol. Oncol. 2023, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X. Metabolic reprogramming in cancer: Mechanisms and therapeutics. Medcomm 2023, 4, e218. [Google Scholar] [CrossRef]
- Kersten, S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001, 2, 282–286. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, W.; Li, S.; Guo, D.; He, J. Acetyl-CoA Carboxylases and Diseases. Front. Oncol. 2022, 12, 836058. [Google Scholar] [CrossRef]
- Pirmoradi, L.; Seyfizadeh, N.; Ghavami, S.; Zeki, A.A.; Shojaei, S. Targeting cholesterol metabolism in glioblastoma: A new therapeutic approach in cancer therapy. J. Investig. Med. 2019, 67, 715–719. [Google Scholar] [CrossRef]
- Jin, U.; Park, S.J.; Park, S.M. Cholesterol Metabolism in the Brain and Its Association with Parkinson’s Disease. Exp. Neurobiol. 2019, 28, 554–567. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, S.; Yang, Z.; Li, Z.-A.; Hu, W.; Dai, L.; Liang, W.; Wang, X. Cholesterol metabolism and its implication in glioblastoma therapy. J. Cancer 2022, 13, 1745–1757. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef] [PubMed]
- Ghannad-Zadeh, K.; Das, S. One-Carbon Metabolism Associated Vulnerabilities in Glioblastoma: A Review. Cancers 2021, 13, 3067. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef]
- Li, J.; Ye, J.; Zhu, S.; Cui, H. Down-Regulation of Phosphoribosyl Pyrophosphate Synthetase 1 Inhibits Neuroblastoma Cell Proliferation. Cells 2019, 8, 955. [Google Scholar] [CrossRef]
- Caniglia, J.L.; Jalasutram, A.; Asuthkar, S.; Sahagun, J.; Park, S.; Ravindra, A.; Tsung, A.J.; Guda, M.R.; Velpula, K.K. Beyond glucose: Alternative sources of energy in glioblastoma. Theranostics 2021, 11, 2048–2057. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 2019, 11, aau4972. [Google Scholar] [CrossRef]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef]
- Kökoglu, E.; Belce, A.; Özyurt, E.; Tepeler, Z. Xanthine oxidase levels in human brain tumors. Cancer Lett. 1990, 50, 179–181. [Google Scholar] [CrossRef]
- Chung, T.; Na, J.; Chang, D.-Y.; Kim, Y.I.; Kim, H.; Moon, H.E.; Kang, K.W.; Lee, D.S.; Chung, J.-K.; Kim, S.-S.; et al. Dihydropyrimidine Dehydrogenase Is a Prognostic Marker for Mesenchymal Stem Cell-Mediated Cytosine Deaminase Gene and 5-Fluorocytosine Prodrug Therapy for the Treatment of Recurrent Gliomas. Theranostics 2016, 6, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, R.; Battaglia-Hsu, S.F.; Hergalant, S.; Quéré, M.; Alberto, J.M.; Chéry, C.; Rouyer, P.; Gauchotte, G.; Guéant, J.L.; Namour, F. Folate can promote the methionine-dependent reprogramming of glioblastoma cells towards pluripotency. Cell Death Dis. 2019, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Spina, R.; Mills, I.; Ahmad, F.; Chen, C.; Ames, H.M.; Winkles, J.A.; Woodworth, G.F.; Bar, E.E. DHODH inhibition impedes glioma stem cell proliferation, induces DNA damage, and prolongs survival in orthotopic glioblastoma xenografts. Oncogene 2022, 41, 5361–5372. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.D.; Savani, M.R.; Levitt, M.M.; Wang, A.C.; Endress, J.E.; Bird, C.E.; Buehler, J.; Stopka, S.A.; Regan, M.S.; Lin, Y.-F.; et al. De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell 2022, 40, 939–956.e16. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Shi, J.; Cao, J.; Dong, B.; Guan, W. Emerging Roles and Therapeutic Interventions of Aerobic Glycolysis in Glioma. OncoTargets Ther. 2020, ume 13, 6937–6955. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Marallano, V.J.; Ughetta, M.E.; Tejero, R.; Nanda, S.; Ramalingam, R.; Stalbow, L.; Sattiraju, A.; Huang, Y.; Ramakrishnan, A.; Shen, L.; et al. Hypoxia drives shared and distinct transcriptomic changes in two invasive glioma stem cell lines. Sci. Rep. 2024, 14, 7246. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, J.; Cui, X.; Zhan, Q.; Yi, K.; Wang, Q.; Xiao, M.; Tan, Y.; Hong, B.; Fang, C.; et al. TCA-phospholipid-glycolysis targeted triple therapy effectively suppresses ATP production and tumor growth in glioblastoma. Theranostics 2022, 12, 7032–7050. [Google Scholar] [CrossRef]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncology 2015, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Reiter-Brennan, C.; Semmler, L.; Klein, A. The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Wspolczesna Onkol. Oncol. 2018, 22, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Maus, A.; Peters, G.J. Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids 2016, 49, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G.; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of Temozolomide Chemoresistance in Gliomas Leads to Remodeling of Mitochondrial Electron Transport Chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol. Lett. 2016, 12, 2283–2288. [Google Scholar] [CrossRef]
- Sanzey, M.; Rahim, S.A.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive Analysis of Glycolytic Enzymes as Therapeutic Targets in the Treatment of Glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Trejo-Solís, C.; Castillo-Rodríguez, R.A.; Serrano-García, N.; Silva-Adaya, D.; Vargas-Cruz, S.; Chávez-Cortéz, E.G.; Gallardo-Pérez, J.C.; Zavala-Vega, S.; Cruz-Salgado, A.; Magaña-Maldonado, R. Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells. Metabolites 2024, 14, 249. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. 2020, 78, 195–206. [Google Scholar] [CrossRef]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Cai, Z.; Li, W.; Brenner, M.; Bahiraii, S.; Heiss, E.H.; Weckwerth, W. Branched-chain ketoacids derived from cancer cells modulate macrophage polarization and metabolic reprogramming. Front. Immunol. 2022, 13, 966158. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Mangraviti, A.; Raghavan, T.; Volpin, F.; Skuli, N.; Gullotti, D.; Zhou, J.; Asnaghi, L.; Sankey, E.; Liu, A.; Wang, Y.; et al. HIF-1α-Targeting Acriflavine Provides Long Term Survival and Radiological Tumor Response in Brain Cancer Therapy. Sci. Rep. 2017, 7, 14978. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Nowosad, A.; Marine, J.-C.; Karras, P. Perivascular niches: Critical hubs in cancer evolution. Trends Cancer 2023, 9, 897–910. [Google Scholar] [CrossRef]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC kinase in Glioblastoma: News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Deng, C.-X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Auffinger, B.; Lesniak, M.S. Understanding glioma stem cells: Rationale, clinical relevance and therapeutic strategies. Expert Rev. Neurother. 2013, 13, 545–555. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro-Oncology 2009, 11, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.A.A.; Hochreiner, E.M.; Benjamin, B.P.; Lawler, S.E.; Zwezdaryk, K.J. Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment. Viruses 2022, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Baggetto, L.G. Biochemical, genetic, and metabolic adaptations of tumor cells that express the typical multidrug-resistance phenotype. Reversion by new therapies. J. Bioenerg. Biomembr. 1997, 29, 401–413. [Google Scholar] [CrossRef]
- Sohrabi, A.; Lefebvre, A.E.; Harrison, M.J.; Condro, M.C.; Sanazzaro, T.M.; Safarians, G.; Solomon, I.; Bastola, S.; Kordbacheh, S.; Toh, N.; et al. Microenvironmental stiffness induces metabolic reprogramming in glioblastoma. Cell Rep. 2023, 42, 113175. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Q.; Zheng, X.; Li, S.; Chen, J.; Zhao, H.; Chen, F.; Cui, L.; Li, W. Decellularized brain extracellular matrix slice glioblastoma culture model recapitulates the interaction between cells and the extracellular matrix without a nutrient-oxygen gradient interference. Acta Biomater. 2023, 158, 132–150. [Google Scholar] [CrossRef]
- Won, W.-J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 2019, 3, 47–65. [Google Scholar] [CrossRef]
- Ye, Z.; Ai, X.; Yang, K.; Yang, Z.; Fei, F.; Liao, X.; Qiu, Z.; Gimple, R.C.; Yuan, H.; Huang, H.; et al. Targeting Microglial Metabolic Rewiring Synergizes with Immune-Checkpoint Blockade Therapy for Glioblastoma. Cancer Discov. 2023, 13, 974–1001. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Evans, K.T.; Blake, K.; Longworth, A.; Coburn, M.A.; Insua-Rodríguez, J.; McMullen, T.P.; Nguyen, Q.H.; Ma, D.; Lev, T.; Hernandez, G.A.; et al. Microglia promote anti-tumour immunity and suppress breast cancer brain metastasis. Nat. Cell Biol. 2023, 25, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, J.; Fang, S.; Wang, J.; Han, X.; Liu, F.; Jin, G. P4HA1 Down-Regulation Inhibits Glioma Invasiveness by Promoting M1 Microglia Polarization. OncoTargets Ther. 2021, 14, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Fain, C.E.; Tritz, Z.P.; Nesvick, C.L.; Jin-Lee, H.J.; Geiger, P.A.; Peterson, T.E.; Jung, M.-Y.; Parney, I.F. Use of heparin to rescue immunosuppressive monocyte reprogramming by glioblastoma-derived extracellular vesicles. J. Neurosurg. 2023, 138, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Lorimer, I.A.J. Potential roles for efferocytosis in glioblastoma immune evasion. Neuro-Oncol. Adv. 2024, 6, vdae012. [Google Scholar] [CrossRef]
- Perelroizen, R.; Philosof, B.; Budick-Harmelin, N.; Chernobylsky, T.; Ron, A.; Katzir, R.; Shimon, D.; Tessler, A.; Adir, O.; Gaoni-Yogev, A.; et al. Astrocyte immunometabolic regulation of the tumour microenvironment drives glioblastoma pathogenicity. Brain 2022, 145, 3288–3307. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Metabolic regulation of macrophages in tumor microenvironment. Curr. Opin. Hematol. 2018, 25, 52–59. [Google Scholar] [CrossRef]
- Safaee, M.M.; Wang, E.J.; Jain, S.; Chen, J.-S.; Gill, S.; Zheng, A.C.; Garcia, J.H.; Beniwal, A.S.; Tran, Y.; Nguyen, A.T.; et al. CD97 is associated with mitogenic pathway activation, metabolic reprogramming, and immune microenvironment changes in glioblastoma. Sci. Rep. 2022, 12, 1464. [Google Scholar] [CrossRef]
- Kesarwani, P.; Prabhu, A.; Kant, S.; Chinnaiyan, P. Metabolic remodeling contributes towards an immune-suppressive phenotype in glioblastoma. Cancer Immunol. Immunother. 2019, 68, 1107–1120. [Google Scholar] [CrossRef]
- Li, S.; Li, Z.; Wang, X.; Zhong, J.; Yu, D.; Chen, H.; Ma, W.; Liu, L.; Ye, M.; Shen, R.; et al. HK3 stimulates immune cell infiltration to promote glioma deterioration. Cancer Cell Int. 2023, 23, 227. [Google Scholar] [CrossRef]
- Notarangelo, G.; Spinelli, J.B.; Perez, E.M.; Baker, G.J.; Kurmi, K.; Elia, I.; Stopka, S.A.; Baquer, G.; Lin, J.R.; Golby, A.J.; et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8. Science 2022, 377, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wang, M.; Zhang, G.; Li, Y.; Wang, L.; Cui, H. POU2F2 regulates glycolytic reprogramming and glioblastoma progression via PDPK1-dependent activation of PI3K/AKT/mTOR pathway. Cell Death Dis. 2021, 12, 433. [Google Scholar] [CrossRef] [PubMed]
- Nyalali, A.M.K.; Leonard, A.U.; Xu, Y.; Li, H.; Zhou, J.; Zhang, X.; Rugambwa, T.K.; Shi, X.; Li, F. CD147: An integral and potential molecule to abrogate hallmarks of cancer. Front. Oncol. 2023, 13, 1238051. [Google Scholar] [CrossRef] [PubMed]
- Pires-Afonso, Y.; Muller, A.; Grzyb, K.; Oudin, A.; Yabo, Y.A.; Sousa, C.; Scafidi, A.; Poli, A.; Cosma, A.; Halder, R.; et al. Elucidating tumour-associated microglia/macrophage diversity along glioblastoma progression and under ACOD1 deficiency. Mol. Oncol. 2022, 16, 3167–3191. [Google Scholar] [CrossRef] [PubMed]
- Geiß, C.; Witzler, C.; Poschet, G.; Ruf, W.; Régnier-Vigouroux, A. Metabolic and inflammatory reprogramming of macrophages by ONC201 translates in a pro-inflammatory environment even in presence of glioblastoma cells. Eur. J. Immunol. 2021, 51, 1246–1261. [Google Scholar] [CrossRef]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Xu, S.; Liu, Z.; Cheng, Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Transduct. Target. Ther. 2021, 6, 124. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Staicu, G.-A.; Sevastre, A.-S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma Stem Cells—Useful Tools in the Battle against Cancer. Int. J. Mol. Sci. 2022, 23, 4602. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef]
- Hulleman, E.; Quarto, M.; Vernell, R.; Masserdotti, G.; Colli, E.; Kros, J.M.; Levi, D.; Gaetani, P.; Tunici, P.; Finocchiaro, G.; et al. A role for the transcription factor HEY1 in glioblastoma. J. Cell. Mol. Med. 2009, 13, 136–146. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Dai, X.-M.; Du, B. Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 2015, 16, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Geribaldi-Doldán, N.; Fernández-Ponce, C.; Quiroz, R.N.; Sánchez-Gomar, I.; Escorcia, L.G.; Velásquez, E.P.; Quiroz, E.N. The Role of Microglia in Glioblastoma. Front. Oncol. 2021, 10, 603495. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2017, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Audia, A.; Bhat, K.P.; Okafo, G.; Eugenin, E.A. Tunneling Nanotubes Mediate Adaptation of Glioblastoma Cells to Temozolomide and Ionizing Radiation Treatment. iScience 2020, 23, 101450. [Google Scholar] [CrossRef]
- Venkatesh, V.S.; Lou, E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019, 2, e1185. [Google Scholar] [CrossRef]
- De Fazio, E.; Pittarello, M.; Gans, A.; Ghosh, B.; Slika, H.; Alimonti, P.; Tyler, B. Intrinsic and Microenvironmental Drivers of Glioblastoma Invasion. Int. J. Mol. Sci. 2024, 25, 2563. [Google Scholar] [CrossRef]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Nakhle, J.; Khattar, K.; Özkan, T.; Boughlita, A.; Abba Moussa, D.; Darlix, A.; Lorcy, F.; Rigau, V.; Bauchet, L.; Gerbal-Chaloin, S.; et al. Mitochondria Transfer from Mesenchymal Stem Cells Confers Chemoresistance to Glioblastoma Stem Cells through Metabolic Rewiring. Cancer Res. Commun. 2023, 3, 1041–1056. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Li, H.-Y.; Feng, Y.-H.; Lin, C.-L.; Hsu, T.-I. Mitochondrial Mechanisms in Temozolomide Resistance: Unraveling the Complex Interplay and Therapeutic Strategies in Glioblastoma. Mitochondrion 2024, 75, 101836. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Shang, E.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D. Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells 2022, 11, 2956. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-H.; Hsueh, W.-T.; Chuang, J.-Y.; Chang, K.-Y. Dissecting the mechanism of temozolomide resistance and its association with the regulatory roles of intracellular reactive oxygen species in glioblastoma. J. Biomed. Sci. 2021, 28, 18. [Google Scholar] [CrossRef]
- Cho, J.-H.; Dimri, M.; Dimri, G.P. A Positive Feedback Loop Regulates the Expression of Polycomb Group Protein BMI1 via WNT Signaling Pathway. J. Biol. Chem. 2013, 288, 3406–3418. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2020, 3, 17–43. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med.-Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef]
- Hao, Z.; Wang, J.; Lv, Y.; Wu, W.; Zhang, S.; Hao, S.; Chu, J.; Wan, H.; Feng, J.; Ji, N. Identification of MGMT promoter methylation as a specific lipid metabolism biomarker, reveals the feasibility of atorvastatin application in glioblastoma. Metabolism 2024, 153, 155794. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Reily Rocha, A.; Molina Silva, M.; Rodrigues Gomes, L.; Teatin Latancia, M.; Andrade Tomaz, M.; de Souza, I.; Karolynne Seregni Monteiro, L.; Menck, C.F.M. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Shang, E.; Shu, C.; Kim, S.; Mela, A.; Humala, N.; Mahajan, A.; Yang, H.W.; Akman, H.O.; Quinzii, C.M.; et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat. Commun. 2021, 12, 5203. [Google Scholar] [CrossRef]
- Gunn, K.; Myllykoski, M.; Cao, J.Z.; Ahmed, M.; Huang, B.; Rouaisnel, B.; Diplas, B.H.; Levitt, M.M.; Looper, R.; Doench, J.G.; et al. (R)-2-Hydroxyglutarate Inhibits KDM5 Histone Lysine Demethylases to Drive Transformation in IDH-Mutant Cancers. Cancer Discov. 2023, 13, 1478–1497. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Nishimura, M.C.; Kharbanda, S.; Peale, F.; Deng, Y.; Daemen, A.; Forrest, W.F.; Kwong, M.; Hedehus, M.; Hatzivassiliou, G.; et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc. Natl. Acad. Sci. USA 2014, 111, 14217–14222. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2018, 57, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Cao, Y.; Zheng, Y.; Bu, W.; Zhang, L.; You, M.J.; Koh, M.Y.; Cote, G.; Aldape, K.; et al. EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol. Cell 2012, 48, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Johnson, J.; John, J.C.S. Global DNA methylation synergistically regulates the nuclear and mitochondrial genomes in glioblastoma cells. Nucleic Acids Res. 2018, 46, 5977–5995. [Google Scholar] [CrossRef]
- Pang, B.; Zheng, X.-R.; Tian, J.-X.; Gao, T.-H.; Gu, G.-Y.; Zhang, R.; Fu, Y.-B.; Pang, Q.; Li, X.-G.; Liu, Q. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 2016, 7, 45134–45143. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Marson, A.G.; Smith, C.T.; Young, B.; Mills, S.J.; Cherry, M.G.; Jenkinson, M.D. Ketogenic diets as an adjuvant therapy for glioblastoma (KEATING): A randomized, mixed methods, feasibility study. J. Neuro-Oncol. 2020, 147, 213–227. [Google Scholar] [CrossRef]
- van der Louw, E.J.T.M.; Olieman, J.F.; Bemt, P.M.L.A.v.D.; Bromberg, J.E.C.; Hoop, E.O.-D.; Neuteboom, R.F.; Catsman-Berrevoets, C.E.; Vincent, A.J.P.E. Ketogenic diet treatment as adjuvant to standard treatment of glioblastoma multiforme: A feasibility and safety study. Ther. Adv. Med Oncol. 2019, 11. [Google Scholar] [CrossRef]
- Bello, S.D.; Valdemarin, F.; Martinuzzi, D.; Filippi, F.; Gigli, G.L.; Valente, M. Ketogenic Diet in the Treatment of Gliomas and Glioblastomas. Nutrients 2022, 14, 3851. [Google Scholar] [CrossRef]
- Donne, R.D.; Iannucci, R.; Rinaldi, L.; Roberto, L.; Oliva, M.A.; Senatore, E.; Borzacchiello, D.; Lignitto, L.; Giurato, G.; Rizzo, F.; et al. Targeted inhibition of ubiquitin signaling reverses metabolic reprogramming and suppresses glioblastoma growth. Commun. Biol. 2022, 5, 780. [Google Scholar] [CrossRef]
- Teng, X.-Q.; Qu, J.; Li, G.-H.; Zhuang, H.-H.; Qu, Q. Small Interfering RNA for Gliomas Treatment: Overcoming Hurdles in Delivery. Front. Cell Dev. Biol. 2022, 10, 824299. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J.; et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef] [PubMed]
- Reckzeh, E.S.; Karageorgis, G.; Schwalfenberg, M.; Ceballos, J.; Nowacki, J.; Stroet, M.C.; Binici, A.; Knauer, L.; Brand, S.; Choidas, A.; et al. Inhibition of Glucose Transporters and Glutaminase Synergistically Impairs Tumor Cell Growth. Cell Chem. Biol. 2019, 26, 1214–1228.e25. [Google Scholar] [CrossRef]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Viola, G.; Della Puppa, A.; Cavallini, L.; Frasson, C.; Persano, L.; Panchision, D.M.; Basso, G. Hypoxia and succinate antagonize 2-deoxyglucose effects on glioblastoma. Biochem. Pharmacol. 2010, 80, 1517–1527. [Google Scholar] [CrossRef]
- Shah, S.S.; Rodriguez, G.A.; Musick, A.; Walters, W.M.; de Cordoba, N.; Barbarite, E.; Marlow, M.M.; Marples, B.; Prince, J.S.; Komotar, R.J.; et al. Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers 2019, 11, 159. [Google Scholar] [CrossRef]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.-Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.-J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Sun, M.; Sheng, H.; Wu, T.; Song, J.; Sun, H.; Wang, Y.; Wang, J.; Li, Z.; Zhao, H.; Tan, J.; et al. PIKE-A promotes glioblastoma growth by driving PPP flux through increasing G6PD expression mediated by phosphorylation of STAT3. Biochem. Pharmacol. 2021, 192, 114736. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.J.; Fu, X.L.; Guang, R.; To, S.T. Advances in the targeting of HIF-1α and future therapeutic strategies for glioblastoma multiforme (Review). Oncol. Rep. 2017, 37, 657–670. [Google Scholar] [CrossRef]
- Boren, J.; Cascante, M.; Marin, S.; Comín-Anduix, B.; Centelles, J.J.; Lim, S.; Bassilian, S.; Ahmed, S.; Lee, W.-N.P.; Boros, L.G. Gleevec (STI571) Influences Metabolic Enzyme Activities and Glucose Carbon Flow toward Nucleic Acid and Fatty Acid Synthesis in Myeloid Tumor Cells. J. Biol. Chem. 2001, 276, 37747–37753. [Google Scholar] [CrossRef] [PubMed]
- Shukla, K.; Ferraris, D.V.; Thomas, A.G.; Stathis, M.; Duvall, B.; Delahanty, G.; Alt, J.; Rais, R.; Rojas, C.; Gao, P.; et al. Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors. J. Med. Chem. 2012, 55, 10551–10563. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, M.; Li, H.; Rao, X.; Ao, L.; Wang, H.; Yao, L.; Wang, X.; Hong, X.; Wang, J.; et al. Therapeutic targeting of glutamate dehydrogenase 1 that links metabolic reprogramming and Snail-mediated epithelial–mesenchymal transition in drug-resistant lung cancer. Pharmacol. Res. 2022, 185, 106490. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Agnihotri, N.; Kumar, S. Targeting fuel pocket of cancer cell metabolism: A focus on glutaminolysis. Biochem. Pharmacol. 2022, 198, 114943. [Google Scholar] [CrossRef]
- Ahluwalia, G.S.; Grem, J.L.; Hao, Z.; Cooney, D.A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271. [Google Scholar] [CrossRef]
- Lessner, H.; Valenstein, S.; Kaplan, R.; Desimone, P.; Yunis, A. Phase II Study of L-Asparaginase in the Treatment of Pancreatic Carcinoma. Cancer Treat. Rep. 1980, 64, 1359–1361. [Google Scholar]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021, 21, 141–162. [Google Scholar] [CrossRef]
- Darwish, A.; Pammer, M.; Gallyas, F.; Vígh, L.; Balogi, Z.; Juhász, K. Emerging Lipid Targets in Glioblastoma. Cancers 2024, 16, 397. [Google Scholar] [CrossRef]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty Acid Synthase As a Potential Therapeutic Target in Cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.; Duque, A.E.D.; Michalek, J.; Konkel, B.; Caflisch, L.; Chen, Y.; Pathuri, S.C.; Madhusudanannair-Kunnuparampil, V.; Floyd, J.; Brenner, A. Phase II Investigation of TVB-2640 (Denifanstat) with Bevacizumab in Patients with First Relapse High-Grade Astrocytoma. Clin. Cancer Res. 2023, 29, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Mangraviti, A.; Gorelick, N.L.; Shapira-Furman, T.; Alomari, S.; Cecia, A.; Darjee, N.; Brem, H.; Rottenberg, Y.; Domb, A.J.; et al. Combined intracranial Acriflavine, temozolomide and radiation extends survival in a rat glioma model. Eur. J. Pharm. Biopharm. 2021, 170, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, X.; Dong, Z.; Luo, Z.; Zhao, Z.; Xu, Y.; Zhang, J.-T. Fatty acid synthase causes drug resistance by inhibiting TNF-α and ceramide production. J. Lipid Res. 2013, 54, 776–785. [Google Scholar] [CrossRef]
- Bisht, P.; Kumar, V.U.; Pandey, R.; Velayutham, R.; Kumar, N. Role of PARP Inhibitors in Glioblastoma and Perceiving Challenges as Well as Strategies for Successful Clinical Development. Front. Pharmacol. 2022, 13, 939570. [Google Scholar] [CrossRef]
- Wang, W.; Cui, J.; Ma, H.; Lu, W.; Huang, J. Targeting Pyrimidine Metabolism in the Era of Precision Cancer Medicine. Front. Oncol. 2021, 11, 684961. [Google Scholar] [CrossRef]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.A.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology 2009, 12, 95–103. [Google Scholar] [CrossRef]
- Arif, S.H.; Pandith, A.A.; Tabasum, R.; Ramzan, A.U.; Singh, S.; Siddiqi, M.A.; Bhat, A.R. Significant effect of anti-tyrosine kinase inhibitor (Gefitinib) on overall survival of the glioblastoma multiforme patients in the backdrop of mutational status of epidermal growth factor receptor and PTEN Genes. Asian J. Neurosurg. 2018, 13, 46–52. [Google Scholar] [CrossRef]
- Kindler, H.L.; Schilsky, R.L. Eniluracil: An irreversible inhibitor of dihydropyrimidine dehydrogenase. Expert Opin. Investig. Drugs 2000, 9, 1635–1649. [Google Scholar] [CrossRef]
- Shen, Q.; Yang, H.; Kong, Q.-P.; Li, G.-H.; Li, L. Metabolic Modeling Identifies a Novel Molecular Type of Glioblastoma Associated with Good Prognosis. Metabolites 2023, 13, 172. [Google Scholar] [CrossRef] [PubMed]
- Restall, I.J.; Cseh, O.; Richards, L.M.; Pugh, T.J.; Luchman, H.A.; Weiss, S. Brain Tumor Stem Cell Dependence on Glutaminase Reveals a Metabolic Vulnerability through the Amino Acid Deprivation Response Pathway. Cancer Res. 2020, 80, 5478–5490. [Google Scholar] [CrossRef] [PubMed]
- Santos-Jiménez, J.D.L.; Rosales, T.; Ko, B.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J.; DeBerardinis, R.J.; Matés, J.M. Metabolic Adjustments following Glutaminase Inhibition by CB-839 in Glioblastoma Cell Lines. Cancers 2023, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-H.; Yang, R.-C.; Chiou, S.-S.; Shieh, T.-M.; Shih, Y.-H.; Lin, P.-C. Curcumin induces apoptosis by inhibiting BCAT1 expression and mTOR signaling in cytarabine-resistant myeloid leukemia cells. Mol. Med. Rep. 2021, 24, 565. [Google Scholar] [CrossRef]
- Grankvist, N.; Lagerborg, K.A.; Jain, M.; Nilsson, R. Gabapentin Can Suppress Cell Proliferation Independent of the Cytosolic Branched-Chain Amino Acid Transferase 1 (BCAT1). Biochemistry 2018, 57, 6762–6766. [Google Scholar] [CrossRef]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef]
- Fuentes-Fayos, A.C.; G-García, M.E.; Pérez-Gómez, J.M.; Montero-Hidalgo, A.J.; Martín-Colom, J.; Doval-Rosa, C.; Blanco-Acevedo, C.; Torres, E.; Toledano-Delgado, Á.; Sánchez-Sánchez, R.; et al. Metformin and simvastatin exert additive antitumour effects in glioblastoma via senescence-state: Clinical and translational evidence. eBioMedicine 2023, 90, 104484. [Google Scholar] [CrossRef]
- Yoon, W.S.; Chang, J.H.; Kim, J.H.; Kim, Y.J.; Jung, T.Y.; Yoo, H.; Kim, S.H.; Ko, Y.C.; Nam, D.H.; Kim, T.M.; et al. Efficacy and safety of metformin plus low-dose temozolomide in patients with recurrent or refractory glioblastoma: A randomized, prospective, multicenter, double-blind, controlled, phase 2 trial (KNOG-1501 study). Discov. Oncol. 2023, 14, 90. [Google Scholar] [CrossRef]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle 2013, 12, 491–500. [Google Scholar] [CrossRef]
- Kennedy, C.R.; Tilkens, S.B.; Guan, H.; Garner, J.A.; Or, P.M.; Chan, A.M. Differential sensitivities of glioblastoma cell lines towards metabolic and signaling pathway inhibitions. Cancer Lett. 2013, 336, 299–306. [Google Scholar] [CrossRef]
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; O Pieper, R. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2006, 14, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.B.; Park, K.R.; Kim, S.Y.; Han, S.-Y. Induction of Nuclear Enlargement and Senescence by Sirtuin Inhibitors in Glioblastoma Cells. Immune Netw. 2016, 16, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Minogue, E.; Cunha, P.P.; Wadsworth, B.J.; Grice, G.L.; Sah-Teli, S.K.; Hughes, R.; Bargiela, D.; Quaranta, A.; Zurita, J.; Antrobus, R.; et al. Glutarate regulates T cell metabolism and anti-tumour immunity. Nat. Metab. 2023, 5, 1747–1764. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Pladna, K.M.; Schramm, N.J.; Wheeler, F.B.; Kridel, S.; Pardee, T.S. Pyruvate Dehydrogenase Inhibition Leads to Decreased Glycolysis, Increased Reliance on Gluconeogenesis and Alternative Sources of Acetyl-CoA in Acute Myeloid Leukemia. Cancers 2023, 15, 484. [Google Scholar] [CrossRef] [PubMed]
- Thang, M.; Mellows, C.; Mercer-Smith, A.; Nguyen, P.; Hingtgen, S. Current approaches in enhancing TRAIL therapies in glioblastoma. Neuro-Oncol. Adv. 2023, 5, vdad047. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes Ballen, A.I.; Amosu, M.; Ravinder, S.; Chan, J.; Derin, E.; Slika, H.; Tyler, B. Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets. Cells 2024, 13, 1574. https://doi.org/10.3390/cells13181574
Cortes Ballen AI, Amosu M, Ravinder S, Chan J, Derin E, Slika H, Tyler B. Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets. Cells. 2024; 13(18):1574. https://doi.org/10.3390/cells13181574
Chicago/Turabian StyleCortes Ballen, Ashley Irin, Maryam Amosu, Surya Ravinder, Joey Chan, Emre Derin, Hasan Slika, and Betty Tyler. 2024. "Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets" Cells 13, no. 18: 1574. https://doi.org/10.3390/cells13181574
APA StyleCortes Ballen, A. I., Amosu, M., Ravinder, S., Chan, J., Derin, E., Slika, H., & Tyler, B. (2024). Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets. Cells, 13(18), 1574. https://doi.org/10.3390/cells13181574