
The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets


Abstract
:1. Introduction
1.1. Inflammatory Arthritis Pathogenesis
1.2. Currently Available Therapeutics
2. The Role of Macrophages in Chronic Inflammation
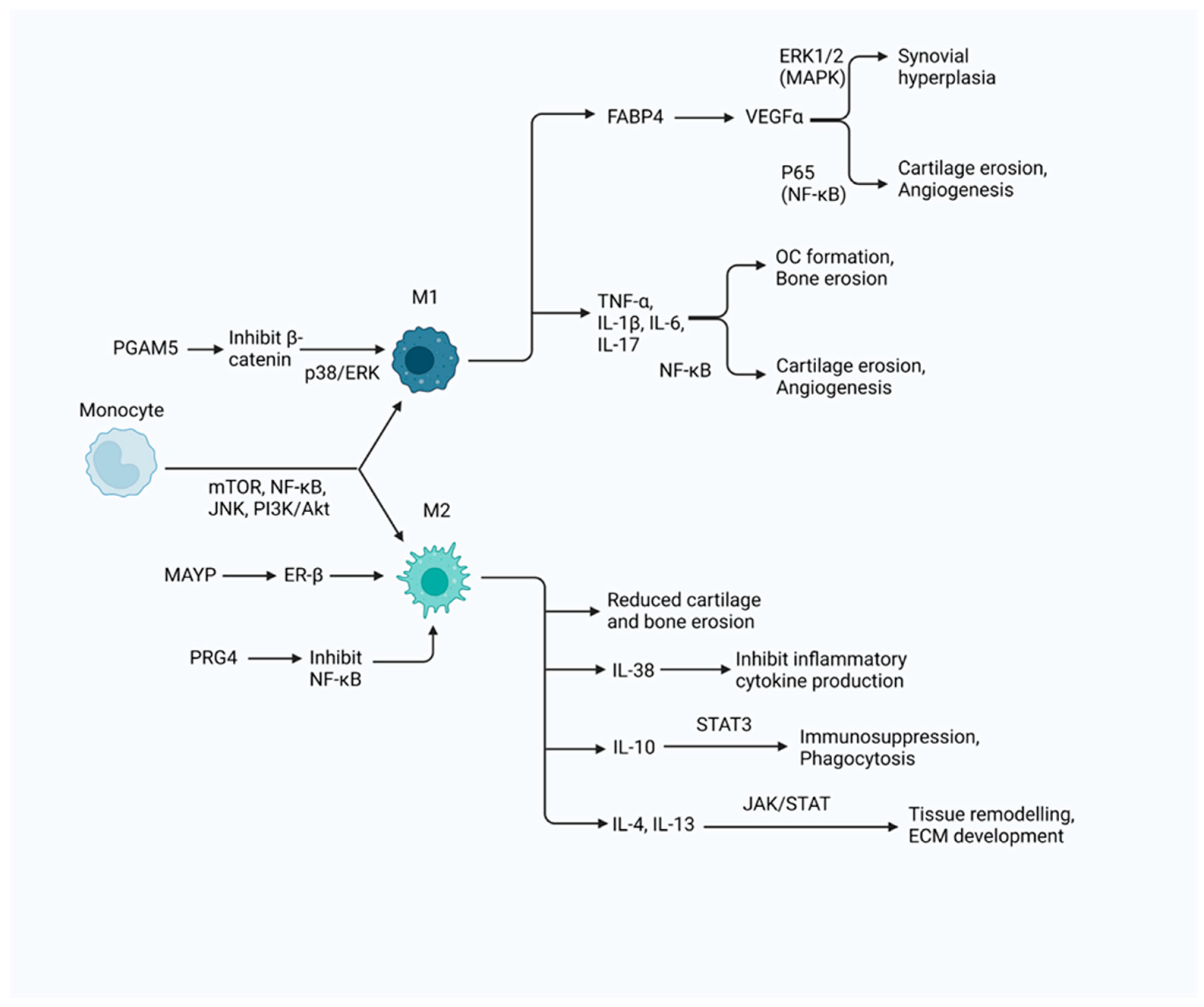
2.1. M1 Macrophages
2.1.1. Phenotype
2.1.2. Polarisation Conditions
2.1.3. Contribution to Inflammation
2.1.4. Metabolism
2.2. M2 Macrophages
2.2.1. Phenotype
2.2.2. Polarisation Conditions
2.2.3. Contribution to Inflammation
2.2.4. Metabolism
2.3. Plasticity
2.3.1. M2a Macrophages
2.3.2. M2b Macrophages
2.3.3. M2c Macrophages
2.3.4. M2d Macrophages
2.3.5. Repolarisation
3. The Role of Macrophages in the Pathogenesis of IA
3.1. Macrophage Correlation with Disease Progression
3.1.1. M1 Macrophages in IA Pathogenesis
3.1.2. M2 Macrophages in IA Pathogenesis
3.2. Predisposition of RA Patient Monocytes to Differentiate into Pro-Inflammatory Macrophages
3.3. Potential Impairment in Phagocytosis
3.4. Protective Role of CX3CR1 Macrophages
4. Therapeutic Approaches Targeting Macrophages for the Management of IA
4.1. Targeting M1 Macrophages
4.2. Targeting M2 Macrophages
4.3. Targeting Plasticity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fleischmann, R.; Schiff, M.; van der Heijde, D.; Ramos-Remus, C.; Spindler, A.; Stanislav, M.; Zerbini, C.A.F.; Gurbuz, S.; Dickson, C.; de Bono, S.; et al. Baricitinib, Methotrexate, or Combination in Patients with Rheumatoid Arthritis and No or Limited Prior Disease-Modifying Antirheumatic Drug Treatment. Arthritis Rheumatol. 2017, 69, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Lin, C.; Lu, Y.; Guan, H.; Qi, W.; Zhang, H.; Shao, Y.; Zeng, C.; Zhang, R.; Zhang, H.; et al. FABP4 secreted by M1-polarized macrophages promotes synovitis and angiogenesis to exacerbate rheumatoid arthritis. Bone Res. 2022, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Noordenbos, T.; de Hair, M.J.; Tak, P.P.; Baeten, D.L. Intimal lining layer macrophages but not synovial sublining macrophages display an IL-10 polarized-like phenotype in chronic synovitis. Arthritis Res. Ther. 2012, 14, R74. [Google Scholar] [CrossRef] [PubMed]
- Söderlin, M.K.; Börjesson, O.; Kautiainen, H.; Skogh, T.; Leirisalo-Repo, M. Annual incidence of inflammatory joint diseases in a population based study in southern Sweden. Ann. Rheum. Dis. 2002, 61, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jonsson, A.H.; Nathan, A.; Millard, N.; Curtis, M.; Xiao, Q.; Gutierrez-Arcelus, M.; Apruzzese, W.; Watts, G.F.M.; Weisenfeld, D.; et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature 2023, 623, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Costenbader, K.H.; Feskanich, D.; Mandl, L.A.; Karlson, E.W. Smoking Intensity, Duration, and Cessation, and the Risk of Rheumatoid Arthritis in Women. Am. J. Med. 2006, 119, 503.e1–503.e9. [Google Scholar] [CrossRef]
- Bouzit, L.; Malspeis, S.; Sparks, J.A.; Cui, J.; Karlson, E.W.; Yoshida, K.; Costenbader, K.H. Assessing improved risk prediction of rheumatoid arthritis by environmental, genetic, and metabolomic factors. Semin. Arthritis Rheum. 2021, 51, 1016–1022. [Google Scholar] [CrossRef]
- Paoletti, A.; Rohmer, J.; Ly, B.; Pascaud, J.; Rivière, E.; Seror, R.; Le Goff, B.; Nocturne, G.; Mariette, X. Monocyte/Macrophage Abnormalities Specific to Rheumatoid Arthritis Are Linked to miR-155 and Are Differentially Modulated by Different TNF Inhibitors. J. Immunol. 2019, 203, 1766–1775. [Google Scholar] [CrossRef]
- Bresnihan, B.; Tak, P.P. Synovial tissue analysis in rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 1999, 13, 645–659. [Google Scholar] [CrossRef]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.Á.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Bisoendial, R.J.; Stroes, E.S.G.; Tak, P.P. Critical determinants of cardiovascular risk in rheumatoid arthritis. Curr. Pharm. Des. 2011, 17, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Remus, C.; Ramirez-Gomez, A.; Brambila-Barba, V.; Barajas-Ochoa, A.; Castillo-Ortiz, J.D.; Adebajo, A.O.; Espinoza, L.R.; Aceves-Avila, F.J.; Sánchez-González, J.M.; Boudersa, N.; et al. Latitude gradient influences the age of onset of rheumatoid arthritis: A worldwide survey. Clin. Rheumatol. 2017, 36, 485–497. [Google Scholar] [CrossRef]
- Truchetet, M.-E.; Dublanc, S.; Barnetche, T.; Vittecoq, O.; Mariette, X.; Richez, C.; Blanco, P.; Mahler, M.; Contin-Bordes, C.; Schaeverbeke, T.; et al. Association of the Presence of Anti-Carbamylated Protein Antibodies in Early Arthritis with a Poorer Clinical and Radiologic Outcome: Data from the French ESPOIR Cohort. Arthritis Rheumatol. 2017, 69, 2292–2302. [Google Scholar] [CrossRef]
- Juarez, M.; Bang, H.; Hammar, F.; Reimer, U.; Dyke, B.; Sahbudin, I.; Buckley, C.D.; Fisher, B.; Filer, A.; Raza, K. Identification of novel antiacetylated vimentin antibodies in patients with early inflammatory arthritis. Ann. Rheum. Dis. 2016, 75, 1099–1107. [Google Scholar] [CrossRef]
- de Moel, E.C.; Derksen, V.F.A.M.; Trouw, L.A.; Bang, H.; Collée, G.; Lard, L.R.; Ramiro, S.; Huizinga, T.W.J.; Allaart, C.F.; Toes, R.E.M.; et al. In rheumatoid arthritis, changes in autoantibody levels reflect intensity of immunosuppression, not subsequent treatment response. Arthritis Res. Ther. 2019, 21, 28. [Google Scholar] [CrossRef]
- Silman, A.J.; MacGregor, A.J.; Thomson, W.; Holligan, S.; Carthy, D.; Farhan, A.; Ollier, W.E. Twin concordance rates for rheumatoid arthritis: Results from a nationwide study. Br. J. Rheumatol. 1993, 32, 903–907. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, Characterization, and Sequence Analysis of a Potential Virulence Factor from Porphyromonas gingivalis, Peptidylarginine Deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.-A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Mitsi, E.; Kamng’ona, R.; Rylance, J.; Solórzano, C.; Jesus Reiné, J.; Mwandumba, H.C.; Ferreira, D.M.; Jambo, K.C. Human alveolar macrophages predominately express combined classical M1 and M2 surface markers in steady state. Respir. Res. 2018, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Azuaga, A.B.; Ramírez, J.; Cañete, J.D. Psoriatic Arthritis: Pathogenesis and Targeted Therapies. Int. J. Mol. Sci. 2023, 24, 4901. [Google Scholar] [CrossRef]
- Shah, K.; Paris, M.; Mellars, L.; Changolkar, A.; Mease, P.J. Real-world burden of comorbidities in US patients with psoriatic arthritis. RMD Open 2017, 3, e000588. [Google Scholar] [CrossRef] [PubMed]
- Torre Alonso, J.C.; Rodriguez Perez, A.; Arribas Castrillo, J.M.; Ballina Garcia, J.; Riestra Noriega, J.L.; Lopez Larrea, C. Psoriatic arthritis (PA): A clinical, immunological and radiological study of 180 patients. Br. J. Rheumatol. 1991, 30, 245–250. [Google Scholar] [CrossRef]
- Abrar, D.B.; Schleich, C.; Müller-Lutz, A.; Frenken, M.; Radke, K.L.; Vordenbäumen, S.; Schneider, M.; Ostendorf, B.; Sewerin, P. Cartilage Degradation in Psoriatic Arthritis Is Associated with Increased Synovial Perfusion as Detected by Magnetic Resonance Imaging. Front. Med. 2020, 7, 539870. [Google Scholar] [CrossRef]
- van Baarsen, L.G.; Lebre, M.C.; van der Coelen, D.; Aarrass, S.; Tang, M.W.; Ramwadhdoebe, T.H.; Gerlag, D.M.; Tak, P.P. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: Possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res. Ther. 2014, 16, 426. [Google Scholar] [CrossRef]
- Sucur, A.; Jajic, Z.; Artukovic, M.; Matijasevic, M.I.; Anic, B.; Flegar, D.; Markotic, A.; Kelava, T.; Ivcevic, S.; Kovacic, N.; et al. Chemokine signals are crucial for enhanced homing and differentiation of circulating osteoclast progenitor cells. Arthritis Res. Ther. 2017, 19, 142. [Google Scholar] [CrossRef]
- Floudas, A.; Smith, C.M.; Tynan, O.; Neto, N.; Krishna, V.; Wade, S.M.; Hanlon, M.; Cunningham, C.; Marzaioli, V.; Canavan, M.; et al. Distinct stromal and immune cell interactions shape the pathogenesis of rheumatoid and psoriatic arthritis. Ann. Rheum. Dis. 2022, 81, 1224–1242. [Google Scholar] [CrossRef]
- Vandooren, B.; Noordenbos, T.; Ambarus, C.; Krausz, S.; Cantaert, T.; Yeremenko, N.; Boumans, M.; Lutter, R.; Tak, P.P.; Baeten, D. Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum. 2009, 60, 966–975. [Google Scholar] [CrossRef]
- Pan, S.; Wu, S.; Wei, Y.; Liu, J.; Zhou, C.; Chen, T.; Zhu, J.; Tan, W.; Huang, C.; Feng, S.; et al. Exploring the causal relationship between inflammatory cytokines and inflammatory arthritis: A Mendelian randomization study. Cytokine 2024, 173, 156446. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, A.C.; Soldano, S.; Contini, P.; Tomatis, V.; Ruaro, B.; Paolino, S.; Brizzolara, R.; Montagna, P.; Sulli, A.; Pizzorni, C.; et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir. Res. 2018, 19, 186. [Google Scholar] [CrossRef] [PubMed]
- Abji, F.; Rasti, M.; Gómez-Aristizábal, A.; Muytjens, C.; Saifeddine, M.; Mihara, K.; Motahhari, M.; Gandhi, R.; Viswanathan, S.; Hollenberg, M.D.; et al. Proteinase-Mediated Macrophage Signaling in Psoriatic Arthritis. Front. Immunol. 2021, 11, 629726. [Google Scholar] [CrossRef]
- Zhai, D.; Fu, Z.; Jia, J.; Lu, Z.; Wang, L. Cyclophilin A Aggravates Collagen-Induced Arthritis via Promoting Classically Activated Macrophages. Inflammation 2017, 40, 1761–1772. [Google Scholar] [CrossRef]
- Lee, M.-S.; Tseng, Y.-H.; Chen, Y.-C.; Kuo, C.-H.; Wang, S.-L.; Lin, M.-H.; Huang, Y.-F.; Wang, Y.-W.; Lin, Y.-C.; Hung, C.-H. M2 macrophage subset decrement is an indicator of bleeding tendency in pediatric dengue disease. J. Microbiol. Immunol. Infect. 2018, 51, 829–838. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Brzustewicz, E.; Henc, I.; Daca, A.; Szarecka, M.; Sochocka-Bykowska, M.; Witkowski, J.; Bryl, E. Autoantibodies, C-reactive protein, erythrocyte sedimentation rate and serum cytokine profiling in monitoring of early treatment. Cent. Eur. J. Immunol. 2017, 42, 259–268. [Google Scholar] [CrossRef]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef]
- Liu, L.; Stokes, J.V.; Tan, W.; Pruett, S.B. An optimized flow cytometry panel for classifying macrophage polarization. J. Immunol. Methods 2022, 511, 113378. [Google Scholar] [CrossRef]
- Bresnihan, B.; Pontifex, E.; Thurlings, R.M.; Vinkenoog, M.; El-Gabalawy, H.; Fearon, U.; Fitzgerald, O.; Gerlag, D.M.; Rooney, T.; van de Sande, M.G.; et al. Synovial tissue sublining CD68 expression is a biomarker of therapeutic response in rheumatoid arthritis clinical trials: Consistency across centers. J. Rheumatol. 2009, 36, 1800–1802. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamamura, F.; Naito, M. Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: A light-microscopic, enzyme-cytochemical, immunohistochemical, and ultrastructural study. J. Leukoc. Biol. 1989, 45, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, T.; Yamamoto, M.; Araumi, S.; Hara, M.; Yogo, K.; Uchida, K.; Kasakura, K.; Nishiyama, C. PU.1 and IRF8 Modulate Activation of NLRP3 Inflammasome via Regulating Its Expression in Human Macrophages. Front. Immunol. 2021, 12, 649572. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-W.; Kwon, M.-J.; Kim, I.-H.; Kim, Y.-M.; Lee, M.-K.; Nam, T.-J. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int. J. Mol. Med. 2016, 38, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, H.; Li, H.; Li, D.; Cai, Z.; Xu, J.; Ma, R. A Single-Cell RNA-Sequencing Analysis of Distinct Subsets of Synovial Macrophages in Rheumatoid Arthritis. DNA Cell Biol. 2023, 42, 212–222. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Schmidt, T.A.; Totonchy, J.; Elsaid, K.A. Proteoglycan-4 is an essential regulator of synovial macrophage polarization and inflammatory macrophage joint infiltration. Arthritis Res. Ther. 2021, 23, 241. [Google Scholar] [CrossRef]
- Kelly, A.; Grabiec, A.M.; Travis, M.A. Culture of Human Monocyte-Derived Macrophages. In Macrophages: Methods and Protocols; Rousselet, G., Ed.; Springer: New York, NY, USA, 2018; pp. 1–11. ISBN 978-1-4939-7837-3. [Google Scholar]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef]
- Saeki, N.; Imai, Y. Reprogramming of synovial macrophage metabolism by synovial fibroblasts under inflammatory conditions. Cell Commun. Signal. 2020, 18, 188. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, R.; Lv, J.; Yuan, J.; Wang, X.; Xu, C.; Ma, D.; Duan, Z.; Zhang, B.; Dai, L.; et al. Targeted knockdown of PGAM5 in synovial macrophages efficiently alleviates osteoarthritis. Bone Res. 2024, 12, 15. [Google Scholar] [CrossRef]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.-H.; De Palma, M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Mily, A.; Kalsum, S.; Loreti, M.G.; Rekha, R.S.; Muvva, J.R.; Lourda, M.; Brighenti, S. Polarization of M1 and M2 Human Monocyte-Derived Cells and Analysis with Flow Cytometry upon Mycobacterium tuberculosis Infection. JoVE (J. Vis. Exp.) 2020, 163, e61807. [Google Scholar] [CrossRef]
- Huang, S.; Yue, Y.; Feng, K.; Huang, X.; Li, H.; Hou, J.; Yang, S.; Huang, S.; Liang, M.; Chen, G.; et al. Conditioned medium from M2b macrophages modulates the proliferation, migration, and apoptosis of pulmonary artery smooth muscle cells by deregulating the PI3K/Akt/FoxO3a pathway. PeerJ 2020, 8, e9110. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Moran, L.B.; Duke, D.C.; Graeber, M.B. The microglial gene regulatory network activated by interferon-gamma. J. Neuroimmunol. 2007, 183, 1–6. [Google Scholar] [CrossRef]
- Schmitt, M.J.; Philippidou, D.; Reinsbach, S.E.; Margue, C.; Wienecke-Baldacchino, A.; Nashan, D.; Behrmann, I.; Kreis, S. Interferon-γ-induced activation of Signal Transducer and Activator of Transcription 1 (STAT1) up-regulates the tumor suppressing microRNA-29 family in melanoma cells. Cell Commun. Signal 2012, 10, 41. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 Signaling to IRF-3/7 and NF-κB Involves the Toll Adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef]
- Luo, P.; Du, M.; Sun, Q.; Zhao, T.; He, H. IL-38 suppresses macrophage M1 polarization to ameliorate synovial inflammation in the TMJ via GLUT-1 inhibition. Int. Immunopharmacol. 2023, 122, 110619. [Google Scholar] [CrossRef]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef]
- de Graaf, D.M.; Maas, R.J.A.; Smeekens, S.P.; Eisenmesser, E.; Redzic, J.S.; Helsen, M.M.; Powers, N.E.; Li, S.; Kalabokis, V.; Gresnigt, M.S.; et al. Human recombinant interleukin-38 suppresses inflammation in mouse models of local and systemic disease. Cytokine 2021, 137, 155334. [Google Scholar] [CrossRef]
- Chen, S.; Du, J.; Zhao, W.; Cao, R.; Wang, N.; Li, J.; Shen, B.; Chen, S. Elevated expression of FABP4 is associated with disease activity in rheumatoid arthritis patients. Biomark. Med. 2020, 14, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Han, B.; Li, H.; Tao, Y.; Wu, W. FABP4 knockdown suppresses inflammation, apoptosis and extracellular matrix degradation in IL-1β-induced chondrocytes by activating PPARγ to regulate the NF-κB signaling pathway. Mol. Med. Rep. 2021, 24, 855. [Google Scholar] [CrossRef] [PubMed]
- Andrés Cerezo, L.; Kuklová, M.; Hulejová, H.; Vernerová, Z.; Pešáková, V.; Pecha, O.; Veigl, D.; Haluzík, M.; Pavelka, K.; Vencovský, J.; et al. The level of fatty acid-binding protein 4, a novel adipokine, is increased in rheumatoid arthritis and correlates with serum cholesterol levels. Cytokine 2013, 64, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef] [PubMed]
- van Hamburg, J.P.; Asmawidjaja, P.S.; Davelaar, N.; Mus, A.M.C.; Colin, E.M.; Hazes, J.M.W.; Dolhain, R.J.E.M.; Lubberts, E. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum. 2011, 63, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Gupta, R.; Malhotra, R.; Sharma, V.; Farooque, K.; Kumar, V.; Chakraborty, S.; Mitra, D.K. Interleukin-9 Facilitates Osteoclastogenesis in Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 10397. [Google Scholar] [CrossRef]
- Iwasaki, M.; Kamiya, K.; Murata, S.; Maekawa, T.; Komine, M.; Ohtsuki, M. Involvement of M1/M2 macrophages in the pathomechanisms of intralymphatic histiocytosis associated with rheumatoid arthritis. J. Dermatol. 2019, 46, e42–e43. [Google Scholar] [CrossRef]
- Fukui, S.; Iwamoto, N.; Takatani, A.; Igawa, T.; Shimizu, T.; Umeda, M.; Nishino, A.; Horai, Y.; Hirai, Y.; Koga, T.; et al. M1 and M2 Monocytes in Rheumatoid Arthritis: A Contribution of Imbalance of M1/M2 Monocytes to Osteoclastogenesis. Front. Immunol. 2017, 8, 1958. [Google Scholar] [CrossRef]
- Fearon, U.; Reece, R.; Smith, J.; Emery, P.; Veale, D.J. Synovial cytokine and growth factor regulation of MMPs/TIMPs: Implications for erosions and angiogenesis in early rheumatoid and psoriatic arthritis patients. Ann. N. Y. Acad. Sci. 1999, 878, 619–621. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Haas-Smith, S.A.; Li, P.; Hicks, D.G.; Schwarz, E.M. Mechanisms of TNF-α– and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J. Clin. Investig. 2003, 111, 821–831. [Google Scholar] [CrossRef]
- Lee, B.-W.; Moon, S.-J. Inflammatory Cytokines in Psoriatic Arthritis: Understanding Pathogenesis and Implications for Treatment. Int. J. Mol. Sci. 2023, 24, 11662. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Renaudin, F.; Orliaguet, L.; Castelli, F.; Fenaille, F.; Prignon, A.; Alzaid, F.; Combes, C.; Delvaux, A.; Adimy, Y.; Cohen-Solal, M.; et al. Gout and pseudo-gout-related crystals promote GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1β activation on macrophages. Ann. Rheum. Dis. 2020, 79, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, e9029327. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Santarsiero, A.; Convertini, P.; Vassallo, A.; Santoro, V.; Todisco, S.; Iacobazzi, D.; Fondufe-Mittendorf, Y.; Martelli, G.; de Oliveira, M.R.; Montanaro, R.; et al. Phenolic Compounds of Red Wine Aglianico del Vulture Modulate the Functional Activity of Macrophages via Inhibition of NF-κB and the Citrate Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 5533793. [Google Scholar] [CrossRef]
- Behmoaras, J. The versatile biochemistry of iron in macrophage effector functions. FEBS J. 2021, 288, 6972–6989. [Google Scholar] [CrossRef]
- Suzuki, H.; Hisamatsu, T.; Chiba, S.; Mori, K.; Kitazume, M.T.; Shimamura, K.; Nakamoto, N.; Matsuoka, K.; Ebinuma, H.; Naganuma, M.; et al. Glycolytic pathway affects differentiation of human monocytes to regulatory macrophages. Immunol. Lett. 2016, 176, 18–27. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed]
- Avila-Ponce de León, U.; Vázquez-Jiménez, A.; Matadamas-Guzman, M.; Pelayo, R.; Resendis-Antonio, O. Transcriptional and Microenvironmental Landscape of Macrophage Transition in Cancer: A Boolean Analysis. Front. Immunol. 2021, 12, 642842. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, A.; Ly, B.; Cailleau, C.; Gao, F.; de Ponfilly-Sotier, M.P.; Pascaud, J.; Rivière, E.; Yang, L.; Nwosu, L.; Elmesmari, A.; et al. Liposomal AntagomiR-155-5p Restores Anti-Inflammatory Macrophages and Improves Arthritis in Preclinical Models of Rheumatoid Arthritis. Arthritis Rheumatol. 2024, 76, 18–31. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Richendrfer, H.; Schmidt, T.A.; Elsaid, K.A. Proteoglycan-4 regulates fibroblast to myofibroblast transition and expression of fibrotic genes in the synovium. Arthritis Res. Ther. 2020, 22, 113. [Google Scholar] [CrossRef]
- Szodoray, P.; Alex, P.; Chappell-Woodward, C.M.; Madland, T.M.; Knowlton, N.; Dozmorov, I.; Zeher, M.; Jarvis, J.N.; Nakken, B.; Brun, J.G.; et al. Circulating cytokines in Norwegian patients with psoriatic arthritis determined by a multiplex cytokine array system. Rheumatology 2007, 46, 417–425. [Google Scholar] [CrossRef]
- Barbarroja, N.; López-Montilla, M.D.; Cuesta-López, L.; Pérez-Sánchez, C.; Ruiz-Ponce, M.; López-Medina, C.; Ladehesa-Pineda, M.L.; López-Pedrera, C.; Escudero-Contreras, A.; Collantes-Estévez, E.; et al. Characterization of the inflammatory proteome of synovial fluid from patients with psoriatic arthritis: Potential treatment targets. Front. Immunol. 2023, 14, 1133435. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Zhang, M.; Zhang, X.; Ren, F.; Zheng, Y.; Fei, W.; Zhao, M.; Zheng, C. PSTPIP2 regulates synovial macrophages polarization and dynamics via ERβ in the joint microenvironment. Arthritis Res. Ther. 2022, 24, 247. [Google Scholar] [CrossRef]
- Xiao, H.; Guo, Y.; Li, B.; Li, X.; Wang, Y.; Han, S.; Cheng, D.; Shuai, X. M2-Like Tumor-Associated Macrophage-Targeted Codelivery of STAT6 Inhibitor and IKKβ siRNA Induces M2-to-M1 Repolarization for Cancer Immunotherapy with Low Immune Side Effects. ACS Cent. Sci. 2020, 6, 1208–1222. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, Y.; Zhang, Y.; Bian, Y.; Zeng, Y.; Yao, X.; Wan, J.; Chen, X.; Li, J.; et al. S100A4 enhances protumor macrophage polarization by control of PPAR-γ-dependent induction of fatty acid oxidation. J. Immunother. Cancer 2021, 9, e002548. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis–specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2008, 58, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Galván-Peña, S.; O’Neill, L.A.J. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef]
- Cao, S.; Li, Y.; Song, R.; Meng, X.; Fuchs, M.; Liang, C.; Kachler, K.; Meng, X.; Wen, J.; Schlötzer-Schrehardt, U.; et al. L-arginine metabolism inhibits arthritis and inflammatory bone loss. Ann. Rheum. Dis. 2024, 83, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Zerrouk, N.; Alcraft, R.; Hall, B.A.; Augé, F.; Niarakis, A. Large-scale computational modelling of the M1 and M2 synovial macrophages in rheumatoid arthritis. NPJ Syst. Biol. Appl. 2024, 10, 10. [Google Scholar] [CrossRef]
- Li, D.; Ma, H.; Shu, Q.; Wang, T.; Li, L.; Huang, P.; Lou, K.; Xu, H. Arsenite inhibits M2a polarization of macrophages through downregulation of peroxisome proliferator-activated receptor gamma. Toxicol. Appl. Pharmacol. 2022, 450, 116142. [Google Scholar] [CrossRef]
- Rodríguez-Ubreva, J.; de la Calle-Fabregat, C.; Li, T.; Ciudad, L.; Ballestar, M.L.; Català-Moll, F.; Morante-Palacios, O.; Garcia-Gomez, A.; Celis, R.; Humby, F.; et al. Inflammatory cytokines shape a changing DNA methylome in monocytes mirroring disease activity in rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1505–1516. [Google Scholar] [CrossRef]
- Davies, L.C.; Rosas, M.; Smith, P.J.; Fraser, D.J.; Jones, S.A.; Taylor, P.R. A quantifiable proliferative burst of tissue macrophages restores homeostatic macrophage populations after acute inflammation. Eur. J. Immunol. 2011, 41, 2155–2164. [Google Scholar] [CrossRef]
- Nakajima, H.; Uchida, K.; Guerrero, A.R.; Watanabe, S.; Sugita, D.; Takeura, N.; Yoshida, A.; Long, G.; Wright, K.T.; Johnson, W.E.B.; et al. Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. J. Neurotrauma 2012, 29, 1614–1625. [Google Scholar] [CrossRef]
- Bianchini, R.; Roth-Walter, F.; Ohradanova-Repic, A.; Flicker, S.; Hufnagl, K.; Fischer, M.B.; Stockinger, H.; Jensen-Jarolim, E. IgG4 drives M2a macrophages to a regulatory M2b-like phenotype: Potential implication in immune tolerance. Allergy 2019, 74, 483–494. [Google Scholar] [CrossRef]
- Vlk, A.M.; Prantner, D.; Shirey, K.A.; Perkins, D.J.; Buzza, M.S.; Thumbigere-Math, V.; Keegan, A.D.; Vogel, S.N. M2a macrophages facilitate resolution of chemically-induced colitis in TLR4-SNP mice. mBio 2023, 14, e01208-23. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sudan, K.; Schmoeckel, E.; Kost, B.P.; Kuhn, C.; Vattai, A.; Vilsmaier, T.; Mahner, S.; Jeschke, U.; Heidegger, H.H. CCL22-Polarized TAMs to M2a Macrophages in Cervical Cancer In Vitro Model. Cells 2022, 11, 2027. [Google Scholar] [CrossRef] [PubMed]
- Lurier, E.B.; Dalton, D.; Dampier, W.; Raman, P.; Nassiri, S.; Ferraro, N.M.; Rajagopalan, R.; Sarmady, M.; Spiller, K.L. Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology 2017, 222, 847–856. [Google Scholar] [CrossRef]
- Xia, T.; Zhang, M.; Lei, W.; Yang, R.; Fu, S.; Fan, Z.; Yang, Y.; Zhang, T. Advances in the role of STAT3 in macrophage polarization. Front. Immunol. 2023, 14, 1160719. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Chen, J.; Heng, B.C.; Jiang, Y.; Hu, X.; Ge, Z. Macrophages promote cartilage regeneration in a time- and phenotype-dependent manner. J. Cell. Physiol. 2022, 237, 2258–2270. [Google Scholar] [CrossRef]
- Junior; Lai, Y.-S.; Nguyen, H.T.; Salmanida, F.P.; Chang, K.-T. MERTK+/hi M2c Macrophages Induced by Baicalin Alleviate Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 10604. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.; Xian, C.; Chen, M.; Luo, D.; Zheng, J.; Zhao, S.; Li, X.-G. Single-cell and bulk transcriptomics reveals M2d macrophages as a potential therapeutic strategy for mucosal healing in ulcerative colitis. Int. Immunopharmacol. 2023, 121, 110509. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef]
- Smith, T.D.; Tse, M.J.; Read, E.L.; Liu, W.F. Regulation of macrophage polarization and plasticity by complex activation signals. Integr. Biol. 2016, 8, 946–955. [Google Scholar] [CrossRef]
- Quero, L.; Hanser, E.; Manigold, T.; Tiaden, A.N.; Kyburz, D. TLR2 stimulation impairs anti-inflammatory activity of M2-like macrophages, generating a chimeric M1/M2 phenotype. Arthritis Res. Ther. 2017, 19, 245. [Google Scholar] [CrossRef] [PubMed]
- Lescoat, A.; Ballerie, A.; Jouneau, S.; Fardel, O.; Vernhet, L.; Jego, P.; Lecureur, V. M1/M2 polarisation state of M-CSF blood-derived macrophages in systemic sclerosis. Ann. Rheum. Dis. 2019, 78, e127. [Google Scholar] [CrossRef]
- Raes, G.; De Baetselier, P.; Noël, W.; Beschin, A.; Brombacher, F.; Hassanzadeh Gh, G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002, 71, 597–602. [Google Scholar] [CrossRef]
- Becker, L.; Nguyen, L.; Gill, J.; Kulkarni, S.; Pasricha, P.J.; Habtezion, A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut 2018, 67, 827–836. [Google Scholar] [CrossRef]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef]
- Udalova, I.A.; Mantovani, A.; Feldmann, M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 472–485. [Google Scholar] [CrossRef]
- Kinne, R.W.; Stuhlmüller, B.; Burmester, G.-R. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res. Ther. 2007, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef]
- Hegarty, C.; Neto, N.; Cahill, P.; Floudas, A. Computational approaches in rheumatic diseases—Deciphering complex spatio-temporal cell interactions. Comput. Struct. Biotechnol. J. 2023, 21, 4009–4020. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Wick, M.C.; Lampa, J.; Lindblad, S.; Nordmark, B.; Klareskog, L.; Vollenhoven, R.F. van Longitudinal analysis of citrullinated protein/peptide antibodies (anti-CP) during 5 year follow up in early rheumatoid arthritis: Anti-CP status predicts worse disease activity and greater radiological progression. Ann. Rheum. Dis. 2005, 64, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Mathsson, L.; Lampa, J.; Mullazehi, M.; Rönnelid, J. Immune complexes from rheumatoid arthritis synovial fluid induce FcγRIIa dependent and rheumatoid factor correlated production of tumour necrosis factor-α by peripheral blood mononuclear cells. Arthritis Res. Ther. 2006, 8, R64. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, Z.; Liu, S.; Liu, J.; Zhang, Z.; Ouyang, Y.; Su, Z.; Chen, D.; Guo, L.; Luo, T. Roles of extracellular vesicles on macrophages in inflammatory bone diseases. Mol. Cell Biochem. 2024, 479, 1401–1414. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, C.; Yang, S.; Yang, L.; Luo, W.; Zhang, W.; Zhang, X.; Chao, J. Macrophage-derived MMP12 promotes fibrosis through sustained damage to endothelial cells. J. Hazard. Mater. 2024, 461, 132733. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Cuda, C.M.; Saber, R.; Turner, J.D.; Gierut, A.K.; Haines, G.K.; Berdnikovs, S.; Filer, A.; Clark, A.R.; Buckley, C.D.; et al. Nonclassical Ly6C− monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 2014, 9, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Alivernini, S.; Ballantine, L.E.; Asquith, D.L.; Millar, N.L.; Gilchrist, D.S.; Reilly, J.; Ierna, M.; Fraser, A.R.; Stolarski, B.; et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 11193–11198. [Google Scholar] [CrossRef]
- Narayan, N.; Mandhair, H.K.; Sekine, T.; Sabokbar, A.; Taylor, P. Predisposition of RA monocytes/macrophages to a pro-inflammatory phenotype through down-regulation of mitochondrial translocator protein. Rheumatology 2018, 57, key075.223. [Google Scholar] [CrossRef]
- Folkersen, L.; Brynedal, B.; Diaz-Gallo, L.M.; Ramsköld, D.; Shchetynsky, K.; Westerlind, H.; Sundström, Y.; Schepis, D.; Hensvold, A.; Vivar, N.; et al. Integration of Known DNA, RNA and Protein Biomarkers Provides Prediction of Anti-TNF Response in Rheumatoid Arthritis: Results from the COMBINE Study. Mol. Med. 2016, 22, 322–328. [Google Scholar] [CrossRef]
- Bugatti, S.; Manzo, A.; Vitolo, B.; Benaglio, F.; Binda, E.; Scarabelli, M.; Humby, F.; Caporali, R.; Pitzalis, C.; Montecucco, C. High expression levels of the B cell chemoattractant CXCL13 in rheumatoid synovium are a marker of severe disease. Rheumatol. 2014, 53, 1886–1895. [Google Scholar] [CrossRef]
- Greisen, S.R.; Schelde, K.K.; Rasmussen, T.K.; Kragstrup, T.W.; Stengaard-Pedersen, K.; Hetland, M.L.; Hørslev-Petersen, K.; Junker, P.; Østergaard, M.; Deleuran, B.; et al. CXCL13 predicts disease activity in early rheumatoid arthritis and could be an indicator of the therapeutic ‘window of opportunity’. Arthritis Res. Ther. 2014, 16, 434. [Google Scholar] [CrossRef]
- Biesemann, N.; Margerie, D.; Asbrand, C.; Rehberg, M.; Savova, V.; Agueusop, I.; Klemmer, D.; Ding-Pfennigdorff, D.; Schwahn, U.; Dudek, M.; et al. Additive efficacy of a bispecific anti–TNF/IL-6 nanobody compound in translational models of rheumatoid arthritis. Sci. Transl. Med. 2023, 15, eabq4419. [Google Scholar] [CrossRef]
- Gabay, C.; Burmester, G.R.; Strand, V.; Msihid, J.; Zilberstein, M.; Kimura, T.; van Hoogstraten, H.; Boklage, S.H.; Sadeh, J.; Graham, N.M.H.; et al. Sarilumab and adalimumab differential effects on bone remodelling and cardiovascular risk biomarkers, and predictions of treatment outcomes. Arthritis Res. Ther. 2020, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, Y.; Zhou, Y.; Xu, Y.; Bo, X.; Wu, J. M1 Macrophages Increase Endothelial Permeability and Enhance p38 Phosphorylation via PPAR-γ/CXCL13-CXCR5 in Sepsis. Int. Arch. Allergy Immunol. 2022, 183, 997–1006. [Google Scholar] [CrossRef]
- Fenyo, I.M.; Gafencu, A.V. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology 2013, 218, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Cailhier, J.F.; Partolina, M.; Vuthoori, S.; Wu, S.; Ko, K.; Watson, S.; Savill, J.; Hughes, J.; Lang, R.A. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 2005, 174, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.; DiPietro, L.A.; Koh, T.J. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 2009, 175, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Gibson, P.G.; Yang, I.A.; Upham, J.; James, A.; Reynolds, P.N.; Hodge, S.; AMAZES Study Research Group. Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin. Exp. Allergy 2013, 43, 29–35. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Zhai, N.; Song, H.; Li, H.; Yang, Y.; Li, T.; Guo, X.; Chi, B.; Niu, J.; et al. HCV core protein inhibits polarization and activity of both M1 and M2 macrophages through the TLR2 signaling pathway. Sci. Rep. 2016, 6, 36160. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef]
- Buch, M.H. Defining refractory rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 966–969. [Google Scholar] [CrossRef]
- Listing, J.; Kekow, J.; Manger, B.; Burmester, G.-R.; Pattloch, D.; Zink, A.; Strangfeld, A. Mortality in rheumatoid arthritis: The impact of disease activity, treatment with glucocorticoids, TNFα inhibitors and rituximab. Ann. Rheum. Dis. 2015, 74, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Yanni, G.; Whelan, A.; Feighery, C.; Bresnihan, B. Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Surace, A.E.A.; Goldmann, K.; Sciacca, E.; Çubuk, C.; Giorli, G.; John, C.R.; Nerviani, A.; Fossati-Jimack, L.; Thorborn, G.; et al. Rituximab versus tocilizumab in rheumatoid arthritis: Synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat. Med. 2022, 28, 1256–1268. [Google Scholar] [CrossRef]
- Pitzalis, C.; Choy, E.H.; Buch, M.H. Transforming clinical trials in rheumatology: Towards patient-centric precision medicine. Nat. Rev. Rheumatol. 2020, 16, 590–599. Available online: https://www.nature.com/articles/s41584-020-0491-4 (accessed on 18 July 2024). [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Chen, Y.; Lai, B.; Wang, J.; Wang, W.; Xiao, W.; Cha, X. Shikonin combined with methotrexate regulate macrophage polarization to treat psoriasis. Bioengineered 2022, 13, 11146–11155. [Google Scholar] [CrossRef] [PubMed]
- Lindström, U.; di Giuseppe, D.; Exarchou, S.; Alenius, G.-M.; Olofsson, T.; Klingberg, E.; Jacobsson, L.; Askling, J.; Wallman, J.K. Methotrexate treatment in early psoriatic arthritis in comparison to rheumatoid arthritis: An observational nationwide study. RMD Open 2023, 9, e002883. [Google Scholar] [CrossRef] [PubMed]
- Palasiewicz, K.; Umar, S.; Romay, B.; Zomorrodi, R.K.; Shahrara, S. Tofacitinib therapy intercepts macrophage metabolic reprogramming instigated by SARS-CoV-2 Spike protein. Eur. J. Immunol. 2021, 51, 2330–2340. [Google Scholar] [CrossRef]
- Degboé, Y.; Rauwel, B.; Baron, M.; Boyer, J.-F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.-L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front. Immunol. 2019, 10, 3. [Google Scholar] [CrossRef]
- Cutolo, M.; Soldano, S.; Gotelli, E.; Montagna, P.; Campitiello, R.; Paolino, S.; Pizzorni, C.; Sulli, A.; Smith, V.; Tardito, S. CTLA4-Ig treatment induces M1–M2 shift in cultured monocyte-derived macrophages from healthy subjects and rheumatoid arthritis patients. Arthritis Res. Ther. 2021, 23, 306. [Google Scholar] [CrossRef]
- Rubbert-Roth, A.; Enejosa, J.; Pangan, A.L.; Haraoui, B.; Rischmueller, M.; Khan, N.; Zhang, Y.; Martin, N.; Xavier, R.M. Trial of Upadacitinib or Abatacept in Rheumatoid Arthritis. N. Engl. J. Med. 2020, 383, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Quero, L.; Tiaden, A.N.; Hanser, E.; Roux, J.; Laski, A.; Hall, J.; Kyburz, D. miR-221-3p Drives the Shift of M2-Macrophages to a Pro-Inflammatory Function by Suppressing JAK3/STAT3 Activation. Front. Immunol. 2020, 10, 3087. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, Y.; Zheng, X.; Jia, M.; Mei, Z.; Wang, Y.; Zhang, Z.; Zhou, M.; Li, C. M2-type exosomes nanoparticles for rheumatoid arthritis therapy via macrophage re-polarization. J. Control. Release 2022, 341, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tang, J.; Hu, X.; Bao, P.; Pan, J.; Ou, Y.; Deng, W.; Liang, Y. Imatinib inhibits CSF1R that stimulates proliferation of rheumatoid arthritis fibroblast-like synoviocytes. Clin. Exp. Immunol. 2019, 195, 237–250. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Liang, C.; Wu, S.; Xia, G.; Huang, J.; Wen, Z.; Zhang, W.; Cao, X. Engineered M2a macrophages for the treatment of osteoarthritis. Front. Immunol. 2022, 13, 1054938. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| RA | PsA | |
|---|---|---|
| Disease Characteristics | Progressive bone and joint destruction [12] | New bone formation via osteoclastogenesis after initial destructive processes [12] |
| Phenotype in Synovial Microenvironment | Predominantly pro-inflammatory M1 [3] | Dysfunctional M2 and pro-inflammatory M1 [30,31] |
| Macrophage Activation | Pathways involving TLR expression (TLR2, TL4) [32,33] | Pathways involving CD206 [34] |
| Signalling Pathways | NF-κB, JAK/STAT and MAPK [35,36] | NF-κB, JAK/STAT, MAPK and IL-17 [31] |
| Cytokine Expression | High IFNγ, TNF-α and IL-1β [30] | High CD163, VEGF, IL-6 and IL-17A [3,30,31] |
| Presence of Autoantibody | RF and ACPA [8] | No autoantibodies [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulakova, K.; Lawal, T.R.; Mccarthy, E.; Floudas, A. The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets. Cells 2024, 13, 1586. https://doi.org/10.3390/cells13181586
Kulakova K, Lawal TR, Mccarthy E, Floudas A. The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets. Cells. 2024; 13(18):1586. https://doi.org/10.3390/cells13181586
Chicago/Turabian StyleKulakova, Karina, Tope Remilekun Lawal, Eoghan Mccarthy, and Achilleas Floudas. 2024. "The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets" Cells 13, no. 18: 1586. https://doi.org/10.3390/cells13181586
APA StyleKulakova, K., Lawal, T. R., Mccarthy, E., & Floudas, A. (2024). The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets. Cells, 13(18), 1586. https://doi.org/10.3390/cells13181586