
Glutamate Transporter 1 as a Novel Negative Regulator of Amyloid β

 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture
2.3. Expression Constructs and Transfections
2.4. ELISA for Aβ Species
2.5. Cytotoxicity Assay
2.6. Western Blotting
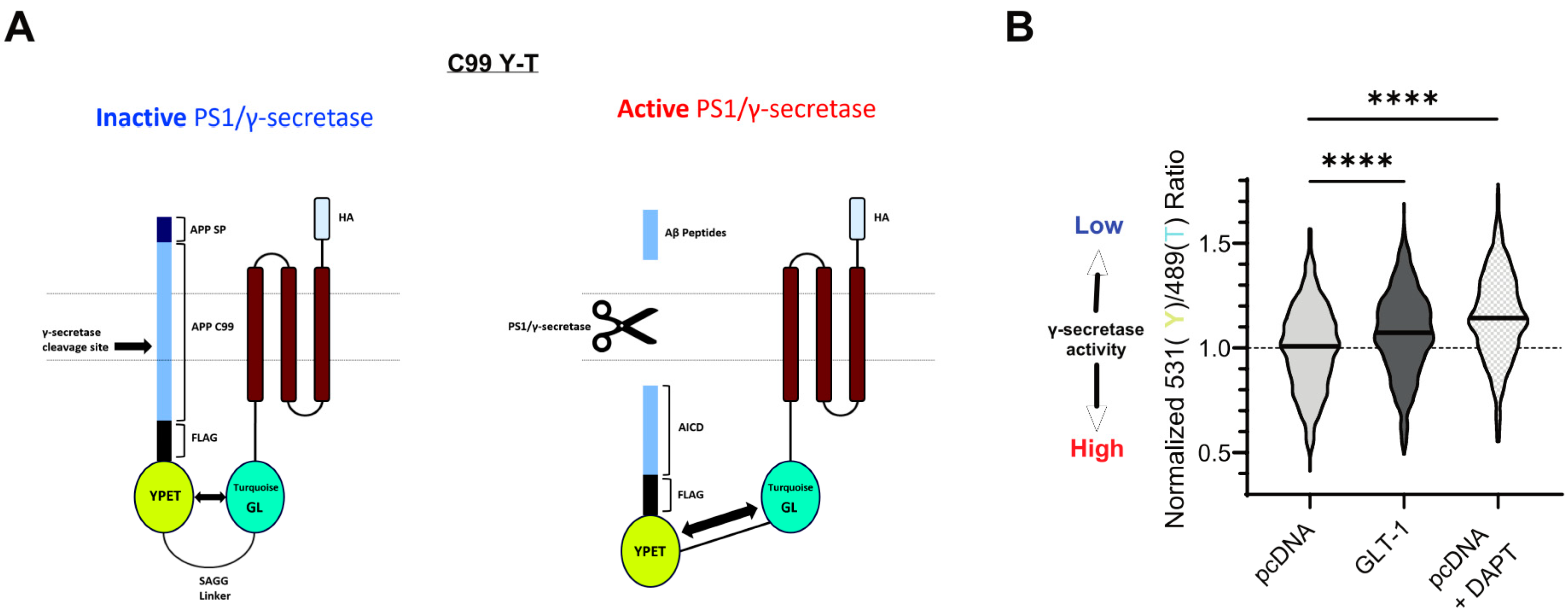
2.7. Spectral Förster Resonance Energy Transfer (FRET) Analysis of γ-Secretase Activity
2.8. Immunocytochemistry
2.9. Fluorescence Lifetime Imaging Microscopy (FLIM)
2.10. Statistics
3. Results
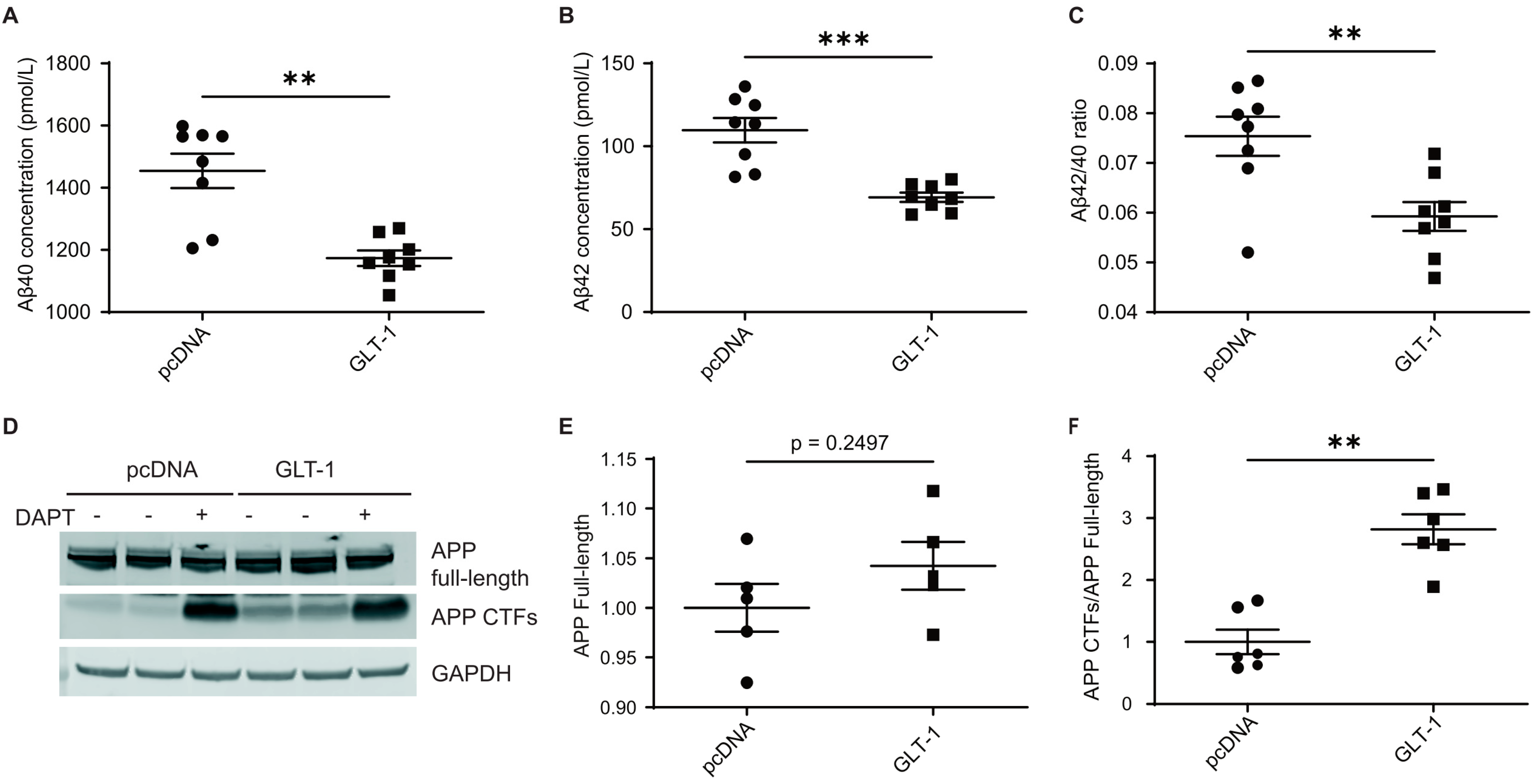
3.1. GLT-1 Overexpression Reduces Aβ40 and Aβ42 Production
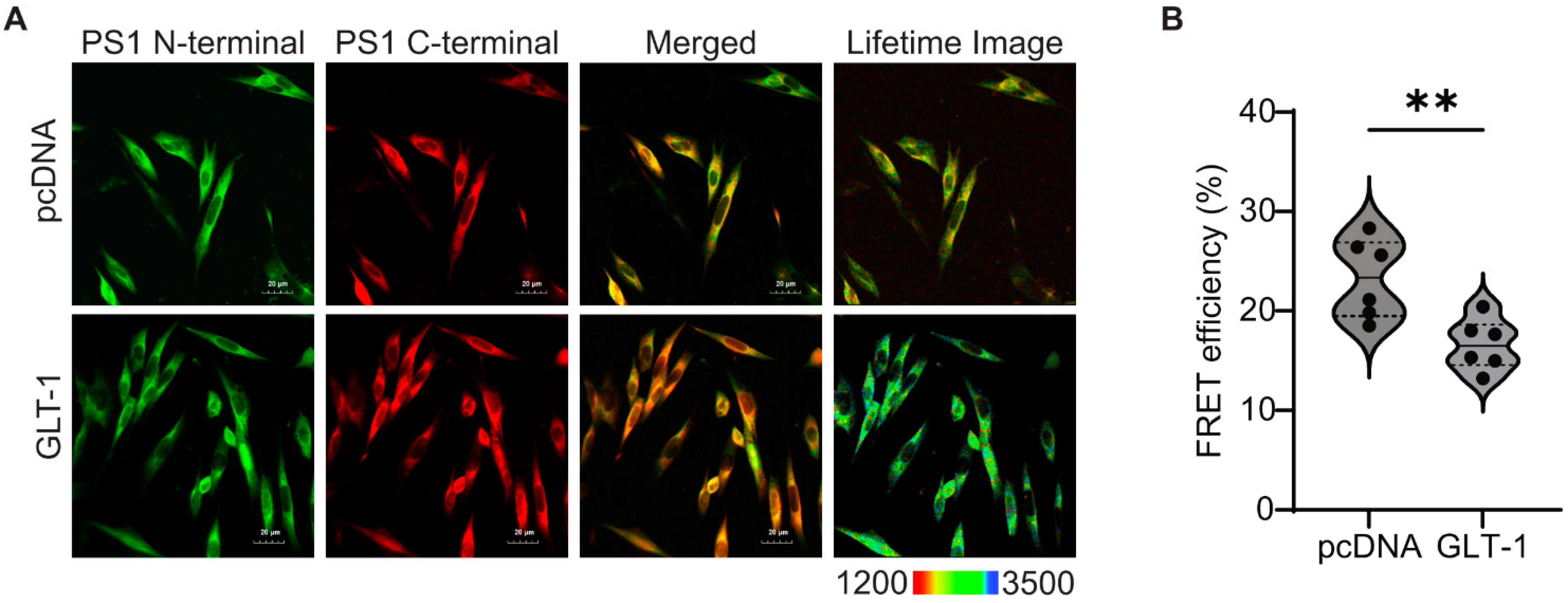
3.2. GLT-1 Promotes “Open” PS1 Conformation
3.3. GLT-1 Overexpression Reduces APP Processing by γ-Secretase
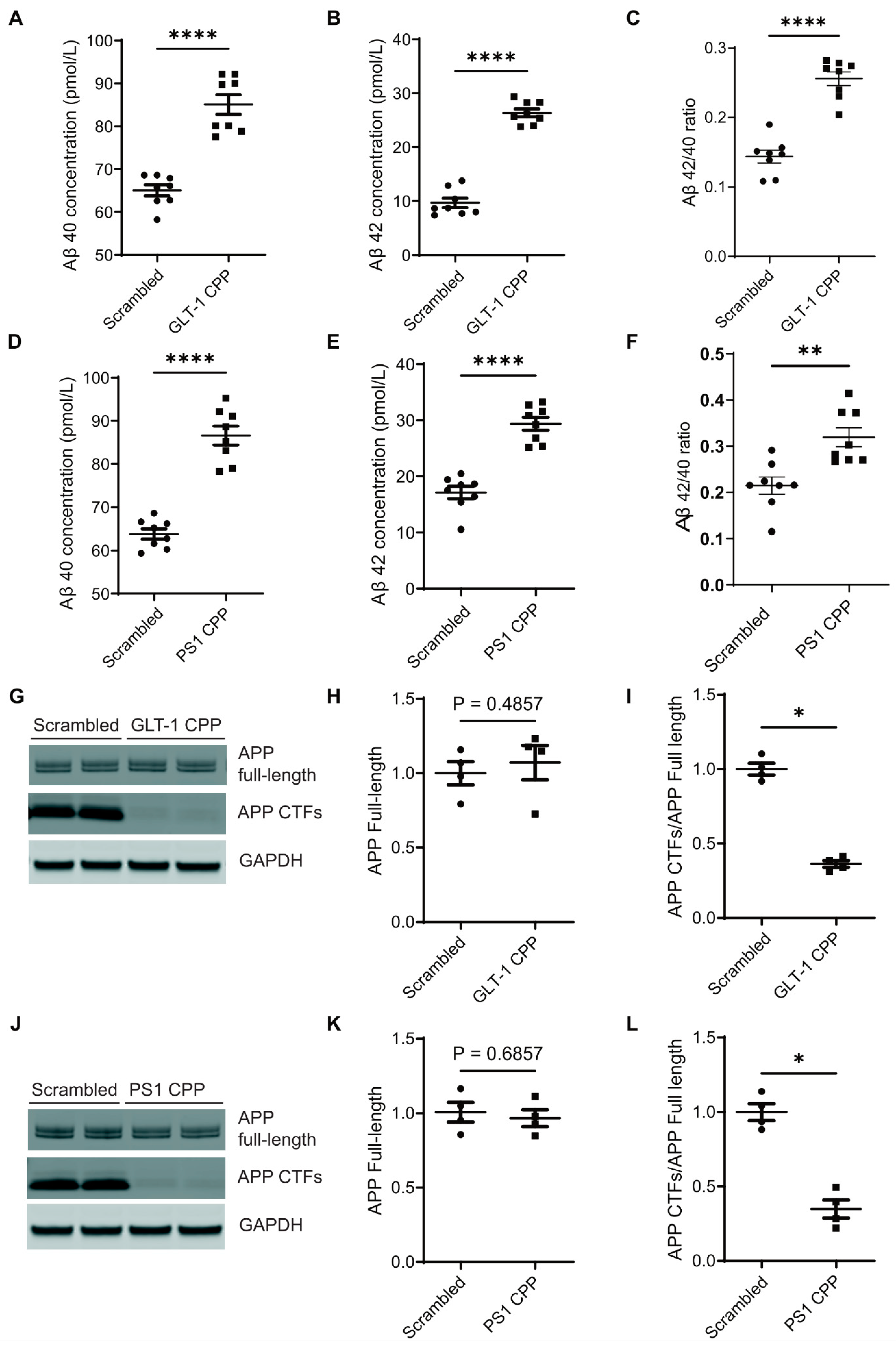
3.4. Disruption of the GLT-1/PS1 Interaction Increases Aβ Production
4. Discussion
Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Morohashi, Y.; Tomita, T.; Iwatsubo, T. Structure of the Catalytic Pore of γ-Secretase Probed by the Accessibility of Substituted Cysteines. J. Neurosci. 2006, 26, 12081–12088. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Farner, K.C.; Nasser-Ghodsi, N.; Jones, P.; Berezovska, O. Reciprocal relationship between APP positioning relative to the membrane and PS1 conformation. Mol. Neurodegener. 2011, 6, 15. [Google Scholar] [CrossRef]
- Elad, N.; De Strooper, B.; Lismont, S.; Hagen, W.; Veugelen, S.; Arimon, M.; Horré, K.; Berezovska, O.; Sachse, C.; Chávez-Gutiérrez, L. The dynamic conformational landscape of γ-secretase. J. Cell Sci. 2014, 128, 589–598. [Google Scholar] [CrossRef]
- Yang, F.; Chen, L.; Yu, Y.; Xu, T.; Chen, L.; Yang, W.; Wu, Q.; Han, Y. Alzheimer’s disease and epilepsy: An increasingly recognized comorbidity. Front. Aging Neurosci. 2022, 14, 940515. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, W.; Yang, F.; Yu, Y.; Xu, T.; Wang, D.; Zhao, Q.; Wu, Q.; Han, Y. The crosstalk between epilepsy and dementia: A systematic review and meta-analysis. Epilepsy Behav. 2024, 152, 109640. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Kim, B.-K.; Kang, K.; Lee, W.-W.; Yoo, I.; Kim, Y.S.; Lee, J.-J. Aphasic Status Epilepticus Associated with Alzheimer’s Disease: Clinical and Electrographic Characteristics. J. Epilepsy Res. 2023, 13, 55–58. [Google Scholar] [CrossRef]
- Nous, A.; Seynaeve, L.; Feys, O.; Wens, V.; De Tiège, X.; Van Mierlo, P.; Baroumand, A.G.; Nieboer, K.; Allemeersch, G.-J.; Mangelschots, S.; et al. Subclinical epileptiform activity in the Alzheimer continuum: Association with disease, cognition and detection method. Alzheimer’s Res. Ther. 2024, 16, 19. [Google Scholar] [CrossRef]
- Vicente, M.; Addo-Osafo, K.; Vossel, K. Latest advances in mechanisms of epileptic activity in Alzheimer’s disease and dementia with Lewy Bodies. Front. Neurol. 2024, 15, 1277613. [Google Scholar] [CrossRef]
- Kalyvas, A.-C.; Dimitriou, M.; Ioannidis, P.; Grigoriadis, N.; Afrantou, T. Alzheimer’s Disease and Epilepsy: Exploring Shared Pathways and Promising Biomarkers for Future Treatments. J. Clin. Med. 2024, 13, 3879. [Google Scholar] [CrossRef]
- Lam, A.D.; Sarkis, R.A.; Pellerin, K.R.; Jing, J.; Dworetzky, B.A.; Hoch, D.B.; Jacobs, C.S.; Lee, J.W.; Weisholtz, D.S.; Zepeda, R.; et al. Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology 2020, 95, e2259–e2270. [Google Scholar] [CrossRef] [PubMed]
- Shea, Y.-F.; Chu, L.-W.; Chan, A.O.-K.; Ha, J.; Li, Y.; Song, Y.-Q. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef]
- Kazim, S.F.; Seo, J.H.; Bianchi, R.; Larson, C.S.; Sharma, A.; Wong, R.K.S.; Gorbachev, K.Y.; Pereira, A.C. Neuronal Network Excitability in Alzheimer’s Disease: The Puzzle of Similar versus Divergent Roles of Amyloid β and Tau. eNeuro 2021, 8, 0418–0420. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid β-Induced Neuronal Hyperexcitability Triggers Progressive Epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef]
- Vossel, K.A.; Ranasinghe, K.G.; Ba, A.J.B.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Vossel, K.; Ranasinghe, K.G.; Beagle, A.J.; La, A.; Pook, K.A.; Castro, M.; Mizuiri, D.; Honma, S.M.; Venkateswaran, N.; Koestler, M.; et al. Effect of Levetiracetam on Cognition in Patients with Alzheimer Disease with and Without Epileptiform Activity. JAMA Neurol. 2021, 78, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Naik, U.P.; Naik, M.U.; Yadav, S.K.; Chaurasia, R.N.; Dash, D. Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications. Biomolecules 2023, 13, 1609. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Hascup, K.N.; Hascup, E.R. Altered Neurotransmission Prior to Cognitive Decline in AβPP/PS1 Mice, a Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 771–776. [Google Scholar] [CrossRef]
- Bragina, L.; Melone, M.; Fattorini, G.; Torres-Ramos, M.; Vallejo-Illarramendi, A.; Matute, C.; Conti, F. GLT-1 down-regulation induced by clozapine in rat frontal cortex is associated with synaptophysin up-regulation. J. Neurochem. 2006, 99, 134–141. [Google Scholar] [CrossRef]
- Mookherjee, P.; Green, P.S.; Watson, G.S.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G.; et al. GLT-1 Loss Accelerates Cognitive Deficit Onset in an Alzheimer’s Disease Animal Model. J. Alzheimer’s Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Huffels, C.F.M.; Middeldorp, J.; Hol, E.M. Aß Pathology and Neuron–Glia Interactions: A Synaptocentric View. Neurochem. Res. 2022, 48, 1026–1046. [Google Scholar] [CrossRef]
- Meeker, K.D.; Meabon, J.S.; Cook, D.G. Partial Loss of the Glutamate Transporter GLT-1 Alters Brain Akt and Insulin Signaling in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 509–520. [Google Scholar] [CrossRef]
- Gao, J.; Liu, L.; Liu, C.; Fan, S.; Liu, L.; Liu, S.; Xian, X.-H.; Li, W.-B. GLT-1 Knockdown Inhibits Ceftriaxone-Mediated Improvements on Cognitive Deficits, and GLT-1 and xCT Expression and Activity in APP/PS1 AD Mice. Front. Aging Neurosci. 2020, 12, 580772. [Google Scholar] [CrossRef]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β1–42Slows Clearance of Synaptically Released Glutamate by Mislocalizing Astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Zoltowska, K.M.; Maesako, M.; Meier, J.; Berezovska, O. Novel interaction between Alzheimer’s disease-related protein presenilin 1 and glutamate transporter 1. Sci. Rep. 2018, 8, 8718. [Google Scholar] [CrossRef] [PubMed]
- Perrin, F.; Sinha, P.; Mitchell, S.P.C.; Sadek, M.; Maesako, M.; Berezovska, O. Identification of PS1/gamma-secretase and glutamate transporter GLT-1 interaction sites. J. Biol. Chem. 2024, 300, 107172. [Google Scholar] [CrossRef]
- Houser, M.C.; Hou, S.S.; Perrin, F.; Turchyna, Y.; Bacskai, B.J.; Berezovska, O.; Maesako, M. A Novel NIR-FRET Biosensor for Reporting PS/γ-Secretase Activity in Live Cells. Sensors 2020, 20, 5980. [Google Scholar] [CrossRef]
- Lehmann, S.; Delaby, C.; Boursier, G.; Catteau, C.; Ginestet, N.; Tiers, L.; Maceski, A.; Navucet, S.; Paquet, C.; Dumurgier, J.; et al. Relevance of Aβ42/40 Ratio for Detection of Alzheimer Disease Pathology in Clinical Routine: The PLMR Scale. Front. Aging Neurosci. 2018, 10, 138. [Google Scholar] [CrossRef]
- Uemura, K.; Lill, C.M.; Li, X.; Peters, J.A.; Ivanov, A.; Fan, Z.; DeStrooper, B.; Bacskai, B.J.; Hyman, B.T.; Berezovska, O. Allosteric Modulation of PS1/γ-Secretase Conformation Correlates with Amyloid β42/40 Ratio. PLoS ONE 2009, 4, e7893. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kong, Q.; Lin, Y.; Stouffer, N.; Schulte, D.A.; Lai, L.; Liu, Q.; Chang, L.-C.; Dominguez, S.; Xing, X.; et al. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J. Exp. Med. 2015, 212, 319–332. [Google Scholar] [CrossRef]
- Mi, D.J.; Dixit, S.; Warner, T.A.; Kennard, J.A.; Scharf, D.A.; Kessler, E.S.; Moore, L.M.; Consoli, D.C.; Bown, C.W.; Eugene, A.J.; et al. Altered glutamate clearance in ascorbate deficient mice increases seizure susceptibility and contributes to cognitive impairment in APP/PSEN1 mice. Neurobiol. Aging 2018, 71, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Berezovska, O.; Lleo, A.; Herl, L.D.; Frosch, M.P.; Stern, E.A.; Bacskai, B.J.; Hyman, B.T. Familial Alzheimer’s Disease Presenilin 1 Mutations Cause Alterations in the Conformation of Presenilin and Interactions with Amyloid Precursor Protein. J. Neurosci. 2005, 25, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Morishima, K.; Takagi-Niidome, S.; Tominaga, A.; Tomita, T. Conformational Dynamics of Transmembrane Domain 3 of Presenilin 1 Is Associated with the Trimming Activity of γ-Secretase. J. Neurosci. 2019, 39, 8600–8610. [Google Scholar] [CrossRef] [PubMed]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.M.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of Intermediate Steps in Amyloid Beta (Aβ) Production under Near-native Conditions. J. Biol. Chem. 2014, 289, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horré, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456. [Google Scholar] [CrossRef]
- Masliah, E.; Hansen, L.; Alford, M.; Deteresa, R.; Mallory, M. Deficient glutamate tranport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1712. [Google Scholar] [CrossRef]
- Perrin, F.; Anderson, L.C.; Mitchell, S.P.C.; Sinha, P.; Turchyna, Y.; Maesako, M.; Houser, M.C.Q.; Zhang, C.; Wagner, S.L.; Tanzi, R.E.; et al. PS1/gamma-secretase acts as rogue chaperone of glutamate transporter EAAT2/GLT-1 in Alzheimer’s disease. Research Square 2023. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, T.S.; Li, S.; Andersen, J.V.; Westi, E.W.; Rotenberg, A.; Wang, J.; Aldana, B.I.; Selkoe, D.J.; Aoki, C.J.; Dulla, C.G.; et al. Neuronal Loss of the Glutamate Transporter GLT-1 Promotes Excitotoxic Injury in the Hippocampus. Front. Cell. Neurosci. 2021, 15, 788262. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Zoltowska, K.M.; Maesako, M.; Lushnikova, I.; Takeda, S.; Keller, L.J.; Skibo, G.; Hyman, B.T.; Berezovska, O. Dynamic presenilin 1 and synaptotagmin 1 interaction modulates exocytosis and amyloid β production. Mol. Neurodegener. 2017, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Islam, S.; Michikawa, M.; Zou, K. Presenilin: A Multi-Functional Molecule in the Pathogenesis of Alzheimer’s Disease and Other Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 1757. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Olivé, M.G.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, P.; Turchyna, Y.; Mitchell, S.P.C.; Sadek, M.; Armagan, G.; Perrin, F.; Maesako, M.; Berezovska, O. Glutamate Transporter 1 as a Novel Negative Regulator of Amyloid β. Cells 2024, 13, 1600. https://doi.org/10.3390/cells13191600
Sinha P, Turchyna Y, Mitchell SPC, Sadek M, Armagan G, Perrin F, Maesako M, Berezovska O. Glutamate Transporter 1 as a Novel Negative Regulator of Amyloid β. Cells. 2024; 13(19):1600. https://doi.org/10.3390/cells13191600
Chicago/Turabian StyleSinha, Priyanka, Yuliia Turchyna, Shane Patrick Clancy Mitchell, Michael Sadek, Gokce Armagan, Florian Perrin, Masato Maesako, and Oksana Berezovska. 2024. "Glutamate Transporter 1 as a Novel Negative Regulator of Amyloid β" Cells 13, no. 19: 1600. https://doi.org/10.3390/cells13191600
APA StyleSinha, P., Turchyna, Y., Mitchell, S. P. C., Sadek, M., Armagan, G., Perrin, F., Maesako, M., & Berezovska, O. (2024). Glutamate Transporter 1 as a Novel Negative Regulator of Amyloid β. Cells, 13(19), 1600. https://doi.org/10.3390/cells13191600