
The Role of CENPK Splice Variant in Abiraterone Response in Metastatic Castration-Resistant Prostate Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
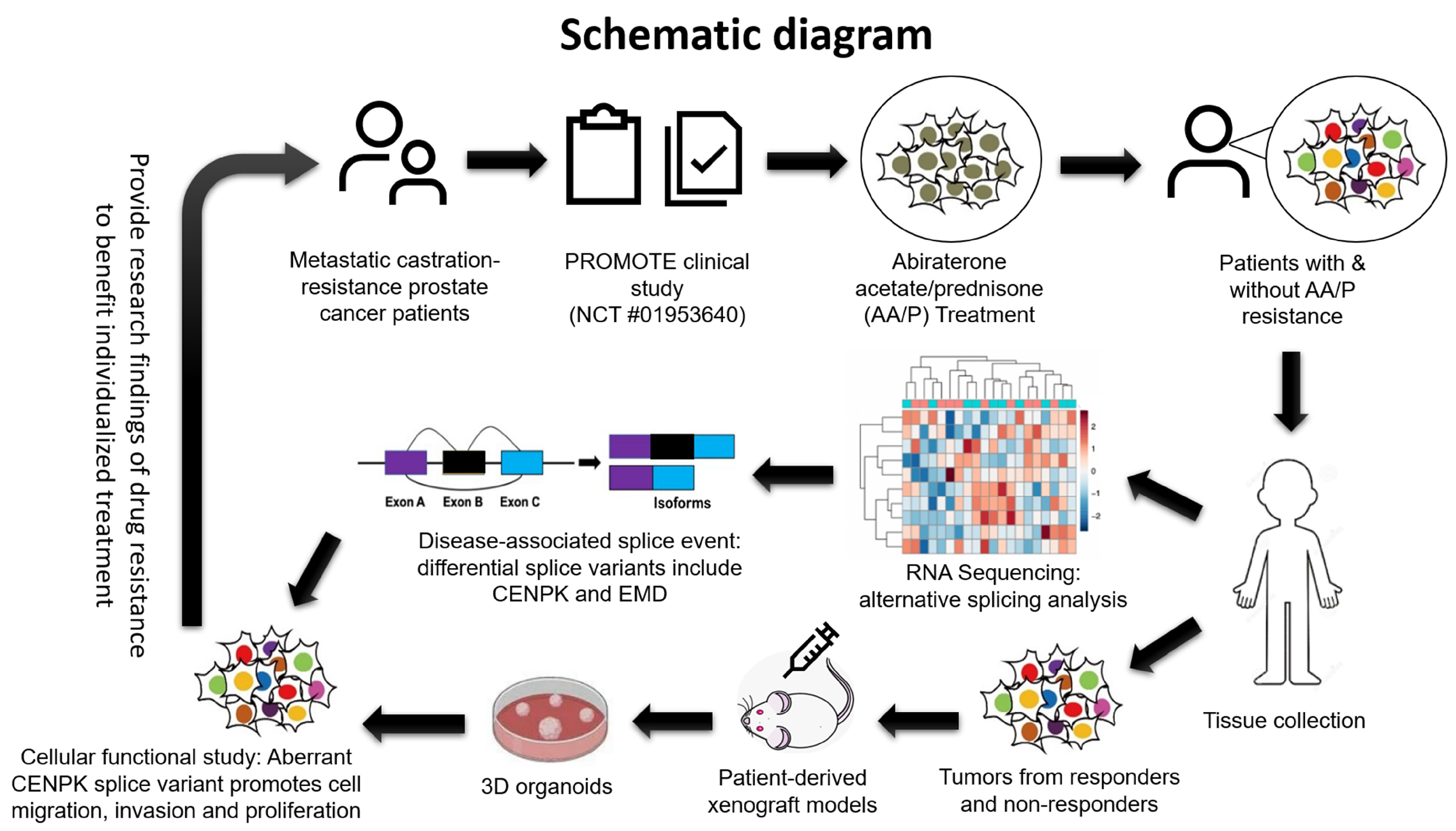
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. PDX Mouse Model Generation and RNA Sequencing
2.3. PDX 3D Organoid Generation
2.4. Cytotoxicity Assay for Cells and 3D Organoids
2.5. Clinical Data and Bioinformatic Analysis
2.6. Plasmids
2.7. Co-Immunoprecipitaion (Co–IP) and Mass Spectrometry
2.8. Transwell Migration and Invasion Assays
2.9. Wound Healing Assay
2.10. Colony Formation Assay
2.11. qRT–PCR
2.12. Western Blot Analysis and Immunofluorescence Staining
2.13. Flow Cytometry
2.14. Statistical Analysis
3. Results
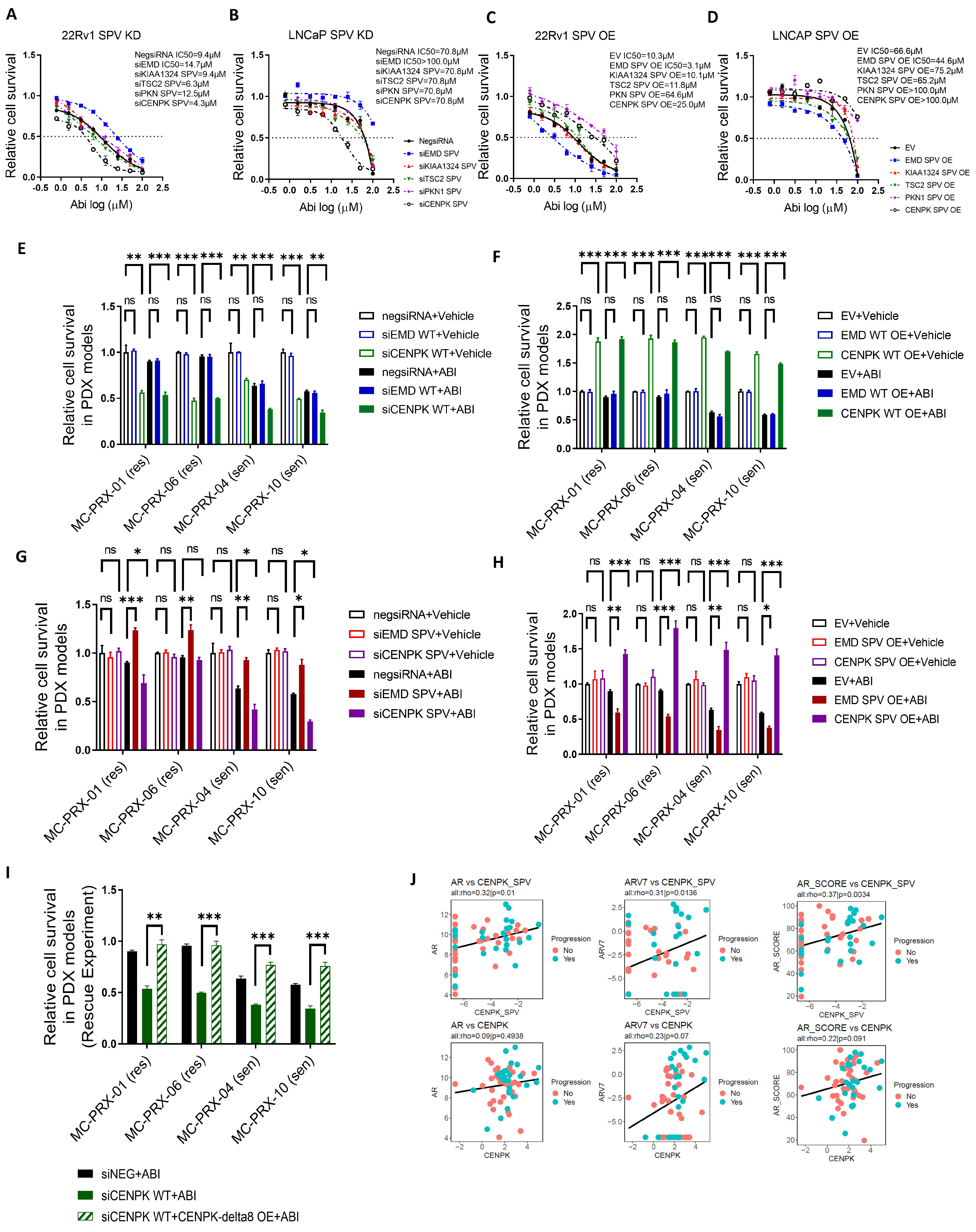
3.1. Splice Variants Are Associated with Abiraterone Response
3.2. CENPK–delta8 as an Abiraterone-Resistant Splice Variant Biomarker in mCRPC Patients
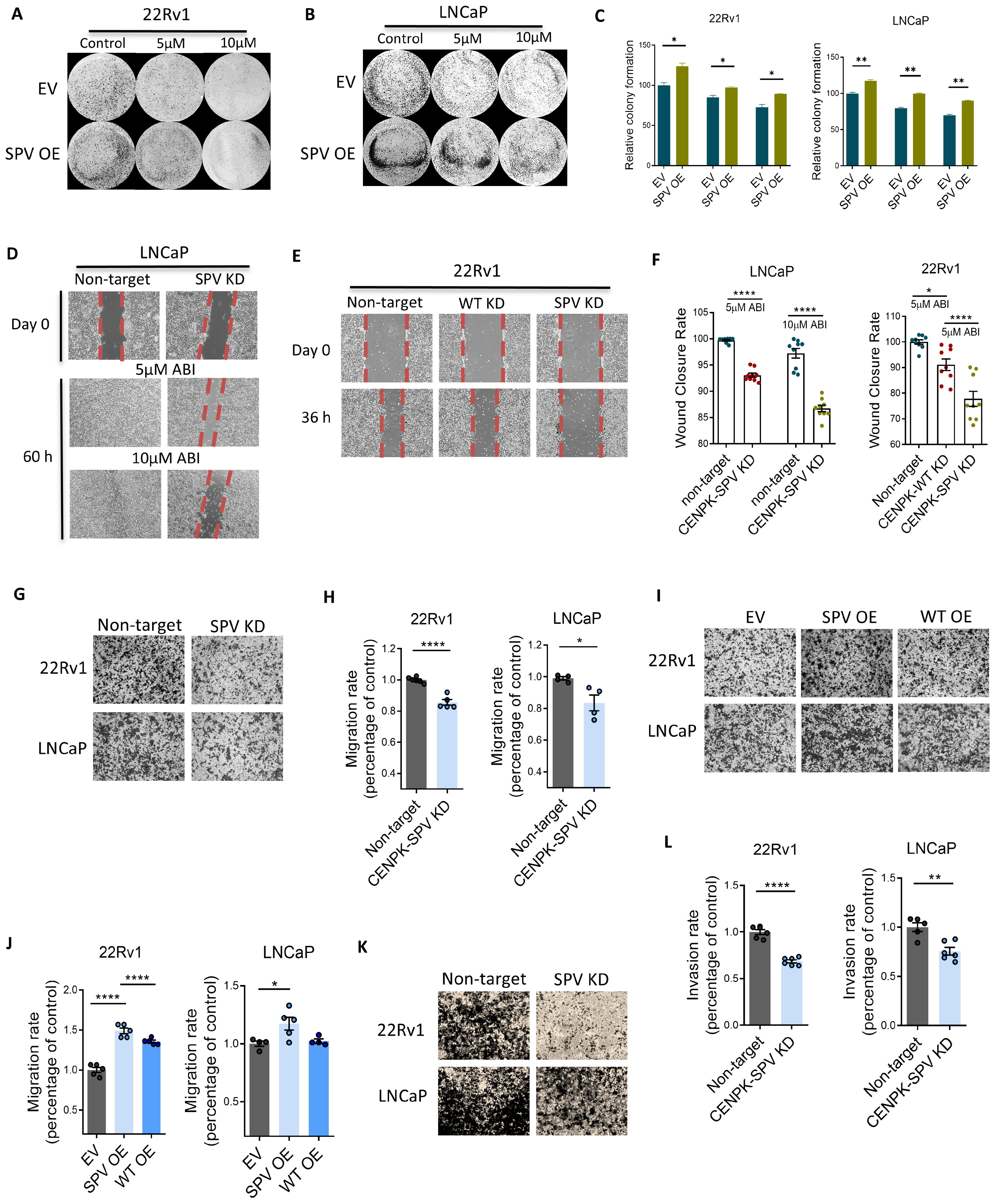
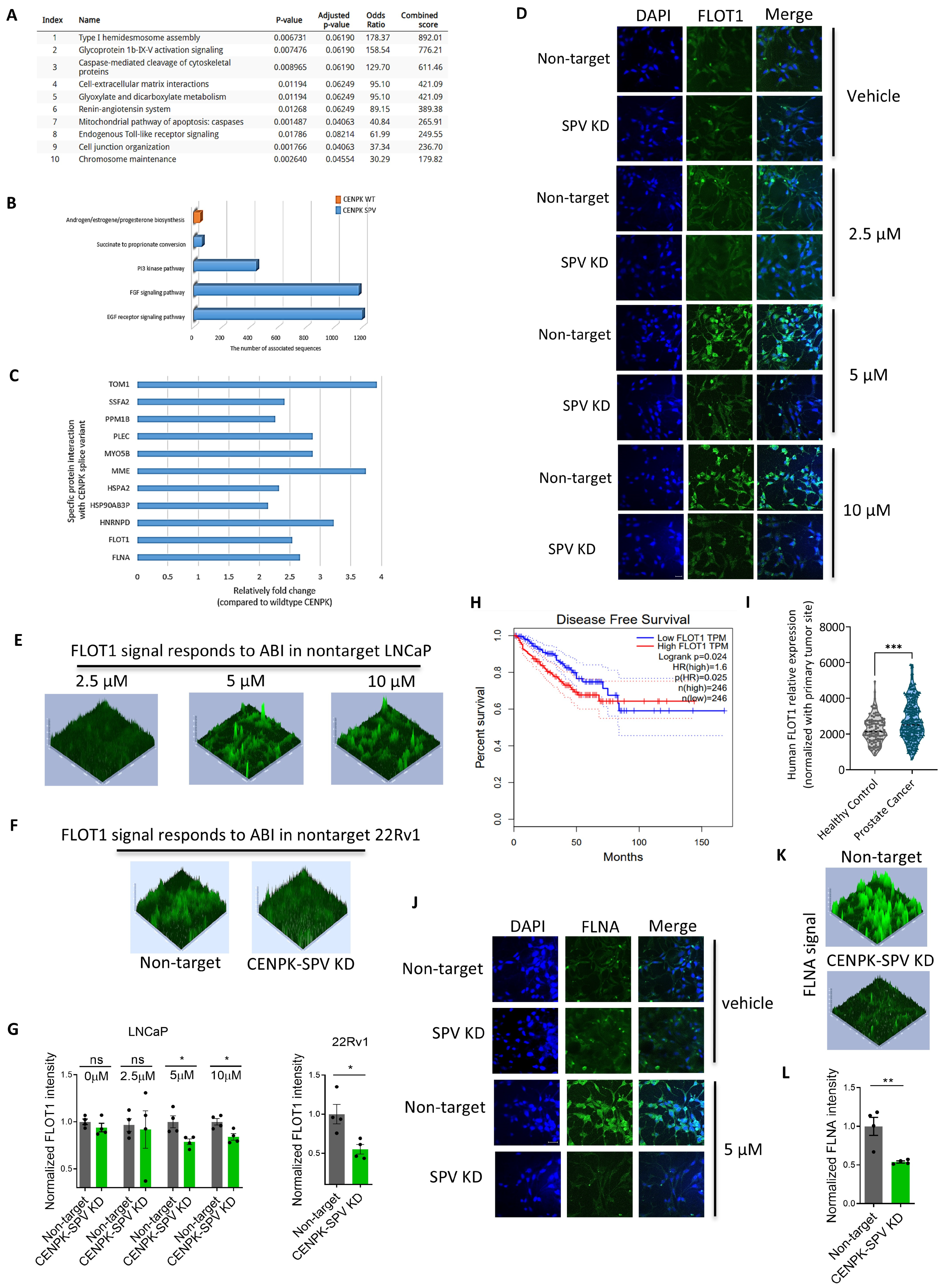
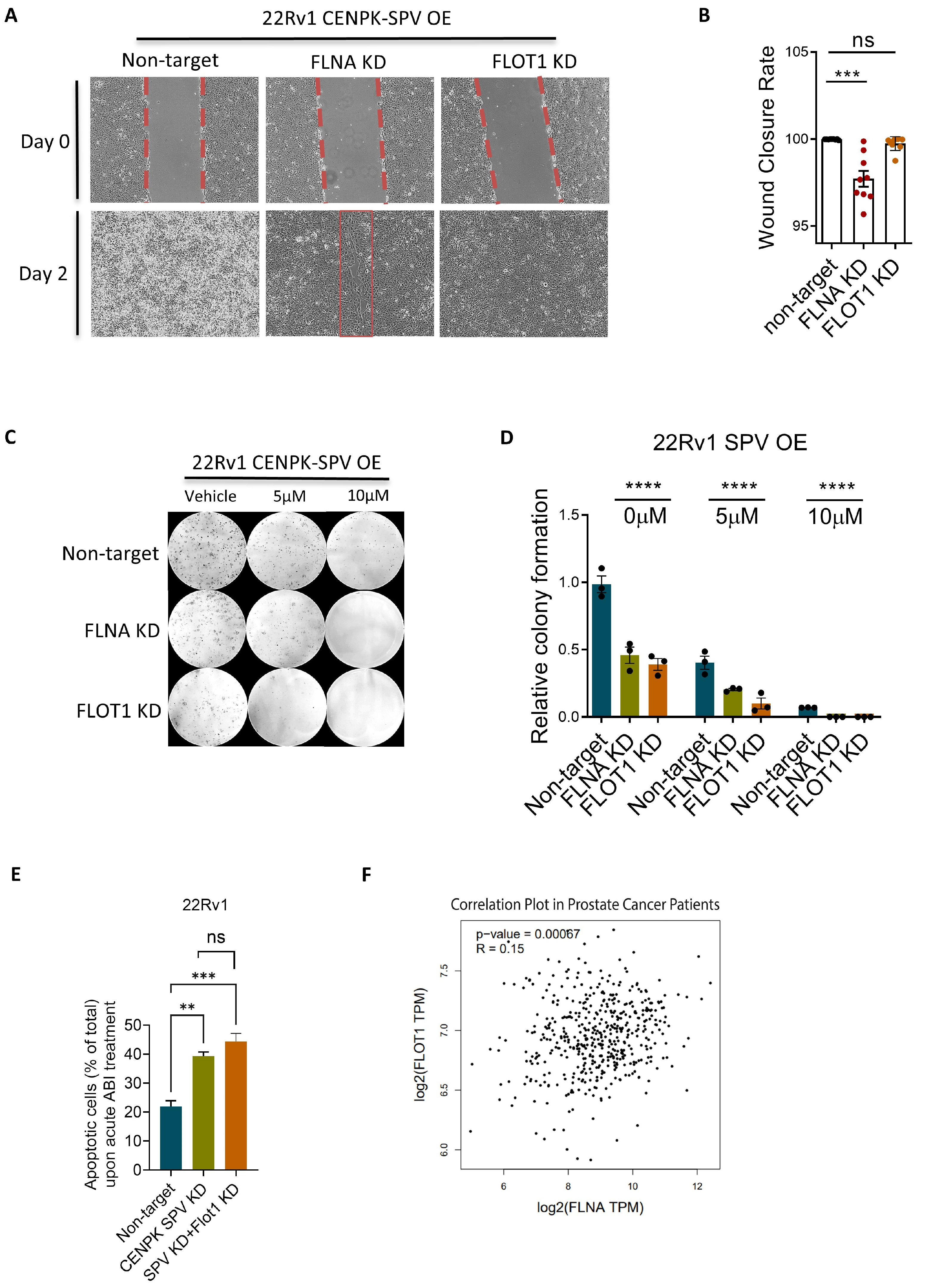
3.3. CENPK–delta8 Contribution to Abiraterone-Resistant Phenotype through Enhanced Cell Migration
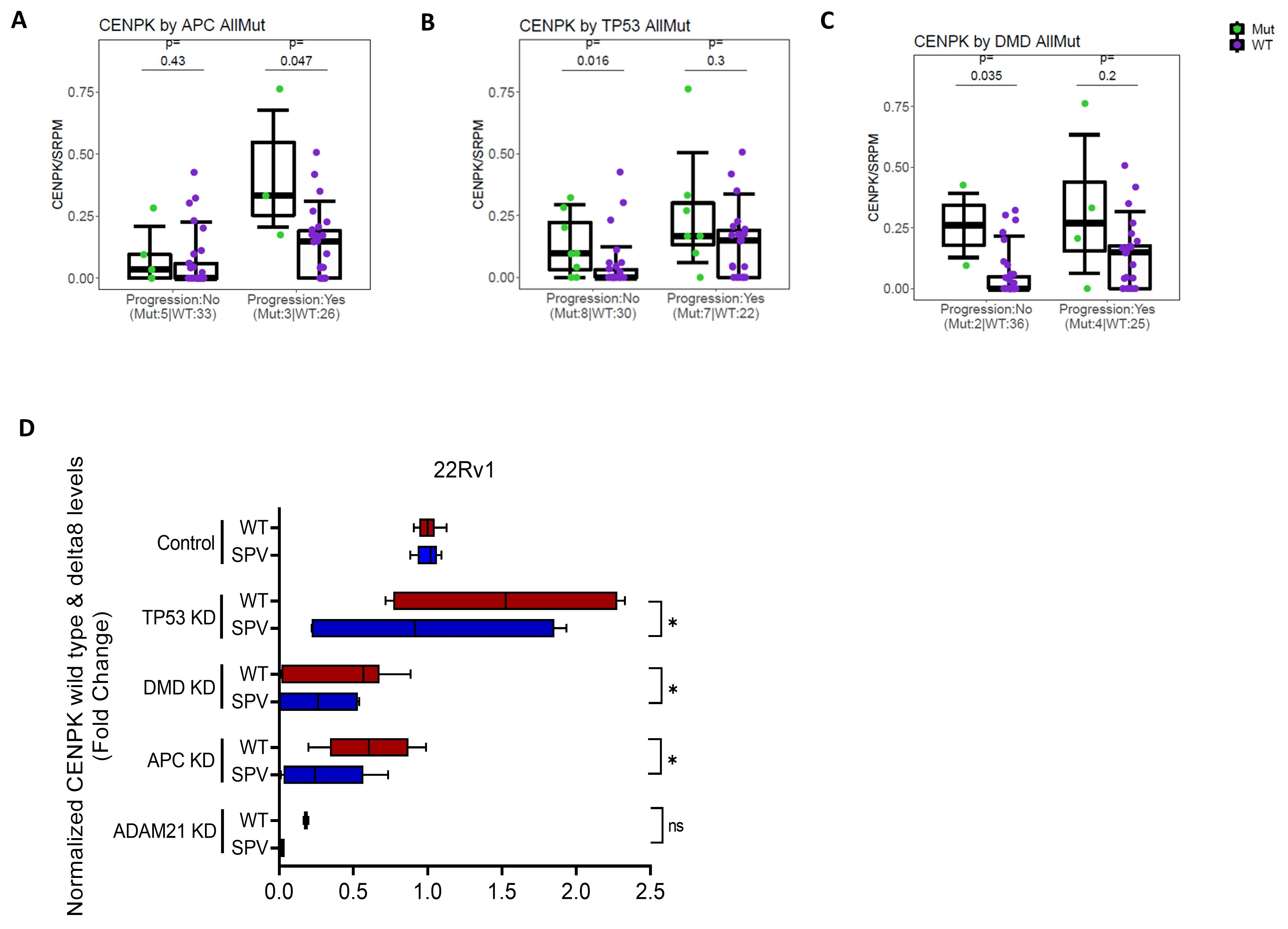
3.4. CENPK–delta8 Is Associated with Known Clinically Relevant Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Elmehrath, A.O.; Afifi, A.M.; Al-Husseini, M.J.; Saad, A.M.; Wilson, N.; Shohdy, K.S.; Pilie, P.; Sonbol, M.B.; Alhalabi, O. Causes of Death Among Patients with Metastatic Prostate Cancer in the US From 2000 to 2016. JAMA Netw. Open 2021, 4, e2119568. [Google Scholar] [CrossRef] [PubMed]
- Chaput, G.; Sumar, N. Endocrine therapies for breast and prostate cancers: Essentials for primary care. Can. Fam. Physician 2022, 68, 271–276. [Google Scholar] [CrossRef]
- DE Nunzio, C.; Presicce, F.; Giacinti, S.; Bassanelli, M.; Tubaro, A. Castration-resistance prostate cancer: What is in the pipeline? Minerva Urol. Nefrol. 2018, 70, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.D.; Ellison, S.J.; Baker, J.G.; Stagg, D.B.; Wardell, S.E.; Park, S.; Alley, H.M.; Baldi, R.M.; Yllanes, A.; Andreano, K.J.; et al. Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J. Clin. Investig. 2017, 127, 2326–2338. [Google Scholar] [CrossRef]
- Kluetz, P.G.; Ning, Y.M.; Maher, V.E.; Zhang, L.; Tang, S.; Ghosh, D.; Aziz, R.; Palmby, T.; Pfuma, E.; Zirkelbach, J.F.; et al. Abiraterone acetate in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. 2013, 19, 6650–6656. [Google Scholar] [CrossRef]
- Ning, Y.M.; Pierce, W.; Maher, V.E.; Karuri, S.; Tang, S.H.; Chiu, H.J.; Palmby, T.; Zirkelbach, J.F.; Marathe, D.; Mehrotra, N.; et al. Enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer who have previously received docetaxel: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. 2013, 19, 6067–6073. [Google Scholar] [CrossRef]
- Jayaram, A.; Wingate, A.; Wetterskog, D.; Wheeler, G.; Sternberg, C.N.; Jones, R.; Berruti, A.; Lefresne, F.; Lahaye, M.; Thomas, S.; et al. Plasma tumor gene conversions after one cycle abiraterone acetate for metastatic castration-resistant prostate cancer: A biomarker analysis of a multicenter international trial. Ann. Oncol. 2021, 32, 726–735. [Google Scholar] [CrossRef]
- Buonerba, C.; Ferro, M.; Dolce, P.; Crocetto, F.; Verde, A.; Lucarelli, G.; Scafuri, L.; Facchini, S.; Vaia, A.; Marinelli, A.; et al. Predictors of efficacy of androgen-receptor-axis-targeted therapies in patients with metastatic castration-sensitive prostate cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2020, 151, 102992. [Google Scholar] [CrossRef]
- Wang, L.; Dehm, S.M.; Hillman, D.W.; Sicotte, H.; Tan, W.; Gormley, M.; Bhargava, V.; Jimenez, R.; Xie, F.; Yin, P.; et al. A prospective genome-wide study of prostate cancer metastases reveals association of wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann. Oncol. 2018, 29, 352–360. [Google Scholar] [CrossRef]
- Mao, S.; Qian, Y.; Wei, W.; Lin, X.; Ling, Q.; Ye, W.; Li, F.; Pan, J.; Zhou, Y.; Zhao, Y.; et al. FLOT1 knockdown inhibits growth of AML cells through triggering apoptosis and pyroptosis. Ann. Hematol. 2023, 102, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Luo, J.; Wang, B.; Wang, D.; Xie, X.; Yuan, L.; Guo, J.; Xi, S.; Gao, J.; Lin, X.; et al. Microrna-124 targets flotillin-1 to regulate proliferation and migration in breast cancer. Mol. Cancer 2013, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhou, Y.; Pan, Y.; Liu, P.; Yu, K.; Chen, S. Long non-coding RNA A1BG-AS1 promotes tumorigenesis in breast cancer by sponging microRNA-485-5p and consequently increasing expression of FLOT1 expression. Hum. Cell 2021, 34, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wu, Z.; Lin, X.; Yu, C.; Shi, T.; Zeng, Y.; Wang, X.; Li, J.; Song, L. Knockdown of FLOT1 impairs cell proliferation and tumorigenicity in breast cancer through upregulation of FOXO3a. Clin. Cancer Res. 2011, 17, 3089–3099. [Google Scholar] [CrossRef]
- Song, L.; Gong, H.; Lin, C.; Wang, C.; Liu, L.; Wu, J.; Li, M.; Li, J. Flotillin-1 promotes tumor necrosis factor-α receptor signaling and activation of NF-κB in esophageal squamous cell carcinoma cells. Gastroenterology 2012, 143, 995–1005. [Google Scholar] [CrossRef]
- Wang, R.; Chen, Z.; Zhang, Y.; Xiao, S.; Zhang, W.; Hu, X.; Xiao, Q.; Liu, Q.; Wang, X. Flotillin-1 is a prognostic biomarker for glioblastoma and promotes cancer development through enhancing invasion and altering tumour microenvironment. J. Cell Mol. Med. 2023, 27, 392–402. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, Y.; Mao, Q.; Fan, W.; Xu, L.; Chen, Y.; Wang, J. FLOT1 promotes tumor development, induces epithelial-mesenchymal transition, and modulates the cell cycle by regulating the Erk/Akt signaling pathway in lung adenocarcinoma. Thorac. Cancer 2019, 10, 909–917. [Google Scholar] [CrossRef]
- Baig, N.; Li, Z.; Lu, J.; Chen, H.; Yu, S.; Li, T.; Niu, Z.; Niu, J. Clinical significance and comparison of flotillin 1 expression in left and right colon cancer. Oncol. Lett. 2019, 18, 997–1004. [Google Scholar] [CrossRef]
- Wei, J.; Wang, R.; Lu, Y.; He, S.; Ding, Y. Flotillin-1 promotes progression and dampens chemosensitivity to cisplatin in gastric cancer via ERK and AKT signaling pathways. Eur. J. Pharmacol. 2022, 916, 174631. [Google Scholar] [CrossRef]
- Zhang, S.H.; Wang, C.J.; Shi, L.; Li, X.H.; Zhou, J.; Song, L.B.; Liao, W.T. High Expression of FLOT1 Is Associated with Progression and Poor Prognosis in Hepatocellular Carcinoma. PLoS ONE 2013, 8, e64709. [Google Scholar] [CrossRef]
- Ye, D.M.; Ye, S.C.; Yu, S.Q.; Shu, F.F.; Xu, S.S.; Chen, Q.Q.; Wang, Y.L.; Tang, Z.T.; Pan, C. Drug-resistance reversal in colorectal cancer cells by destruction of flotillins, the key lipid rafts proteins. Neoplasma 2019, 66, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Jiang, Y.; Yang, H.; Zhao, D.; Li, L.; Liu, X. FLNA promotes chemoresistance of colorectal cancer through inducing epithelial-mesenchymal transition and smad2 signaling pathway. Am. J. Cancer Res. 2020, 10, 403–423. [Google Scholar] [PubMed]
- Yue, J.; Huhn, S.; Shen, Z. Complex roles of filamin-A mediated cytoskeleton network in cancer progression. Cell Biosci. 2013, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, L.; Xu, P.; Li, Y.; Ji, Z.; Kang, X. Filamin A Is a Potential Driver of Breast Cancer Metastasis. Front. Oncol. 2022, 12, 836126. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.S. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Sinnwell, J.P.; Thompson, K.J.; Copland, J.A.; Marlow, L.A.; Miller, J.L.; Yin, P.; Gao, B.; et al. Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res. 2017, 19, 130. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, H.; Gu, Y.; John, A.; Wei, L.; Huang, M.; Yu, J.; Adeosun, A.A.; Weinshilboum, R.M.; Wang, L. 3D CRISPR screen in prostate cancer cells reveals PARP inhibitor sensitization through TBL1XR1-SMC3 interaction. Front. Oncol. 2022, 12, 999302. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Huang, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Mitochondrial stress-induced H4K12 hyperacetylation dysregulates transcription in Parkinson’s disease. Front. Cell Neurosci. 2024, 18, 1422362. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Huang, M.; Lou, D.; Charli, A.; Kong, D.; Jin, H.; Zenitsky, G.; Anantharam, V.; Kanthasamy, A.; Wang, Z.; Kanthasamy, A.G. Mitochondrial dysfunction-induced H3K27 hyperacetylation perturbs enhancers in Parkinson’s disease. JCI Insight 2021, 6, e138088. [Google Scholar] [CrossRef] [PubMed]
- Van Moerbeke, M.; Kasim, A.; Shkedy, Z. The Usage of Exon-Exon Splice Junctions for the Detection of Alternative Splicing using the REIDS model. Sci. Rep. 2018, 8, 8331. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Malovic, E.; Ealy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Microglial immune regulation by epigenetic reprogramming through histone H3K27 acetylation in neuroinflammation. Front. Immunol. 2023, 14, 1052925. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Fasoulaki, V.; Kalamida, D.; Mitrakas, A.; Kakouratos, C.; Lialiaris, T.; Koukourakis, M.I. CYP17A1 and Androgen-Receptor Expression in Prostate Carcinoma Tissues and Cancer Cell Lines. Curr. Urol. 2019, 13, 157–165. [Google Scholar] [CrossRef]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2013, 20, R49–R64. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Chen, X.; Shao, Y.; Li, Y.; Yang, Z.; Chen, Y.; Yu, W.; Shang, Z.; Wei, W. The cell cycle gene centromere protein K. Transl. Cancer Res. 2022, 11, 1099–1111. [Google Scholar] [CrossRef]
- Fei, D.L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C.L.; Bradley, R.K.; Varmus, H. Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet. 2016, 12, e1006384. [Google Scholar] [CrossRef]
- Qin, S.; Gao, H.; Kim, W.; Zhang, H.; Gu, Y.; Kalari, K.R.; Sinnwell, J.P.; Scholz, J.A.; Xie, F.; Yin, P.; et al. Biomarkers for Predicting Abiraterone Treatment Outcome and Selecting Alternative Therapies in Castration-Resistant Prostate Cancer. Clin. Pharmacol. Ther. 2022, 111, 1296–1306. [Google Scholar] [CrossRef]
- Corn, P.G.; Wang, F.; McKeehan, W.L.; Navone, N. Targeting fibroblast growth factor pathways in prostate cancer. Clin. Cancer Res. 2013, 19, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Yamamoto, Y.; Tsubaki, M.; Matsuda, T.; Kimura, A.; Shimo, N.; Nishida, S. PI3K/Akt/YAP signaling promotes migration and invasion of DLD-1 colorectal cancer cells. Oncol. Lett. 2022, 23, 106. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef]
- Zhao, T.; Mu, X.; You, Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget 2017, 8, 53819–53828. [Google Scholar] [CrossRef]
- Di Donato, M.; Zamagni, A.; Galasso, G.; Di Zazzo, E.; Giovannelli, P.; Barone, M.V.; Zanoni, M.; Gunelli, R.; Costantini, M.; Auricchio, F.; et al. The androgen receptor/filamin A complex as a target in prostate cancer microenvironment. Cell Death Dis. 2021, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.; Kwon, H.; Choi, M.; Lee, J.; Pak, Y. Sumoylation of Flotillin-1 promotes EMT in metastatic prostate cancer by suppressing Snail degradation. Oncogene 2019, 38, 3248–3260. [Google Scholar] [CrossRef]
- Koh, M.; Yong, H.Y.; Kim, E.S.; Son, H.; Jeon, Y.R.; Hwang, J.S.; Kim, M.O.; Cha, Y.; Choi, W.S.; Noh, D.Y.; et al. A novel role for flotillin-1 in H-Ras-regulated breast cancer aggressiveness. Int. J. Cancer 2016, 138, 1232–1245. [Google Scholar] [CrossRef]
- Planchon, D.; Rios Morris, E.; Genest, M.; Comunale, F.; Vacher, S.; Bièche, I.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Linder, S.; et al. MT1-MMP targeting to endolysosomes is mediated by upregulation of flotillins. J. Cell Sci. 2018, 131, jcs218925. [Google Scholar] [CrossRef]
- Zhang, P.F.; Zeng, G.Q.; Hu, R.; Li, C.; Yi, H.; Li, M.Y.; Li, X.H.; Qu, J.Q.; Wan, X.X.; He, Q.Y.; et al. Identification of flotillin-1 as a novel biomarker for lymph node metastasis and prognosis of lung adenocarcinoma by quantitative plasma membrane proteome analysis. J. Proteomics 2012, 77, 202–214. [Google Scholar] [CrossRef]
- Tomiyama, A.; Uekita, T.; Kamata, R.; Sasaki, K.; Takita, J.; Ohira, M.; Nakagawara, A.; Kitanaka, C.; Mori, K.; Yamaguchi, H.; et al. Flotillin-1 regulates oncogenic signaling in neuroblastoma cells by regulating ALK membrane association. Cancer Res. 2014, 74, 3790–3801. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Y.; Chen, S.W.; Li, F.J.; Zhuang, S.M.; Wang, L.P.; Zhang, J.; Song, M. Prognostic significance of Flotillin1 expression in clinically N0 tongue squamous cell cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 996–1003. [Google Scholar] [PubMed]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; de Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Rawat, C.; Heemers, H.V. Alternative splicing in prostate cancer progression and therapeutic resistance. Oncogene 2024, 43, 1655–1668. [Google Scholar] [CrossRef] [PubMed]
- Olender, J.; Lee, N.H. Role of Alternative Splicing in Prostate Cancer Aggressiveness and Drug Resistance in African Americans. Adv. Exp. Med. Biol. 2019, 1164, 119–139. [Google Scholar] [CrossRef]
- Bradley, R.K.; Anczuków, O. RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer 2023, 23, 135–155. [Google Scholar] [CrossRef]
- Urbanski, L.M.; Leclair, N.; Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, W.; Wang, Y.; Yao, M.; Du, L.; Zhang, X.; Huang, Y.; Wang, J.; Yu, H.; Bian, X. Comprehensive analysis of alternative splicing profiling reveals novel events associated with prognosis and the infiltration of immune cells in prostate cancer. Transl. Androl. Urol. 2021, 10, 3056–3068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, J.; Ren, S.; Han, B.; Tian, R.; Zuo, X.; Liu, H.; Wang, Z.; Cui, Y.; Liu, L.; et al. Comprehensively analysis of splicing factors to construct prognosis prediction classifier in prostate cancer. J. Cell Mol. Med. 2023, 27, 2684–2700. [Google Scholar] [CrossRef] [PubMed]
- Mou, Z.; Spencer, J.; McGrath, J.S.; Harries, L.W. Comprehensive analysis of alternative splicing across multiple transcriptomic cohorts reveals prognostic signatures in prostate cancer. Hum. Genom. 2023, 17, 97. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef]
- Antonopoulou, E.; Ladomery, M. Targeting Splicing in Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1287. [Google Scholar] [CrossRef]
- Zhao, S.; Liao, J.; Zhang, S.; Shen, M.; Li, X.; Zhou, L. The positive relationship between androgen receptor splice variant-7 expression and the risk of castration-resistant prostate cancer: A cumulative analysis. Front. Oncol. 2023, 13, 1053111. [Google Scholar] [CrossRef]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef]
- Giacinti, S.; Bassanelli, M.; Aschelter, A.M.; Milano, A.; Roberto, M.; Marchetti, P. Resistance to abiraterone in castration-resistant prostate cancer: A review of the literature. Anticancer. Res. 2014, 34, 6265–6269. [Google Scholar]
- Carceles-Cordon, M.; Kelly, W.K.; Gomella, L.; Knudsen, K.E.; Rodriguez-Bravo, V.; Domingo-Domenech, J. Cellular rewiring in lethal prostate cancer: The architect of drug resistance. Nat. Rev. Urol. 2020, 17, 292–307. [Google Scholar] [CrossRef]
- Melnyk, J.E.; Steri, V.; Nguyen, H.G.; Hwang, Y.C.; Gordan, J.D.; Hann, B.; Feng, F.Y.; Shokat, K.M. Targeting a splicing-mediated drug resistance mechanism in prostate cancer by inhibiting transcriptional regulation by PKCβ1. Oncogene 2022, 41, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Bhatt, R.; Brown, C.; Brown, E.A.; Buhr, D.L.; Chantranuvatana, K.; Danaher, P.; Dunaway, D.; Garrison, R.G.; Geiss, G.; et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 2022, 40, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Cheeseman, I.M.; Hori, T.; Okawa, K.; McLeod, I.X.; Yates, J.R.; Desai, A.; Fukagawa, T. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat. Cell Biol. 2006, 8, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cao, L.; Ke, L.; Yan, Y.; Luo, H.; Hu, X.; Niu, J.; Li, H.; Xu, H.; Chen, W.; et al. Knockdown of CENPK inhibits cell growth and facilitates apoptosis via PTEN-PI3K-AKT signalling pathway in gastric cancer. J. Cell Mol. Med. 2021, 25, 8890–8903. [Google Scholar] [CrossRef]
- Tian, H.; Wang, F.; Deng, Y.; Ying, L.; Fang, W.; Chen, D.; Miao, C.; Li, H.; Sun, S.; Ma, Y.; et al. Centromeric protein K (CENPK) promotes gastric cancer proliferation and migration via interacting with XRCC5. Gastric Cancer 2022, 25, 879–895. [Google Scholar] [CrossRef]
- Thalwieser, Z.; Király, N.; Fonódi, M.; Csortos, C.; Boratkó, A. Protein phosphatase 2A-mediated flotillin-1 dephosphorylation up-regulates endothelial cell migration and angiogenesis regulation. J. Biol. Chem. 2019, 294, 20196–20206. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, L.; Meng, S.; Huang, L.; Luo, S.; Wu, X.; Gong, X. Flotillin-1 enhances radioresistance through reducing radiation-induced DNA damage and promoting immune escape via STING signaling pathway in non-small cell lung cancer. Cancer Biol. Ther. 2023, 24, 2203332. [Google Scholar] [CrossRef]
- Cannaerts, E.; Shukla, A.; Hasanhodzic, M.; Alaerts, M.; Schepers, D.; Van Laer, L.; Girisha, K.M.; Hojsak, I.; Loeys, B.; Verstraeten, A. FLNA mutations in surviving males presenting with connective tissue findings: Two new case reports and review of the literature. BMC Med. Genet. 2018, 19, 140. [Google Scholar] [CrossRef]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Yu, M.; Hong, W.; Ruan, S.; Guan, R.; Tu, L.; Huang, B.; Hou, B.; Jian, Z.; Ma, L.; Jin, H. Genome-Wide Profiling of Prognostic Alternative Splicing Pattern in Pancreatic Cancer. Front. Oncol. 2019, 9, 773. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Qin, S.; Gao, H.; Kim, W.; Xie, F.; Yin, P.; John, A.; Weinshilboum, R.M.; Wang, L. The Role of CENPK Splice Variant in Abiraterone Response in Metastatic Castration-Resistant Prostate Cancer. Cells 2024, 13, 1622. https://doi.org/10.3390/cells13191622
Huang M, Qin S, Gao H, Kim W, Xie F, Yin P, John A, Weinshilboum RM, Wang L. The Role of CENPK Splice Variant in Abiraterone Response in Metastatic Castration-Resistant Prostate Cancer. Cells. 2024; 13(19):1622. https://doi.org/10.3390/cells13191622
Chicago/Turabian StyleHuang, Minhong, Sisi Qin, Huanyao Gao, Wootae Kim, Fang Xie, Ping Yin, August John, Richard M. Weinshilboum, and Liewei Wang. 2024. "The Role of CENPK Splice Variant in Abiraterone Response in Metastatic Castration-Resistant Prostate Cancer" Cells 13, no. 19: 1622. https://doi.org/10.3390/cells13191622