
β-Amyloids and Immune Responses Associated with Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Introduction
2. The Role of β-Amyloid and Its Forms in the Pathogenesis of AD

{kind=link}
| Mutation in Aβ | Effects |
|---|---|
| A2V | Increased Aβ production [63,64] Acceleration of Aβ aggregation [64,65,66] |
| English (H6R) | Increased Zn2+-dependent oligomerization of Aβ [23] Increased neurotoxicity of Aβ oligomers [24,25] Acceleration of Aβ fibril formation [26] |
| Tottori (D7N) | Acceleration of Aβ aggregation [27] Increased oligomerization of Aβ [24] Increased neurotoxicity of Aβ oligomers [24] Acceleration of Aβ fibril formation [26] |
| Taiwan (D7H) | Increased Aβ production [28,29] Increased Aβ42/40 ratio [28] Increased Zn2+/Cu2+-dependent oligomerization of Aβ [28,29] Increased neurotoxicity of Aβ oligomers [28,29] |
| Post-translational modification of Aβ | Effects |
| Iso-D7-Aβ (isomerized Asp7 residue) | Increased Zn2+-dependent oligomerization of Aβ [34,36,38,43,48,49] Increased neurotoxicity of Aβ oligomers [43] Age-related accumulation [44] |
| 3pE-Aβ(pyroglutamate modified glutamic acid residue) | Acceleration of Aβ aggregation [31,46] Increased oligomerization of Aβ [31,45,47] Increased neurotoxicity of Aβ oligomers [31,35,45,46] Age-related accumulation [32] |
| pS8-Aβ(phosphorylated Ser8 residue) | Acceleration of Aβ aggregation [39,40,41] Increased oligomerization of Aβ [39,40,41] Increased neurotoxicity of Aβ oligomers [39,40,41] Age-related accumulation in APP-PS1 tg mice [39,40] |
| Inhibition of Zn2+-dependent oligomerization of Aβ [30] Prevention of Na+/K+ ATPase inhibition [30] Reduction of amyloid plaques in APP-PS1 tg mice [30,37] |
2.1. Possible Mechanisms of Aβ Oligomer Toxicity
2.1.1. Disruption of Cellular Membranes
2.1.2. Dysregulation of Ion Flows
2.1.3. Pathological Engagement of Membrane Receptors
2.1.4. Induction of Oxidative Stress
2.1.5. AD-Related Target Protein Phosphorylation
2.2. Scenarios of Aβ Oligomerization and Post-Translational Modifications
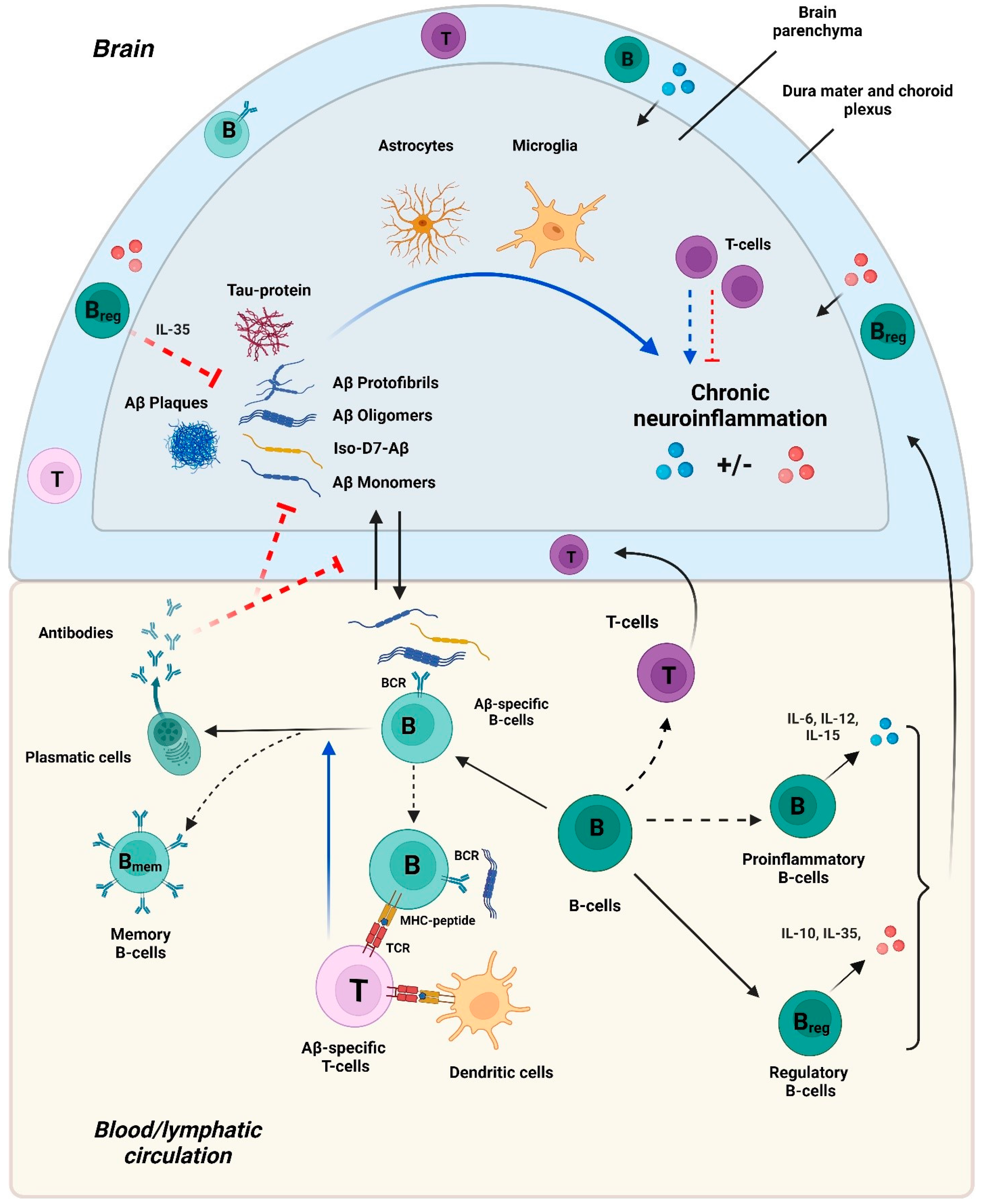
3. The Interaction of Aβ with the Innate Immune System and the Pathogenesis of Alzheimer’s Disease
4. The Interaction of Aβ with the Adaptive Immune System and the Pathogenesis of Alzheimer’s Disease
4.1. Aβ-Specific T Cells in AD
4.2. Aβ-Specific and Other B Cells in AD
4.2.1. Aβ-Specific Antibodies
4.2.2. B Cells
5. Concluding Notes
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 20 June 2024).
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The Role of Tau in Alzheimer’s Disease and Related Disorders. CNS Neurosci. Ther. 2010, 17, 514–524. [Google Scholar] [CrossRef]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef]
- Weber, C.; Dilthey, A.; Finzer, P. The role of microbiome-host interactions in the development of Alzheimer’s disease. Front. Cell. Infect. Microbiol. 2023, 13, 1151021. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Asamu, M.O.; Oladipo, O.O.; Abayomi, O.A.; Adebayo, A.A. Alzheimer’s disease: The role of T lymphocytes in neuroinflammation and neurodegeneration. Brain Res. 2023, 1821, 148589. [Google Scholar] [CrossRef]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Holtzman, D.M. Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity 2022, 55, 2236–2254. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- McManus, R.M. The Role of Immunity in Alzheimer’s Disease. Adv. Biol. 2022, 6, e2101166. [Google Scholar] [CrossRef]
- McManus, R.M.; Mills, K.H.G.; Lynch, M.A. T Cells-Protective or Pathogenic in Alzheimer’s Disease? J. Neuroimmune Pharmacol. 2015, 10, 547–560. [Google Scholar] [CrossRef]
- Wu, K.-M.; Zhang, Y.-R.; Huang, Y.-Y.; Dong, Q.; Tan, L.; Yu, J.-T. The role of the immune system in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101409. [Google Scholar] [CrossRef] [PubMed]
- Wyatt-Johnson, S.K.; Brutkiewicz, R.R. The Complexity of Microglial Interactions With Innate and Adaptive Immune Cells in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 592359. [Google Scholar] [CrossRef]
- Erten-Lyons, D.; Woltjer, R.L.; Dodge, H.; Nixon, R.; Vorobik, R.; Calvert, J.F.; Leahy, M.; Montine, T.; Kaye, J. Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 2009, 72, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.-E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Kulikova, A.A.; Istrate, A.N.; Tsvetkov, P.O.; Zhokhov, S.S.; Mezentsev, Y.V.; Kechko, O.I.; Ivanov, A.S.; Polshakov, V.I.; Makarov, A.A. The English (H6R) familial Alzheimer’s disease mutation facilitates zinc-induced dimerization of the amyloid-β metal-binding domain†. Metallomics 2015, 7, 422–425. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (H6R) and Tottori (D7N) Familial Alzheimer Disease Mutations on Amyloid β-Protein Assembly and Toxicity. J. Biol. Chem. 2010, 285, 23186–23197. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yamada, M. Low-n oligomers as therapeutic targets of Alzheimer’s disease. J. Neurochem. 2011, 117, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Abeta fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923. [Google Scholar] [CrossRef]
- Xu, L.; Chen, Y.; Wang, X. Dual effects of familial Alzheimer’s disease mutations (D7H, D7N, and H6R) on amyloid β peptide: Correlation dynamics and zinc binding. Proteins 2014, 82, 3286–3297. [Google Scholar] [CrossRef]
- Chen, W.-T.; Hong, C.-J.; Lin, Y.-T.; Chang, W.-H.; Huang, H.-T.; Liao, J.-Y.; Chang, Y.-J.; Hsieh, Y.-F.; Cheng, C.-Y.; Liu, H.-C.; et al. Amyloid-Beta (Aβ) D7H Mutation Increases Oligomeric Aβ42 and Alters Properties of Aβ-Zinc/Copper Assemblies. PLoS ONE 2012, 7, e35807. [Google Scholar] [CrossRef]
- Kechko, O.I.; Adzhubei, A.A.; Tolstova, A.P.; Indeykina, M.I.; Popov, I.A.; Zhokhov, S.S.; Gnuchev, N.V.; Mitkevich, V.A.; Makarov, A.A.; Kozin, S.A. Molecular Mechanism of Zinc-Dependent Oligomerization of Alzheimer’s Amyloid-β with Taiwan (D7H) Mutation. Int. J. Mol. Sci. 2023, 24, 11241. [Google Scholar] [CrossRef]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11, 302. [Google Scholar] [CrossRef]
- Cruceta, L.; Sun, Y.; Kenyaga, J.M.; Ostrovsky, D.; Rodgers, A.; Vugmeyster, L.; Yao, L.; Qiang, W. Modulation of aggregation and structural polymorphisms of β-amyloid fibrils in cellular environments by pyroglutamate-3 variant cross-seeding. J. Biol. Chem. 2023, 299, 105196. [Google Scholar] [CrossRef]
- De Kimpe, L.; van Haastert, E.S.; Kaminari, A.; Zwart, R.; Rutjes, H.; Hoozemans, J.J.M.; Scheper, W. Intracellular accumulation of aggregated pyroglutamate amyloid beta: Convergence of aging and Aβ pathology at the lysosome. Age 2013, 35, 673–687. [Google Scholar] [CrossRef]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Faller, P.; Hureau, C. Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-beta peptide. Dalton Trans. Camb. Engl. 2009, 7, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Abedin, F.; Tatulian, S.A. Structure of unmodified and pyroglutamylated amyloid beta peptides in lipid membranes. Biophys. J. 2022, 121, 328a. [Google Scholar] [CrossRef]
- Istrate, A.N.; Tsvetkov, P.O.; Mantsyzov, A.B.; Kulikova, A.A.; Kozin, S.A.; Makarov, A.A.; Polshakov, V.I. NMR solution structure of rat aβ(1-16): Toward understanding the mechanism of rats’ resistance to Alzheimer’s disease. Biophys. J. 2012, 102, 136–143. [Google Scholar] [CrossRef]
- Kozin, S.A.; Barykin, E.P.; Telegin, G.B.; Chernov, A.S.; Adzhubei, A.A.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Intravenously Injected Amyloid-β Peptide With Isomerized Asp7 and Phosphorylated Ser8 Residues Inhibits Cerebral β-Amyloidosis in AβPP/PS1 Transgenic Mice Model of Alzheimer’s Disease. Front. Neurosci. 2018, 12, 518. [Google Scholar] [CrossRef]
- Kozin, S.A.; Zirah, S.; Rebuffat, S.; Hoa, G.H.; Debey, P. Zinc binding to Alzheimer’s Abeta(1-16) peptide results in stable soluble complex. Biochem. Biophys. Res. Commun. 2001, 285, 959–964. [Google Scholar] [CrossRef]
- Kumar, S.; Wirths, O.; Theil, S.; Gerth, J.; Bayer, T.A.; Walter, J. Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer’s disease. Acta Neuropathol. 2013, 125, 699–709. [Google Scholar] [CrossRef]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef]
- Kumar, S.; Walter, J. Phosphorylation of amyloid beta (Aβ) peptides—A trigger for formation of toxic aggregates in Alzheimer’s disease. Aging 2011, 3, 803–812. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 2013, 4, e939. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like Behavior and Tau-dependent Cytotoxicity of Pyroglutamylated β-Amyloid. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Violani, E.; Salis, S.; Venezia, V.; Dolcini, V.; Damonte, G.; Benatti, U.; D’Arrigo, C.; Patrone, E.; Carlo, P.; et al. Pyroglutamate-modified amyloid beta-peptides—AbetaN3(pE)—Strongly affect cultured neuron and astrocyte survival. J. Neurochem. 2002, 82, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Schlenzig, D.; Rönicke, R.; Cynis, H.; Ludwig, H.-H.; Scheel, E.; Reymann, K.; Saido, T.; Hause, G.; Schilling, S.; Demuth, H.-U. N-Terminal pyroglutamate formation of Aβ38 and Aβ40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J. Neurochem. 2012, 121, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.O.; Kulikova, A.A.; Golovin, A.V.; Tkachev, Y.V.; Archakov, A.I.; Kozin, S.A.; Makarov, A.A. Minimal Zn2+ binding site of amyloid-β. Biophys. J. 2010, 99, L84–L86. [Google Scholar] [CrossRef] [PubMed]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Ségalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef]
- Mittal, K.; Eremenko, E.; Berner, O.; Elyahu, Y.; Strominger, I.; Apelblat, D.; Nemirovsky, A.; Spiegel, I.; Monsonego, A. CD4 T Cells Induce A Subset of MHCII-Expressing Microglia that Attenuates Alzheimer Pathology. iScience 2019, 16, 298–311. [Google Scholar] [CrossRef]
- McQuillan, K.; Lynch, M.A.; Mills, K.H.G. Activation of mixed glia by Aβ-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain. Behav. Immun. 2010, 24, 598–607. [Google Scholar] [CrossRef]
- Sigurdsson, E.M.; Scholtzova, H.; Mehta, P.D.; Frangione, B.; Wisniewski, T. Immunization with a Nontoxic/Nonfibrillar Amyloid-β Homologous Peptide Reduces Alzheimer’s Disease-Associated Pathology in Transgenic Mice. Am. J. Pathol. 2001, 159, 439–447. [Google Scholar] [CrossRef]
- Bard, F.; Barbour, R.; Cannon, C.; Carretto, R.; Fox, M.; Games, D.; Guido, T.; Hoenow, K.; Hu, K.; Johnson-Wood, K.; et al. Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc. Natl. Acad. Sci. USA 2003, 100, 2023–2028. [Google Scholar] [CrossRef]
- Park, J.-C.; Noh, J.; Jang, S.; Kim, K.H.; Choi, H.; Lee, D.; Kim, J.; Chung, J.; Lee, D.Y.; Lee, Y.; et al. Association of B cell profile and receptor repertoire with the progression of Alzheimer’s disease. Cell Rep. 2022, 40, 111391. [Google Scholar] [CrossRef]
- Cras, P.; Kawai, M.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1991, 88, 7552. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Ge, Y.-W. Role of the APP promoter in Alzheimer’s disease: Cell type-specific expression of the beta-amyloid precursor protein. Ann. N. Y. Acad. Sci. 2004, 1030, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. Neuromolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Andreasen, N.; Hesse, C.; Davidsson, P.; Minthon, L.; Wallin, A.; Winblad, B.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: Differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch. Neurol. 1999, 56, 673–680. [Google Scholar] [CrossRef]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Hata, S.; Suzuki, T. Alternative Selection of β-Site APP-Cleaving Enzyme 1 (BACE1) Cleavage Sites in Amyloid β-Protein Precursor (APP) Harboring Protective and Pathogenic Mutations within the Aβ Sequence. J. Biol. Chem. 2016, 291, 24041–24053. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Gallardo, R.; Ungureanu, A.-A.; Castillo Cano, V.; Snellinx, A.; Ramakers, M.; Bartic, C.; Rousseau, F.; Schymkowitz, J.; De Strooper, B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 2014, 289, 30977–30989. [Google Scholar] [CrossRef]
- Hatami, A.; Monjazeb, S.; Milton, S.; Glabe, C.G. Familial Alzheimer’s Disease Mutations within the Amyloid Precursor Protein Alter the Aggregation and Conformation of the Amyloid-β Peptide. J. Biol. Chem. 2017, 292, 3172–3185. [Google Scholar] [CrossRef]
- Miners, J.S.; Barua, N.; Kehoe, P.G.; Gill, S.; Love, S. Aβ-degrading enzymes: Potential for treatment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2011, 70, 944–959. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-S.; Jo, S.A. Mechanisms of Amyloid-β Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. 2012, 20, 245–255. [Google Scholar] [CrossRef]
- Mucke, L. Neuroscience: Alzheimer’s disease. Nature 2009, 461, 895–897. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Viles, J.H. Imaging Amyloid-β Membrane Interactions: Ion-Channel Pores and Lipid-Bilayer Permeability in Alzheimer’s Disease. Angew. Chem. Int. Ed. 2023, 62, e202215785. [Google Scholar] [CrossRef]
- Paranjape, G.S.; Gouwens, L.K.; Osborn, D.C.; Nichols, M.R. Isolated Amyloid-β(1–42) Protofibrils, But Not Isolated Fibrils, Are Robust Stimulators of Microglia. ACS Chem. Neurosci. 2012, 3, 302–311. [Google Scholar] [CrossRef]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S67–S78. [Google Scholar] [CrossRef]
- Jang, H.; Connelly, L.; Arce, F.T.; Ramachandran, S.; Kagan, B.L.; Lal, R.; Nussinov, R. Mechanisms for the Insertion of Toxic, Fibril-like β-Amyloid Oligomers into the Membrane. J. Chem. Theory Comput. 2013, 9, 822–833. [Google Scholar] [CrossRef]
- La Rosa, C.; Scalisi, S.; Lolicato, F.; Pannuzzo, M.; Raudino, A. Lipid-assisted protein transport: A diffusion-reaction model supported by kinetic experiments and molecular dynamics simulations. J. Chem. Phys. 2016, 144, 184901. [Google Scholar] [CrossRef]
- Eskandari, H.; Ghanadian, M.; Noleto-Dias, C.; Lomax, C.; Tawfike, A.; Christiansen, G.; Sutherland, D.S.; Ward, J.L.; Mohammad-Beigi, H.; Otzen, D.E. Inhibitors of α-Synuclein Fibrillation and Oligomer Toxicity in Rosa damascena: The All-Pervading Powers of Flavonoids and Phenolic Glycosides. ACS Chem. Neurosci. 2020, 11, 3161–3173. [Google Scholar] [CrossRef]
- Scollo, F.; Tempra, C.; Lolicato, F.; Sciacca, M.F.M.; Raudino, A.; Milardi, D.; La Rosa, C. Phospholipids Critical Micellar Concentrations Trigger Different Mechanisms of Intrinsically Disordered Proteins Interaction with Model Membranes. J. Phys. Chem. Lett. 2018, 9, 5125–5129. [Google Scholar] [CrossRef]
- Tempra, C.; Scollo, F.; Pannuzzo, M.; Lolicato, F.; La Rosa, C. A unifying framework for amyloid-mediated membrane damage: The lipid-chaperone hypothesis. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140767. [Google Scholar] [CrossRef]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef]
- Griffioen, G. Calcium Dyshomeostasis Drives Pathophysiology and Neuronal Demise in Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 13243. [Google Scholar] [CrossRef]
- Hattori, N.; Kitagawa, K.; Higashida, T.; Yagyu, K.; Shimohama, S.; Wataya, T.; Perry, G.; Smith, M.A.; Inagaki, C. Cl−-ATPase and Na+/K+-ATPase activities in Alzheimer’s disease brains. Neurosci. Lett. 1998, 254, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.M.; Garg, S.K.; Keep, R.F.; Albin, R.L.; Banerjee, R. Na+ and K+ ion imbalances in Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Gordon, M.N.; Wilcock, D.M.; Herber, D.L.; Freeman, M.J.; Morgan, D. Dysregulation of Na+/K+ ATPase by amyloid in APP+PS1 transgenic mice. BMC Neurosci. 2005, 6, 7. [Google Scholar] [CrossRef]
- Kairane, C.; Mahlapuu, R.; Ehrlich, K.; Zilmer, M.; Soomets, U. The effects of different antioxidants on the activity of cerebrocortical MnSOD and Na,K-ATPase from post mortem Alzheimer’s disease and age-matched normal brains. Curr. Alzheimer Res. 2014, 11, 79–85. [Google Scholar] [CrossRef]
- Kreutz, F.; Scherer, E.B.; Ferreira, A.G.K.; Petry, F.D.S.; Pereira, C.L.; Santana, F.; de Souza Wyse, A.T.; Salbego, C.G.; Trindade, V.M.T. Alterations on Na+,K+-ATPase and acetylcholinesterase activities induced by amyloid-β peptide in rat brain and GM1 ganglioside neuroprotective action. Neurochem. Res. 2013, 38, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-N.; Sun, Y.-J.; Pan, S.; Li, J.-X.; Qu, Y.-E.; Li, Y.; Wang, Y.-L.; Gao, Z.-B. Na+-K+-ATPase, a potent neuroprotective modulator against Alzheimer disease. Fundam. Clin. Pharmacol. 2013, 27, 96–103. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Liu, R.; Collier, J.M.; Abdul-Rahman, N.-H.; Capuk, O.; Zhang, Z.; Begum, G. Dysregulation of Ion Channels and Transporters and Blood-Brain Barrier Dysfunction in Alzheimer’s Disease and Vascular Dementia. Aging Dis. 2024, 15, 1748–1770. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na, K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E4465–E4474. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Adzhubei, A.A.; Burnysheva, K.M.; Lakunina, V.A.; Kamanina, Y.V.; Dergousova, E.A.; Lopina, O.D.; Ogunshola, O.O.; et al. Direct interaction of beta-amyloid with Na,K-ATPase as a putative regulator of the enzyme function. Sci. Rep. 2016, 6, 27738. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.B.; Zhao, J.X.; Fei, J.; Schwarz, W. Modulation of Na+,K+ pumping and neurotransmitter uptake by beta-amyloid. Neuroscience 2004, 126, 61–67. [Google Scholar] [CrossRef]
- Mark, R.J.; Hensley, K.; Butterfield, D.A.; Mattson, M.P. Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 1995, 15, 6239–6249. [Google Scholar] [CrossRef]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Tikhonova, L.A.; Kosenko, E.A. Critical analysis of Alzheimer’s amyloid-beta toxicity to mitochondria. Front. Biosci. Landmark Ed. 2015, 20, 173–197. [Google Scholar] [CrossRef]
- Morkuniene, R.; Cizas, P.; Jankeviciute, S.; Petrolis, R.; Arandarcikaite, O.; Krisciukaitis, A.; Borutaite, V. Small Aβ1-42 oligomer-induced membrane depolarization of neuronal and microglial cells: Role of N-methyl-D-aspartate receptors. J. Neurosci. Res. 2015, 93, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Sayehmiri, F.; Motamedi, F.; Batool, Z.; Naderi, N.; Shaerzadeh, F.; Zoghi, A.; Rezaei, O.; Khodagholi, F.; Pourbadie, H.G. Mitochondrial plasticity and synaptic plasticity crosstalk; in health and Alzheimer’s disease. CNS Neurosci. Ther. 2024, 30, e14897. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Parri, H.R.; Hernandez, C.M.; Dineley, K.T. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem. Pharmacol. 2011, 82, 931–942. [Google Scholar] [CrossRef]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Freir, D.B.; Nicoll, A.J.; Klyubin, I.; Panico, S.; Mc Donald, J.M.; Risse, E.; Asante, E.A.; Farrow, M.A.; Sessions, R.B.; Saibil, H.R.; et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2011, 2, 336. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Origlia, N.; Bonadonna, C.; Rosellini, A.; Leznik, E.; Arancio, O.; Yan, S.S.; Domenici, L. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 2010, 30, 11414–11425. [Google Scholar] [CrossRef]
- Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. [Google Scholar] [CrossRef] [PubMed]
- Varshavskaya, K.B.; Petrushanko, I.Y.; Mitkevich, V.A.; Barykin, E.P.; Makarov, A.A. Post-translational modifications of beta-amyloid alter its transport in the blood-brain barrier in vitro model. Front. Mol. Neurosci. 2024, 17, 1362581. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Ebenezer, P.J.; Weidner, A.M.; LeVine, H.; Markesbery, W.R.; Murphy, M.P.; Zhang, L.; Dasuri, K.; Fernandez-Kim, S.O.; Bruce-Keller, A.J.; Gavilán, E.; et al. Neuron specific toxicity of oligomeric amyloid-β: Role for JUN-kinase and oxidative stress. J. Alzheimer’s Dis. 2010, 22, 839–848. [Google Scholar] [CrossRef]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Chiang, G.C.; Mao, X.; Kang, G.; Chang, E.; Pandya, S.; Vallabhajosula, S.; Isaacson, R.; Ravdin, L.D.; Alzheimer’s Disease Neuroimaging Initiative; Shungu, D.C. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with 1H-MRS and Pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol. 2017, 38, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels—A novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; He, Z.; Hussain, Z.; Farooq, A.; McIntyre, A.; Laughton, C.A.; Carvalho, A.N.; Finelli, M.J. Protein Oxidative Modifications in Neurodegenerative Diseases: From Advances in Detection and Modelling to Their Use as Disease Biomarkers. Antioxidants 2024, 13, 681. [Google Scholar] [CrossRef] [PubMed]
- Dyer, R.R.; Ford, K.I.; Robinson, R.A.S. The roles of S-nitrosylation and S-glutathionylation in Alzheimer’s disease. Methods Enzymol. 2019, 626, 499–538. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef]
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Petrushanko, I.Y.; Burnysheva, K.M.; Makarov, A.A.; Mitkevich, V.A. Isomerization of Asp7 increases the toxic effects of amyloid β and its phosphorylated form in SH-SY5Y neuroblastoma cells. Mol. Biol. 2016, 50, 863–869. [Google Scholar] [CrossRef]
- Petrovskaya, A.V.; Tverskoi, A.M.; Barykin, E.P.; Varshavskaya, K.B.; Dalina, A.A.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. Distinct Effects of Beta-Amyloid, Its Isomerized and Phosphorylated Forms on the Redox Status and Mitochondrial Functioning of the Blood-Brain Barrier Endothelium. Int. J. Mol. Sci. 2022, 24, 183. [Google Scholar] [CrossRef]
- Henriques, A.G.; Müller, T.; Oliveira, J.M.; Cova, M.; da Cruz E Silva, C.B.; da Cruz E Silva, O.A.B. Altered protein phosphorylation as a resource for potential AD biomarkers. Sci. Rep. 2016, 6, 30319. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Zatsepina, O.G.; Kechko, O.I.; Mitkevich, V.A.; Kozin, S.A.; Yurinskaya, M.M.; Vinokurov, M.G.; Serebryakova, M.V.; Rezvykh, A.P.; Evgen’ev, M.B.; Makarov, A.A. Amyloid-β with isomerized Asp7 cytotoxicity is coupled to protein phosphorylation. Sci. Rep. 2018, 8, 3518. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.G.; Oliveira, J.M.; Carvalho, L.P.; da Cruz E Silva, O.A.B. Aβ Influences Cytoskeletal Signaling Cascades with Consequences to Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 1391–1407. [Google Scholar] [CrossRef] [PubMed]
- Busch, L.; Eggert, S.; Endres, K.; Bufe, B. The Hidden Role of Non-Canonical Amyloid β Isoforms in Alzheimer’s Disease. Cells 2022, 11, 3421. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, J.; Li, X.; Ma, L.; Hou, M.; Zhou, H.; Zhou, R. Based on molecular structures: Amyloid-β generation, clearance, toxicity and therapeutic strategies. Front. Mol. Neurosci. 2022, 15, 927530. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-H.; Wang, J.; Li, Q.-X.; Fowler, C.J.; Zeng, F.; Deng, J.; Xu, Z.-Q.; Zhou, H.-D.; Doecke, J.D.; Villemagne, V.L.; et al. Association of naturally occurring antibodies to β-amyloid with cognitive decline and cerebral amyloidosis in Alzheimer’s disease. Sci. Adv. 2021, 7, eabb0457. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A. Metal Binding of Alzheimer’s Amyloid-β and Its Effect on Peptide Self-Assembly. Acc. Chem. Res. 2023, 56, 2653–2663. [Google Scholar] [CrossRef]
- Chia, S.; Cataldi, R.L.; Ruggeri, F.S.; Limbocker, R.; Condado-Morales, I.; Pisani, K.; Possenti, A.; Linse, S.; Knowles, T.P.J.; Habchi, J.; et al. A Relationship between the Structures and Neurotoxic Effects of Aβ Oligomers Stabilized by Different Metal Ions. ACS Chem. Neurosci. 2024, 15, 1125–1134. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Finder, V.H.; Glockshuber, R. Amyloid-beta aggregation. Neurodegener. Dis. 2007, 4, 13–27. [Google Scholar] [CrossRef]
- Suzuki, N.; Cheung, T.T.; Cai, X.D.; Odaka, A.; Otvos, L.; Eckman, C.; Golde, T.E.; Younkin, S.G. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef]
- Kozin, S.A.; Mezentsev, Y.V.; Kulikova, A.A.; Indeykina, M.I.; Golovin, A.V.; Ivanov, A.S.; Tsvetkov, P.O.; Makarov, A.A. Zinc-induced dimerization of the amyloid-β metal-binding domain 1-16 is mediated by residues 11-14. Mol. Biosyst. 2011, 7, 1053–1055. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Barykin, E.P.; Eremina, S.; Pani, B.; Katkova-Zhukotskaya, O.; Polshakov, V.I.; Adzhubei, A.A.; Kozin, S.A.; Mironov, A.S.; Makarov, A.A.; et al. Zn-dependent β-amyloid aggregation and its reversal by the tetrapeptide HAEE. Aging Dis. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally applied synthetic peptide isoAsp7-Aβ(1-42) triggers cerebral β-amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Head, E.; Velazquez, P.; Cotman, C.W.; Tenner, A.J. The presence of isoaspartic acid in beta-amyloid plaques indicates plaque age. Exp. Neurol. 1999, 157, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Gnoth, K.; Piechotta, A.; Kleinschmidt, M.; Konrath, S.; Schenk, M.; Taudte, N.; Ramsbeck, D.; Rieckmann, V.; Geissler, S.; Eichentopf, R.; et al. Targeting isoaspartate-modified Aβ rescues behavioral deficits in transgenic mice with Alzheimer’s disease-like pathology. Alzheimer’s Res. Ther. 2020, 12, 149. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Renno, T.; Taupin, V.; Bourbonnière, L.; Verge, G.; Tran, E.; De Simone, R.; Krakowski, M.; Rodriguez, M.; Peterson, A.; Owens, T. Interferon-gamma in progression to chronic demyelination and neurological deficit following acute EAE. Mol. Cell. Neurosci. 1998, 12, 376–389. [Google Scholar] [CrossRef]
- Monsonego, A.; Imitola, J.; Petrovic, S.; Zota, V.; Nemirovsky, A.; Baron, R.; Fisher, Y.; Owens, T.; Weiner, H.L. Aβ-induced meningoencephalitis is IFN-γ-dependent and is associated with T cell-dependent clearance of Aβ in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5048–5053. [Google Scholar] [CrossRef]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimer’s Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sobue, A.; Komine, O.; Yamanaka, K. Neuroinflammation in Alzheimer’s disease: Microglial signature and their relevance to disease. Inflamm. Regen. 2023, 43, 26. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef]
- Kasus-Jacobi, A.; Washburn, J.L.; Land, C.A.; Pereira, H.A. Neutrophil Granule Proteins Inhibit Amyloid Beta Aggregation and Neurotoxicity. Curr. Alzheimer Res. 2021, 18, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Jairani, P.S.; Aswathy, P.M.; Krishnan, D.; Menon, R.N.; Verghese, J.; Mathuranath, P.S.; Gopala, S. Apolipoprotein E Polymorphism and Oxidative Stress in Peripheral Blood-Derived Macrophage-Mediated Amyloid-Beta Phagocytosis in Alzheimer’s Disease Patients. Cell. Mol. Neurobiol. 2019, 39, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Prà, I.; Rossi, F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Pan, S. Discovery and Validation of Key Biomarkers Based on Immune Infiltrates in Alzheimer’s Disease. Front. Genet. 2021, 12, 658323. [Google Scholar] [CrossRef]
- Qi, C.; Liu, F.; Zhang, W.; Han, Y.; Zhang, N.; Liu, Q.; Li, H. Alzheimer’s disease alters the transcriptomic profile of natural killer cells at single-cell resolution. Front. Immunol. 2022, 13, 1004885. [Google Scholar] [CrossRef]
- NKGen Biotech’s SNK01 NK Cell Therapy Cleared to Start Phase 2 Clinical Trial in Alzheimer’s Disease | NKGen Biotech. 2024. Available online: https://nkgenbiotech.com/nkgen-biotechs-snk01-nk-cell-therapy-cleared-to-start-phase-2-clinical-trial-in-alzheimers-disease/ (accessed on 24 September 2024).
- Shi, M.; Chu, F.; Zhu, F.; Zhu, J. Peripheral blood amyloid-β involved in the pathogenesis of Alzheimer’s disease via impacting on peripheral innate immune cells. J. Neuroinflamm. 2024, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. A Bridge Between the Innate Immunity System and Amyloid-β Production in Alzheimer’s Disease. Neurosci. Bull. 2021, 37, 898–901. [Google Scholar] [CrossRef]
- Xin, S.-H.; Tan, L.; Cao, X.; Yu, J.-T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.S.; Li, E.; Scharnagl, L.; Poupardin, R.; Altendorfer, B.; Mrowetz, H.; Hutter-Paier, B.; Weiger, T.M.; Heneka, M.T.; Attems, J.; et al. CD8+ T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain. Behav. Immun. 2020, 89, 67–86. [Google Scholar] [CrossRef]
- McManus, R.M.; Heneka, M.T. T cells in Alzheimer’s disease: Space invaders. Lancet Neurol. 2020, 19, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.e24. [Google Scholar] [CrossRef]
- Lanuti, P.; Ciccocioppo, F.; Bonanni, L.; Marchisio, M.; Lachmann, R.; Tabet, N.; Pierdomenico, L.; Santavenere, E.; Catinella, V.; Iacone, A.; et al. Amyloid-specific T-cells differentiate Alzheimer’s disease from Lewy body dementia. Neurobiol. Aging 2012, 33, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Monsonego, A.; Nemirovsky, A.; Harpaz, I. CD4 T cells in immunity and immunotherapy of Alzheimer’s disease. Immunology 2013, 139, 438–446. [Google Scholar] [CrossRef]
- Monsonego, A.; Zota, V.; Karni, A.; Krieger, J.I.; Bar-Or, A.; Bitan, G.; Budson, A.E.; Sperling, R.; Selkoe, D.J.; Weiner, H.L. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J. Clin. Investig. 2003, 112, 415–422. [Google Scholar] [CrossRef]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Calvo, M.G.; Nemni, R.; Clerici, M. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 21, 927–938. [Google Scholar] [CrossRef]
- Machhi, J.; Yeapuri, P.; Lu, Y.; Foster, E.; Chikhale, R.; Herskovitz, J.; Namminga, K.L.; Olson, K.E.; Abdelmoaty, M.M.; Gao, J.; et al. CD4+ effector T cells accelerate Alzheimer’s disease in mice. J. Neuroinflamm. 2021, 18, 272. [Google Scholar] [CrossRef]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.-A.; Mills, K.H.G.; Lynch, M.A. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.M.; Higgins, S.C.; Mills, K.H.G.; Lynch, M.A. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol. Aging 2014, 35, 109–121. [Google Scholar] [CrossRef]
- Orgogozo, J.-M.; Gilman, S.; Dartigues, J.-F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef]
- Song, M.S.; Mook-Jung, I.; Lee, H.J.; Min, J.Y.; Park, M.H. Serum anti-amyloid-beta antibodies and Alzheimer’s disease in elderly Korean patients. J. Int. Med. Res. 2007, 35, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef]
- Nagele, E.P.; Han, M.; Acharya, N.K.; DeMarshall, C.; Kosciuk, M.C.; Nagele, R.G. Natural IgG Autoantibodies Are Abundant and Ubiquitous in Human Sera, and Their Number Is Influenced By Age, Gender, and Disease. PLoS ONE 2013, 8, e60726. [Google Scholar] [CrossRef]
- Britschgi, M.; Olin, C.E.; Johns, H.T.; Takeda-Uchimura, Y.; LeMieux, M.C.; Rufibach, K.; Rajadas, J.; Zhang, H.; Tomooka, B.; Robinson, W.H.; et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 12145–12150. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.; Relkin, N.; Weksler, M.E. Natural human antibodies to amyloid beta peptide. Autoimmun. Rev. 2008, 7, 415–420. [Google Scholar] [CrossRef]
- Esposito, M.; Luccarini, I.; Cicatiello, V.; De Falco, D.; Fiorentini, A.; Barba, P.; Casamenti, F.; Prisco, A. Immunogenicity and therapeutic efficacy of phage-displayed beta-amyloid epitopes. Mol. Immunol. 2008, 45, 1056–1062. [Google Scholar] [CrossRef]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- Suzuki, K.; Iwata, A.; Iwatsubo, T. The past, present, and future of disease-modifying therapies for Alzheimer’s disease. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Spencer, B.; Masliah, E. Immunotherapeutic Approaches Targeting Amyloid-β, α-Synuclein, and Tau for the Treatment of Neurodegenerative Disorders. Neurother. J. Am. Soc. Exp. Neurother. 2016, 13, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A.R. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain J. Neurol. 2008, 131, 3299–3310. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Alzheimer’s disease drug development and the problem of the blood-brain barrier. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2009, 5, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Pocevičiūtė, D.; Nuñez-Diaz, C.; Roth, B.; Janelidze, S.; Giannisis, A.; Hansson, O.; Wennström, M. Increased plasma and brain immunoglobulin A in Alzheimer’s disease is lost in apolipoprotein E ε4 carriers. Alzheimer’s Res. Ther. 2022, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef]
- Wirths, O.; Erck, C.; Martens, H.; Harmeier, A.; Geumann, C.; Jawhar, S.; Kumar, S.; Multhaup, G.; Walter, J.; Ingelsson, M.; et al. Identification of Low Molecular Weight Pyroglutamate Aβ Oligomers in Alzheimer Disease. J. Biol. Chem. 2010, 285, 41517–41524. [Google Scholar] [CrossRef] [PubMed]
- Antonios, G.; Borgers, H.; Richard, B.C.; Brauß, A.; Meißner, J.; Weggen, S.; Pena, V.; Pillot, T.; Davies, S.L.; Bakrania, P.; et al. Alzheimer therapy with an antibody against N-terminal Abeta 4-X and pyroglutamate Abeta 3-X. Sci. Rep. 2015, 5, 17338. [Google Scholar] [CrossRef]
- Bakrania, P.; Hall, G.; Bouter, Y.; Bouter, C.; Beindorff, N.; Cowan, R.; Davies, S.; Price, J.; Mpamhanga, C.; Love, E.; et al. Discovery of a novel pseudo β-hairpin structure of N-truncated amyloid-β for use as a vaccine against Alzheimer’s disease. Mol. Psychiatry 2022, 27, 840–848. [Google Scholar] [CrossRef]
- Janssens, J.; Hermans, B.; Vandermeeren, M.; Barale-Thomas, E.; Borgers, M.; Willems, R.; Meulders, G.; Wintmolders, C.; Van den Bulck, D.; Bottelbergs, A.; et al. Passive immunotherapy with a novel antibody against 3pE-modified Aβ demonstrates potential for enhanced efficacy and favorable safety in combination with BACE inhibitor treatment in plaque-depositing mice. Neurobiol. Dis. 2021, 154, 105365. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.; Liu, B.; Rahfeld, J.-U.; Kleinschmidt, M.; O’nuallain, B.; Le, K.; Lues, I.; Caldarone, B.; Schilling, S.; Demuth, H.-U.; et al. An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol. Aging 2015, 36, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; DeLong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A Plaque-Specific Antibody Clears Existing β-amyloid Plaques in Alzheimer’s Disease Mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Liu, B.; Kleinschmidt, M.; Schilling, S.; Demuth, H.-U.; Lemere, C.A. Passive Immunization against Pyroglutamate-3 Amyloid-β Reduces Plaque Burden in Alzheimer-Like Transgenic Mice: A Pilot Study. Neurodegener. Dis. 2012, 10, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Grill, J.; Sajjadi, S.A.; Sultzer, D. FDA Approves Donanemab—UCI MIND. Available online: https://mind.uci.edu/donanemab/ (accessed on 26 August 2024).
- Busse, M.; Michler, E.; von Hoff, F.; Dobrowolny, H.; Hartig, R.; Frodl, T.; Busse, S. Alterations in the Peripheral Immune System in Dementia. J. Alzheimer’s Dis. 2017, 58, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Pellicanò, M.; Bulati, M.; Buffa, S.; Barbagallo, M.; Di Prima, A.; Misiano, G.; Picone, P.; Di Carlo, M.; Nuzzo, D.; Candore, G.; et al. Systemic immune responses in Alzheimer’s disease: In vitro mononuclear cell activation and cytokine production. J. Alzheimer’s Dis. 2010, 21, 181–192. [Google Scholar] [CrossRef]
- Kim, K.; Wang, X.; Ragonnaud, E.; Bodogai, M.; Illouz, T.; DeLuca, M.; McDevitt, R.A.; Gusev, F.; Okun, E.; Rogaev, E.; et al. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat. Commun. 2021, 12, 2185. [Google Scholar] [CrossRef] [PubMed]
- Shekari, A.; Fahnestock, M. Cholinergic neurodegeneration in Alzheimer disease mouse models. Handb. Clin. Neurol. 2021, 182, 191–209. [Google Scholar] [CrossRef]
- Li, H.; Wei, Y.; Wang, Z.; Wang, Q. Application of APP/PS1 transgenic mouse model for Alzheimers disease. J Alzheimer’s Park. 2015, 5, 10–4172. [Google Scholar]
- Van Meerhaeghe, T.; Néel, A.; Brouard, S.; Degauque, N. Regulation of CD8 T cell by B-cells: A narrative review. Front. Immunol. 2023, 14, 1125605. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.J.; Abu-Rub, M.; Miller, R.H. B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights. Cells 2021, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Ding, S.; Chen, S.; Wang, T.; Wang, Z.; Zou, Y.; Sheng, C.; Chen, Y.; Pang, Y.; et al. B lymphocytes ameliorate Alzheimer’s disease-like neuropathology via interleukin-35. Brain. Behav. Immun. 2023, 108, 16–31. [Google Scholar] [CrossRef]
| Mouse Model NR2B-Containing NMDA Receptor | Gene (Mutation) | Aβ Pathology | Tau Pathology | Neuronal Loss | Cognitive Impairment | First Described |
|---|---|---|---|---|---|---|
| PDAPP | APP (V717F) | 6 mo. + | - | - | 3 mo. + | 1995 [140] |
| APP-Tg J20 | APP (K670/671NL, V717F) | 8 mo. + | - | 3 mo. + | 4 mo. + | [141] |
| APP/PS1 | APP (KM670/671NL) | 6 mo. + | - | 8 mo. + | 12 mo. + | 2006 [142] |
| PSEN1 (delta9) | ||||||
| 5xFAD | APP (KM670/671NL, I716V, V717I) | 8 mo. + | - | 6 mo. + | 4 mo. + | 2006 [143] |
| PSEN1 (M146L, L286V) | ||||||
| 3xTgAD | APP (KM670/671NL) | 6 mo. + | 12 mo. + | unknown | 4 mo. + | 2003 [144] |
| MAPT 0N4R (P301L) | ||||||
| Psen (M146V knock-in) | ||||||
| SJL mice | expressing IFN-γ under the MBP promoter | [145] | ||||
| APP/IFN-γ Tg | homozygous IFN-γ-Tg mice were bred with APP-Tg J20 mice | [146] | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolobova, E.; Petrushanko, I.; Mitkevich, V.; Makarov, A.A.; Grigorova, I.L. β-Amyloids and Immune Responses Associated with Alzheimer’s Disease. Cells 2024, 13, 1624. https://doi.org/10.3390/cells13191624
Kolobova E, Petrushanko I, Mitkevich V, Makarov AA, Grigorova IL. β-Amyloids and Immune Responses Associated with Alzheimer’s Disease. Cells. 2024; 13(19):1624. https://doi.org/10.3390/cells13191624
Chicago/Turabian StyleKolobova, Elizaveta, Irina Petrushanko, Vladimir Mitkevich, Alexander A Makarov, and Irina L Grigorova. 2024. "β-Amyloids and Immune Responses Associated with Alzheimer’s Disease" Cells 13, no. 19: 1624. https://doi.org/10.3390/cells13191624
APA StyleKolobova, E., Petrushanko, I., Mitkevich, V., Makarov, A. A., & Grigorova, I. L. (2024). β-Amyloids and Immune Responses Associated with Alzheimer’s Disease. Cells, 13(19), 1624. https://doi.org/10.3390/cells13191624