

The Interplay between Liver and Adipose Tissue in the Onset of Liver Diseases: Exploring the Role of Vitamin Deficiency

 , , , , ,
, , , , ,  , ,
, , 
Abstract
:1. Introduction
2. Liver Disease: The Link between Adipose Tissue and the Liver

{kind=link}
{kind=link}
{kind=link}
| Vitamin | Ref. | Year | Highlights |
|---|---|---|---|
| A | [40] | 2022 | β-carotene 15,15′-monooxygenase 1 (BCMO1) is a crucial enzyme that converts β-carotene into vitamin A. It is considered an important regulator of lipid metabolism in adipocytes and has the effect of preserving liver functions. |
| [41] | 2022 | Retinoic acid (RA) reveals a therapeutic effect on NAFLD by increasing fatty-acid (FA) oxidation in the liver and thermogenesis and white adipose tissue (WAT) browning in adipose tissue. | |
| [42] | 2023 | Adipose-derived mesenchymal stem cells (ADMSCs) have shown significant therapeutic potential in treating liver fibrosis by upregulating the expression of various genes to promote retinol metabolism. | |
| [43] | 2024 | Retinol-binding protein 4 (RBP4) produced by visceral adipocytes contributes to the transport and mobilization of hepatic retinol storage. In a high-fat diet (HFD), increased plasma retinol levels are correlated with high serum RBP4 levels, which could be associated with an increased risk of NAFLD since RBP4 contributes to free fatty acid (FFA) mobilization from adipose tissue to the liver. | |
| B3 | [44] | 2019 | High niacin supplementation plays a pivotal role in the human NAFLD pathogenesis by inhibiting lipolysis in the adipose tissue of humans, which thereby decreases the amount of free-flowing FAs into the liver. |
| [45] | 2021 | Nicotinamide (NAM) administration increases the mitochondrial β-oxidation of FAs in brown adipose tissue (BAT), and triggers a browning process in WAT, which enhances its energy expenditure. NAM also keeps a check on hepatic steatosis. | |
| [46] | 2022 | N1-Methylnicotinamide (mNAM) induces lipolysis in adipose tissue and gluconeogenesis in hepatocytes under physiological conditions, and releases ketone bodies and glucose as metabolic substrates in skeletal muscle. | |
| [47] | 2024 | mNAM decreases hepatic lipid accumulation and reduces inflammation in WAT. | |
| [48] | 2022 | Supplementation with nicotinamide riboside (NR) exerted an anti-obesity effect and prevented the development of inflammation and fibrosis in the WAT of old, but not young female mice with diet-induced obesity, protecting the liver from obesity effects. | |
| [49] | 2022 | NR reduces lipogenesis in the liver and increases lipolysis in WAT, in high-fructose and high-fat induced models, suggesting a possible therapeutic application in lipid metabolic disorders. | |
| B5 | [50] | 2022 | Pantothenate enhances BAT energy expenditure in an uncoupling protein 1 (UCP1)-dependent manner and reduces adiposity and, thereby indirectly, hepatic steatosis. |
| B7 | [51] | 2022 | In rats, high doses of dietary biotin intake can activate FA oxidation due to the increased hepatic β-oxidation, which, in turn, may contribute to the reduction in the hepatic triglyceride (TG) concentration and WAT weight. |
| B9 | [52] | 2023 | There is a relationship between folic acid, gut microbiota, liver and adipose tissue. |
| C | [53] | 2020 | Vitamin C administration at a medium dose is beneficial for prophylaxis and therapy of HFD-induced NAFLD, while at a low dose it prevents the development of HFD-induced NAFLD and aids in its management. Moreover, it could prevent HFD-fed mice from weight and visceral fat gain. Conversely, a high dose may be risky. |
| [54] | 2022 | Vitamin C modulates hepatic expression and secretion of growth factor 21 (Fgf21) which, in turn, enhances BAT thermogenesis and regulates lipid metabolism. | |
| [55] | 2021 | Supplementation of vitamin C can dysregulate WAT hyperplasia and hepatic steatosis by reversing the hypermethylation due to Tet1 haploinsufficiency, resulting in FA oxidation lipolysis and thermogenic upregulation. | |
| D | [56] | 2020 | In humans and mice, obesity suppresses the vitamin D 25-Hydroxylase (Cytochrome P450 2R1, CYP2R1) in mouse liver and BAT and in human subcutaneous WAT (sWAT), leading to vitamin D deficiency. |
| [57] | 2022 | Vitamin D deficiency promotes NAFLD due to adipose tissue metabolism dysfunction, thereby altering the crosstalk between the liver and adipose tissue. | |
| [58] | 2022 | Vitamin D deficiency promotes NAFLD by trigging WAT-associated macrophage infiltration and secretion of bioactive inflammatory adipokines and resulting in extracellular matrix (ECM) remodeling, which ultimately causes fibrosis. | |
| [59] | 2020 | Supplementation of vitamin D reduces WAT inflammation by downregulating the related markers (such as Mcp1 and Ccl5) and reduces de novo lipogenesis by downregulating the FA synthase (Fasn) and acetyl-CoA carboxylase 1 (Acaca), which, in turn, decreases the hepatic lipid droplets (LDs) in the liver. | |
| [60] | 2021 | The high-fat/sucrose-induced inflammation in inguinal adipose tissue and hepatic steatosis was reduced by the synergetic effect due to the combination of physical exercise and vitamin D supplementation, leading to a reduction of inflammation in WAT and in the liver. | |
| [61] | 2022 | Supplementation of vitamin D improved the HFD-induced weight gain, hepatic steatosis, serum lipid profile, degree of inflammation, and serum adipokine levels. | |
| [62] | 2023 | Calcifediol is considered suitable for all patients with vitamin D deficiency since it is better absorbed, has higher biological activity, and is less prone to sequestration in adipose tissue and may be preferred over vitamin D3 for patients with obesity, liver disease, and malabsorption and those who require a rapid increase in 25-hydroxyvitamin D3 concentrations. | |
| E | [63] | 2021 | Supplementation of α- and γ-tocopherol in the ratio 1:5 reduces and attenuates adipocyte enlargement, hepatic steatosis, and metabolic inflammation (induced by HFD). |
| [64] | 2022 | Prolonged vitamin E supplementation can dysregulate interrelated miRNA profiles in the liver and WAT through negative feedback regulation, negatively impacting lipid metabolism in both the liver and WAT. | |
| K | [65] | 2024 | In an HFD-induced NAFLD mouse model, vitamin K2 reduced the visceral fat burden without reducing the lean mass and free body fluid, and prevented hepatic steatosis, inflammation, and fibrosis. |
| Adipose Tissue | Functions | References | |
|---|---|---|---|
| White Adipose Tissue (WAT) | Femoral-gluteal WAT | Protects against insulin resistance and cardiovascular diseases (CVDs) | [67] |
| Subcutaneous and abdominal WAT (sWAT) | Associated with insulin resistance, metabolic syndrome, type 2 diabetes mellitus (T2DM), and CVDs | [68] | |
| Gonadal (gWAT) (epididymal fat in males, eWAT, and periovarian fat in females) | Regulates gametogenesis via modulation of neuroendocrine signaling and lipid deposition and supports lipid metabolism | [69] | |
| Mesenteric Adipose Tissue (MAT) | Lipid storage, upholding the intestinal barrier, regulation of immune function, and intestinal flora intestinal permeability | [70,71] | |
| Epicardial Adipose Tissue (EpAT) | EpAT has physiological and pathological properties that vary depending on its location. It can be highly protective for the adjacent myocardium through dynamic brown fat-like thermogenic function and harmful via paracrine or endocrine secretion of pro-inflammatory and profibrotic cytokines | [72] | |
| Omental WAT (oWAT) | It presents immunomodulatory functions | [73] | |
| Retroperitoneal WAT (rWAT) | It has positive effects on cardiovascular, metabolic, inflammatory, and hormonal changes induced in high-fat conditions | [74] | |
| Brown Adipose Tissue (BAT) | Paravertebral and supraclavicular BAT (scBAT) | Protect against hypothermia to maintain optimal function and nerve conduction in the central and autonomic nervous system and immune functions. Thermogenic and cardiometabolic function | [75,76,77,78] |
| Cervical BAT | Associated with cardiometabolic homeostasis depending on gender and metabolic status | [79] | |
| Perirenal BAT (PRAT) | It influences metabolic, renal, and cardiovascular homeostasis, and controls the plasticity of brown/white adipose phenotypes | [80,81] | |
| Mediastinal BAT (MAT) | Prognostic biomarker of cardiovascular diseases | [82,83] | |
3. Vitamin A
| Compound | Effects on Liver | Effects on Adipose Tissue | Mechanisms | Models | Treatment | Reference |
|---|---|---|---|---|---|---|
| Retinoic acid (RA) | Reduction in fat deposition, and hepatic triglyceride (TG) and total cholesterol (TC) levels | Decrease in white adipose tissues (WATs) and interscapular brown adipose tissue (iBAT) weight; promotion of WAT browning and thermogenesis | In adipose tissue: upregulation of fatty acid oxidation genes (Cpt1B, Acox1, Pgc1), thermogenesis-related genes (Ucp1 and Pparγ) and markers of adipose tissue browning | Animal (mouse) | 50 mg/kg (high-fat diet, HFD) | [41] |
| β-carotene | Preservation of liver functions | Regulation of lipid metabolism | Regulation of expression of genes involved in Pparα, Acly, and Fabp5 pathways in dorsolumbar and inguinal WAT (iWAT) | Animal (mouse) | 150 mg/Kg (control diet) | [40] |
| Promotion of thermogenesis in adipocytes | Activation of the β3-AR, cAMP, and α1-AR receptor pathways and increase in cytosolic Ca2+ | Cell line (mouse 3T3-L1 preadipocytes) | 20 μM | [111] | ||
| Vitamin A carrier RBP4 | Induction of hepatic steatosis and mitochondrial dysfunction | Suppression of SIRT3-dependent long-chain acyl-CoA dehydrogenase (LCAD) deacetylation | Animal (transgenic mouse) | None (HFD) | [113] | |
| Promotion of de novo lipogenesis and lipid accumulation in hepatocytes and inflammation | Induction of M1-like polarization of Kupffer cells (KCs) by mediating the NOX2/ROS/NF-κB pathway | Cell lines (human Kupffer and hepatic LO2 cells) | 25–100 ng/mL | [114] | ||
| Animal (mouse) | 50 μg/kg (HFD) Intravenous | |||||
| Stimulation of basal lipolysis and inflammation | Increase of TNF-α production | Cell lines (human primary adipocytes and mouse macrophage RAW 264.7 cells) | 50 µg/mL | [114] |
4. Group B Vitamin
4.1. Vitamin B1
4.2. Vitamin B2
4.3. Vitamin B3
4.4. Vitamin B5
4.5. Vitamin B6
4.6. Vitamin B7
4.7. Vitamin B9
4.8. Vitamin B12
| Compound | Effects on Liver | Effects on Adipose Tissue | Mechanisms | Models | Treatment | Reference |
|---|---|---|---|---|---|---|
| Vitamin B1 | Reduction of hepatic steatosis, increased hepatic glycogen content | Increase of MTP, PLIN2, and SOD2 gene expression; inhibition of TNF-α production | Animal (lambs) | 300 mg/animal (high-calorie, HC, diet) Intravenous | [120] | |
| Increase of thermogenesis | Increase the expression of thermogenesis-related genes | Cell line (human primary adipocytes) | 25 μM | [113] | ||
| Vitamin B2 deficiency | Alteration of lipid metabolism (with lipid accumulation) and antioxidant functions | Downregulation of ATGL | Upregulation of FASN, CPT1, and PPARγ protein expression; downregulation of ATGL expression; impaired antioxidant mechanisms, including GR, SOD, and GSH-Px | Animal (mouse) | Riboflavin deprivation, High-fat diet (HFD) | [123] |
| Cell line (human hepatoma cell line HepG2) | 0 and 3 nM | |||||
| Vitamin B3 (Niacin) | Regression of hepatic steatosis, reduction of cholesterol and triglyceride accumulation | Inhibition of hepatic gene and protein expression and activity of DGAT2 | Animal (rats) | 0.5% and 1.0% (HFD) | [126] | |
| Anti-inflammatory effect in epidydimal white adipose tissue (eWAT) | Partially through the increase of adiponectin expression | Animal (mouse) | 360 mg/kg/d (HFD) | [128] | ||
| Vitamin B3 (nicotinamide, NAM) | Prevention of hepatic steatosis | Reduction of inflammation; shift into a brown-like phenotype; increase of mitochondrial β-oxidation of fatty acids (FAs) in inguinal white adipose tissue (iWAT). Reduction in lipid vesicle accumulation and increase of mitochondrial β-oxidation of FAs in interscapular brown adipose tissue (iBAT) | In the liver: downregulation of inflammatory (Tnf-α, Ccl2, and Il6) and fibrosis (Col1a1 and Mmp9) gene expression. In adipose tissue: gene expression upregulation of the anti-inflammatory cytokine Il-10, Ucp1, NAD+ consuming enzyme Sirt1, genes involved in mitochondrial homeostasis (Pgc1a and Pgc1b, Mfn2, Plin1 and Cpt1b) and genes involved in white adipose tissue (WAT) beiging (Ppargc1a, Ppargc1b, Prdm16); reduction of inflammatory (i.e., Tnf-α, Il6, and Ccl2) and fibrosis (i.e., Col1a1 and Mmp9) gene expression. Increase of Ucp1 protein. Activation of AMPK. Decreasing macrophage infiltration | Animal (mouse) | 1% (HFD) | [45] |
| Vitamin B3 (N1-methyl nicotinamide, mNAM) | Reduction of lipid accumulation | Reduction of inflammation in gonadal white adipose tissue (gWAT) | In liver: upregulation of genes (Pck1 and G6pc) related to gluconeogenesis in a NAD+/SIRT1-dependent manner. In gWAT: downregulation of Il1b and Il6 | Animal (pregnant mouse) | 0.3 and 1% (HFD) | [47] |
| Vitamin B3 (nicotinamide riboside, NR) | Liver protection from obesity effects | Reduction in fat mass of gWAT and iWAT, reduction of inflammation and fibrosis in gWAT | In WAT: reduction of expression of macrophages markers (Adgre1, Cd68) and M1 macrophages genes (Itgax, Tnf-α), M2 macrophages genes (Mrc1), and crow-like structures (CLS) | Animal (old female mouse) | 400 mg/kg/d (HFD) | [48] |
| Reduction of TG levels, fat deposition, and lipid synthesis. Anti-inflammatory effect | Increase of lipolysis in WAT | In liver: increase of NAD+/NADH redox imbalance and subsequent SIRT1/NF-κB pathway activation and IL-1β, IL-6, IL-18, and TNF-α downregulation; increase of FGF21 pathway activation. In WAT: Increase of FGF21 path activation | Animal (mouse) | 400 mg/kg/d (high-fructose diet) | [49] | |
| Vitamin B5 | Indirectly, reduction of steatosis | Reduction of adipocyte lipide deposit in BAT, sWAT, and eWAT | Activation of BAT-inducing energy expenditure, and beige adipocyte promotion by phosphorylation of AMPK, which leads to induction of UCP1 expression by PGC1a | Animal (mouse) | 10 mg/Kg (HFD) | [50] |
| Cell line (human primary brown adipocytes) | 1–5 mM | |||||
| Vitamin B6 | Decrease of liver lipid deposition, moderating steatosis | Decrease of the adipocyte size in WAT | In liver: activation of hepatic mitochondrial β-oxidation by upregulation of the expression of liver lipase (Hl), Sirt1, and Pparα; inhibition of the lipogenesis pathway by decreasing the expression of Srebp1c and its downstream lipogenic enzymes Acc and Fas | Animal (rats) | 2–3 mg/kg (HFD) | [137] |
| Vitamin B7 | Reduction of hepatic triglyceride storage | Reduction of WAT weight | Activation of hepatic mitochondrial β-oxidation via upregulation of CPT activity; and inhibition of fatty-acid (FA) synthesis via downregulation of Acc2 | Animal (rats) | 37.9 mg/day (HFD) | [51] |
| Vitamin B9 deficiency | Steatosis | Genetic variations (SNPs) (rs1051266 and rs3788200) within SLC19A1 are associated with MALFD. SLC19A1-knockdown in the human cell line determines the downregulation of pathways controlling non-esterified fatty acid pathways, fatty amides, sterols, glycerophospholipids, and amino acid concentrations | Cell line (human liver THL2) | 0.1 mg/ml | [145] | |
| Vitamin B9 | Impediment of fibrosis resolution | Activation of mitochondrial folate metabolism via upregulation of Shmt2 and Mthfd2 maintain profibrotic TGF-β1 signaling and polyunsaturated FA metabolism for hepatic stellate cells (HSCs) viable activation | Animal (mouse) | 103 mg/Kg (normal chew) | [146] | |
| Cell lines (human LX-2 and mouse primary liver cell line) | 10 mM | |||||
| Normal hepatocytes in contact with sinusoids, central vein, and minimal number of apoptotic figures by impairing lipogenesis, insulin resistance, and imbalanced cytokine production | In a dose-dependent manner, restoring the physiological expression of hepatic miRNA via downregulation of miR-21 and miR-34, and upregulation of their related genes, Hbp1 and Sirt1, respectively; and upregulation of miR-122 causes downregulation of Srebp-1 | Animal (rats) | 75 mg/kg (HFD) | [150] | ||
| Decreased inflammation and fibrosis | Suppression of adipocyte proliferation, differentiation, and adipogenesis via downregulation of IGF1, EGF, and TGF-β | Increased intestinal folic acid transport carriers (RFC) is associated with the increment of Bacteroidetes (Alistipes, Oscillospira, Ruminococcus, Clostridium, Dehalo-bacterium, and Parabacteroides) and caecal short-chain fatty acids (SCFAs) (acetic acid, propionic acid, and isobutyric acid). Each caecal microbiota is positively correlated with the specific acetic acid content | Animal (broilers) | 1.3 mg/kg (normal chew) | [52] | |
| Vitamin B9 + Vitamin B12 | Decreased inflammation and fibrosis | Impairment of STX17 proteasomal degradation recovers autophagy and restoration of homocysteine metabolism via upregulation of related genes (Mat1a, Mthfr, Cbs, Mtr, Pon1, Pon2, Pon3). Consequently, increased β-oxidation of FAs leading to decreased hepatic inflammation (IL6, IL1b, TNF-α) and chemokine (Ccl2, Ccl5, Cxcl10, Cx3cl1, Cxcl16) and fibrosis (Tgf-β, Col1a1, Col1a2, Col3a1, Acta2, Ctgf) genes | Animal (mouse) | B12 30 μg/~4700 kcal and Folate 6 μg/~4700 kcal (fructose in drinking water) | [172] |
5. Vitamin C
| Compound | Effects on Liver | Effects on Adipose Tissue | Mechanisms | Models | Treatment | Reference |
|---|---|---|---|---|---|---|
| Vitamin C | Modulation of gene expression | Induction of white-to-brown conversion, energy expenditure | In the liver: activation of the transcription factor Pparα leading to the secretion of thermogenic hormone Fgf21. In adipose tissue: Fgf21 controls thermogenic energy expenditure via Ucp1 upregulation. | Animal (mouse) | 2 g/L oral gavage, high-fat diet (HFD) | [54] |
| Reduction of hepatic steatosis | Decrement of fat mass in epidydimal and inguinal white adipose tissue (eWAT and iWAT) and brown adipose tissue (BAT) | In iWAT: downregulation of lipogenic genes (Srebf1, Fasn, and Acaca) and upregulation of thermogenic genes (Ucp1, ELovl3, Cox7a1, Dio2, and Cox8b). In liver: upregulation of Hsl, Ppara, Acox1, and Cpt1 and increases methylation of Hsl and Ppara promoters. | Animal (mouse) | 0.36 g/kg (HFD) | [55] | |
| Hypermethylation of the HSL and PPARα promoters and upregulation of genes involved in fatty acid oxidation and lipolysis via reversion of TET1 haplo-insufficiency. | Primary human hepatocytes | 200 mM |
6. Vitamin D
| Compound | Effects on Liver | Effects on Adipose Tissue | Mechanisms | Models | Treatment | Reference |
|---|---|---|---|---|---|---|
| Vitamin D | Inhibition of inflammatory pathway and adipokine expression | Anti-inflammatory activity: decrease of IL-6 and leptin protein expression through suppression of NF-kB and MAPK pathways via vitamin D receptor (VDR) | Human adipose tissue and adipocytes | 10−8 M | [197] | |
| Protection from HFD effects | In epidydimal white adipose tissue (eWAT) suppression of adipogenesis, inflammatory responses, macrophage infiltration, and their phenotypic switch to M1 polarization | Inhibition of the transcription factor PPARγ and AP2, and decrease of the gene expression of Tnf-α, Il-6, and Mcp via inhibiting NF-kB inhibition and AMPK pathway activation | Animal (mouse) | 1000 IU/kg (high-fat diet, HFD) | [58] | |
| Inhibition of browning of white adipose tissue (WAT) | Activation of p53 and inactivation of P13K/Akt/mTOR signaling leading to autophagy, impairment of brow-like adipocyte formation by downregulating the WAT browing markers (UPI1, PPARγ, PGCα) | Animal (mouse) | 50 mg/kg (HFD) | [201] | ||
| Cell line (mouse 3T3-L1 preadipocytes) | 1–100 nM | |||||
| Decrease lipid accumulation | Decrease inflammation | In the liver: suppression of de novo lipogenesis (Fasn and Acaca) and fatty acid oxidation (Acox) | Animal (mouse) | 15,000 IU/Kg (high-fat/sucrose diet) | [59] | |
| Decrease inflammation and lipid accumulation | Decrease inflammation | In the liver: suppression of de novo lipogenesis (Fasn and Acaca) and chemokines Mcp1 In inguinal white adipose tissue (iWAT): strong suppression of Ccl5 but slight suppression of Tgfb1 and Mcp1 | Animal (mouse) | 15,000 IU/Kg (high-fat/sucrose diet) | [60] | |
| Enhancement in fatty degeneration | Prevention of hypertrophy of adipocytes | In liver: Reduction of FATP4 in liver. In liver and adipose tissue: decrease of TLR-4 in both liver and adipose tissue | Animal (rat) | 500 IU/Kg (HFD) | [61] | |
| Enhancement of brown adipogeneis | Stimulation of brown adipogenesis program via Prdm16 and Pgc1α upregulation and inhibition of white adipocyte differentiation via Cebpb, Cebpa, and Pparγ downregulation | Cell lines (mouse C3H10T1/2, 3T3-L1) | 100 pM | [205] | ||
| Reduction of fat vacuoles and inflammation | Inhibition of NLRP3 and pyroptosis, downregulation of ASC, cleaved-caspase-1, pro-IL-1β, IL-1β and GSDMD-N in liver tissues and BRL-3 | Animal (rat) | 5 mg/kg (HFD) | [217] | ||
| Cell line (human hepatocellular HepG2) | 10−6 mol/L | |||||
| Enhancement of hepatic steatosis and systemic inflammation | In cells and rats, upregulation of β-oxidation by increasing expression of Pparα and Cpt1a and downregulation of fatty acid translocation (Fat/Cd36) | Animal (rat) | 12.5 μg/Kg (HFD) | [218] | ||
| Cell line (human hepatocellular HepG2) | 25–200 nM |
7. Vitamin E
| Compound | Effects on Liver | Effects on Adipose Tissue | Mechanisms | Models | Treatment | Reference |
|---|---|---|---|---|---|---|
| Vitamin E | Reduction of steatosis inhibiting lipogenesis | Impairment of intrahepatic triglyceride (IHTG) accumulation by inhibition of maturation of the transcription factor SREBP-1, downregulating de novo lipogenesis genes (FASN and SCD) | Human (liver biopsies) | 100 mmol/L | [232] | |
| Cell lines (human hepatocellular HepG2) | ||||||
| Vitamin E (α- and γ-tocopherol) | Prevention of steatosis, oxidative stress, and inflammation | Reduction of adipocyte size and inflammation | In adipose tissue and the liver: inhibition of NF-κB nuclear translocation decreasing Il-1β and Tnf-α In the liver: positive modulation of Pparα, enhancing the expression of peroxisomal Acox | Animal (mouse) | 0.7 mg/kg (high-fat diet, HFD) | [63] |
| Vitamin E | Impairment of lipid synthesis and activation of FA oxidation | Decreased expression of the dehydrogenases Raldh1 and Raldh2, and the transcription factor Srebp-1c, inhibiting hepatic FA synthesis and transporter CD36 | Animal (mouse) | 0.7 mg/kg (HFD) | [235] | |
| Increase of triglycerides (TGs) | Moderated inflammation and mild increase in fat cell size | In the liver and white adipose tissue (WAT): dose-dependently suppresses the expression of Pgc-1a and Srebp-1c and Srebp-2c upregulation. In the liver and WAT: dose-dependent suppression of miRNAs (miR-22/miR-27) expression implicated in lipid metabolism | Animal (mouse) | 100, 200, and 500 mg/kg oral gavage/day (normal chew) | [64] |
8. Vitamin K
| Compound | Effects on Liver | Effects on Adipose Tissue | Mechanisms | Models | Treatment | Reference |
|---|---|---|---|---|---|---|
| Vitamin K | Reduction of steatosis | Increase of AMPK phosphorylation, and downregulation of Srebp1 and Fas and upregulation Pparα, Cpt1a and Ucp2 via activating Gla-Gas6 | Animal (mouse) | 5 mg/kg (high-fat diet, HFD) | [249] |
9. Conclusions
Funding
Conflicts of Interest
Gene and Protein Abbreviations
| ACACA | Acetyl-CoA Carboxylase 1 |
| ACC2 | Acetyl-CoA carboxylase beta |
| Acly | ATP-citrate Lyase |
| Acox1 | Acyl-CoA Oxidase 1 |
| Acta2 | Actin alpha 2, smooth muscle |
| AFABP | Adipocyte Fatty Acid-Binding Protein |
| Akt | Protein kinase B |
| AMPK | AMP-activated Protein Kinase |
| aP2 | Adipocyte protein 2 |
| apoB | Apolipoprotein B |
| ASC | PYD and CARD domain containing |
| ATGL | Adipose Triglyceride Lipase |
| ATP | Adenosine Triphosphate |
| BCKDH | Branched-Chain Ketoacid Dehydrogenase |
| BCMO1 | β-carotene 15,15′-monooxygenase 1 |
| BRL-3 | BRI1-like 3 |
| Cbs | Cystathionine beta-synthase |
| CCDC80 | Coiled-Coil Domain Containing 80 |
| Ccl2 | C-C Motif Chemokine Ligand 2 |
| Ccl5 | C-C Motif Chemokine Ligand 5 |
| Cebpa | CCAAT enhancer binding protein alpha |
| Cebpb | CCAAT enhancer binding protein beta |
| Col1a1 | Collagen type I alpha 1 chain |
| Col1a2 | Collagen type I alpha 2 chain |
| Col3a1 | Collagen type III alpha 1 chain |
| COL6A2 | Collagen type VI alpha 2 chain |
| Cox7a | Cytochrome c oxidase subunit 7A1 |
| Cox8b | Cytochrome c oxidase subunit 8B |
| Cpt1 | Carnitine O-palmitoyltransferase |
| CPT1A | Carnitine palmitoyltransferase 1A |
| Cpt1B | Carnitine palmitoyltransferase 1B |
| Ctgf | Connective tissue growth factor |
| CX3CL1 | C-X3-C motif chemokine ligand 1 |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| CXCL16 | C-X-C motif chemokine ligand 16 |
| CYP | Cytochrome P450 |
| CYP27B1 | Cytochrome P450 27B1 |
| CYP2R1 | Cytochrome P450 2R1, Vitamin D 25-Hydroxylase |
| Dio2 | Iodothyronine deiodinase 2 |
| DUSP1 | Dual-specific phosphatase |
| EGF | Epidermal growth factor |
| ELOVL3 | ELOVL fatty acid elongase 3 |
| ERβ | Estrogen receptor beta |
| Fabp5 | Fatty Acid Binding Protein 5 |
| FAD | Flavin Adenine Dinucleotide |
| FASN | Fatty Acid Synthase |
| FATP4 | Fatty acid transport protein-4 |
| FGF21 | Fibroblast Growth Factor 21 |
| FMN | Flavin Mononucleotide |
| G6pc | Glucose-6-phosphatase catalytic subunit |
| GADD45B | Growth arrest and DNA-damage-inducible 45 beta |
| GGCX | γ-glutamyl Carboxylase |
| GAS6 | Growth Arrest-Specific protein 6 |
| GLUT4 | Glucose transporter type 4 |
| GR | Glutathione Reductase |
| GSDMD-N | Gasdermin D |
| GSH-Px | Glutathione Peroxidase |
| HBP1 | HMG-box transcription factor 1 |
| HDL | High-density lipoprotein |
| Hsl | Hormone-sensitive lipase |
| IGF1 | Insulin-like growth factor 1 |
| IL-10 | Interleukin-10 |
| Il-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| KGDHC | α-Ketoglutarate Dehydrogenase Complex |
| LCAD | Long-chain acyl-CoA dehydrogenase |
| LDL | Low-density lipoprotein |
| MAPK | Mitogen-activated Protein Kinase |
| Mat1a | Methionine adenosyltransferase 1A |
| MCM | Methylmalonyl-CoA mutase |
| MCP1 | Monocyte Chemoattractant Protein 1 |
| Mmp9 | Matrix metalloproteinase 9 |
| MS | Methionine synthase |
| MTHFD2 | Methylenetetrahydrofolate dehydrogenase 2 |
| Mthfr | Methylenetetrahydrofolate reductase |
| mTOR | Mammalian target of rapamycin kinase |
| MTP | Microsomal Transfer Protein |
| Mtr | 5-methyltetrahydrofolate-homocysteine methyltransferase |
| NADP | Nicotinamide Adenine Dinucleotide Phosphate |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NFκB | Nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain containing 3 |
| NOX2 | NADPH Oxidase 2 |
| NRF2 | Nuclear factor erythroid-derived 2 |
| PANK | Pantothenate Kinase |
| Pck1 | Phosphoenolpyruvate Carboxykinase 1 |
| PDC | Pyruvate Dehydrogenase Complex già esteso |
| Pgc1 | Peroxisome Proliferator-activated Receptor Gamma Coactivator 1 |
| PGC1-α | Peroxisome Proliferator-activated Receptor Gamma Coactivator 1-alpha |
| PI3K | Phosphatidylinositol 3-kinases |
| PIVKA-II | Protein Induced by Vitamin K Absence-II |
| Pon1/2/3 | paraoxonase 1/2/3 |
| PPAR-α | Peroxisome Proliferator-Activated Receptor Alpha |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| Prdm16 | PR domain containing 16 |
| PXR | Pregnane X receptor |
| RALDH 1/2 | Retinaldehyde dehydrogenases 1/2 |
| RBP4 | Retinol-Binding Protein 4 |
| RFC | Reduced folate transporter |
| RORα | RAR-related orphan receptor alpha |
| SCD | Stearoyl-CoA Desaturase |
| SCD1 | Stearoyl-CoA Desaturase 1 |
| SHMT2 | Serine hydroxymethyltransferase 2 |
| Shmt2 | Serine hydroxymethyltransferase 2 |
| SIK1 | Salt inducible kinase 1 |
| SIRT | Sirtuin |
| SIRT1 | Sirtuin 1 |
| SLC19A1 | Solute carrier family 19 member 1 |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SOD | Superoxide Dismutase |
| SOD2 | Superoxide Dismutase 2 |
| SOD3 | Superoxide Dismutase 3 |
| SREBF1 | Sterol regulatory element-binding transcription factor 1 |
| SREBP-1 | Sterol regulatory element-binding protein 1 |
| SREBP-1c | Sterol regulatory element-binding protein 1c |
| Stx17 | Syntaxin 17 |
| TET | Ten-Eleven Translocation enzymes |
| TGF-β1 | Transforming Growth Factor-beta 1 |
| TK | Transketolase |
| TLR-4 | Toll-like receptor 4 |
| TNF-α | Tumor Necrosis Factor-alpha |
| TPP | Thiamine Pyrophosphate |
| TTR | Transthyretin |
| UCP1 | Uncoupling Protein 1 |
| VKOR | Vitamin K Epoxide Reductase |
| VLDL | Very Low-Density Lipoprotein |
| α-KGDH | Alpha-ketoglutarate dehydrogenase |
| Adgre1 | Adhesion G protein-coupled receptor E1 |
| CD68 | Cluster of Differentiation 68 |
| Mrc1 | Mannose Receptor C-Type 1 |
References
- Lowe, N.M. The Global Challenge of Hidden Hunger: Perspectives from the Field. Proc. Nutr. Soc. 2021, 80, 283–289. [Google Scholar] [CrossRef]
- van den Berg, H.; van der Gaag, M.; Hendriks, H. Influence of Lifestyle on Vitamin Bioavailability. Int. J. Vitam. Nutr. Res. 2002, 72, 53–59. [Google Scholar] [CrossRef]
- Pham, V.T.; Dold, S.; Rehman, A.; Bird, J.K.; Steinert, R.E. Vitamins, the Gut Microbiome and Gastrointestinal Health in Humans. Nutr. Res. 2021, 95, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Gudan, A.; Kozłowska-Petriczko, K.; Wunsch, E.; Bodnarczuk, T.; Stachowska, E. Small Intestinal Bacterial Overgrowth and Non-Alcoholic Fatty Liver Disease: What Do We Know in 2023? Nutrients 2023, 15, 1323. [Google Scholar] [CrossRef]
- Juszczak, A.B.; Kupczak, M.; Konecki, T. Does Vitamin Supplementation Play a Role in Chronic Kidney Disease? Nutrients 2023, 15, 2847. [Google Scholar] [CrossRef]
- Mathew, A.R.; Matteo, G.D.; Rosa, P.L.; Barbati, S.A.; Mannina, L.; Moreno, S.; Tata, A.M.; Cavallucci, V.; Fidaleo, M. Vitamin B12 Deficiency and the Nervous System: Beyond Metabolic Decompensation—Comparing Biological Models and Gaining New Insights into Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2024, 25, 590. [Google Scholar] [CrossRef]
- Ekenedilichukwu, O.J.; Solomon, I.C.; Chukwuemeka, O.E.; Adamma, A.R.; Athanatius, O.O.; Nwabunwanne, O.V.; Kalu, A.A.; Uzoma, M.T.; Chukwuemeka, M.S. The Effects of Cigarette Smoking on The Serum Levels of Some Antioxidants (Vitamin C and E) amongst Male Smokers In Levels in College of Health Sciences, Nnamdi Azikiwe University, Nnewi Campus, Anambra State. Int. J. Sci. Res. Manag. 2018, 6–11. [Google Scholar] [CrossRef]
- Sohrabi, M.; Djalali, M.; Javanbakht, M.H.; Shekoohi, N.; Ghavami, A.; Mohammadi, H. Association of Cigarette Smoking and Serum Concentrations of Vitamins A and E in Men: A Case-Control Study. J. Nutr. Sci. Diet. 2021, 5, 14–18. [Google Scholar] [CrossRef]
- Chen, Q.; Kord-Varkaneh, H.; Santos, H.O.; Genario, R.; Dang, M. Higher Intakes of Dietary Caffeine Are Associated with 25-Hydroxyvitamin D Deficiency: A Cross-Sectional Study from the NHANES. Int. J. Vitam. Nutr. Res. 2022, 92, 85–90. [Google Scholar] [CrossRef]
- Sandoval, C.; Farías, J.; Zamorano, M.; Herrera, C. Vitamin Supplements as a Nutritional Strategy against Chronic Alcohol Consumption? An Updated Review. Antioxidants 2022, 11, 564. [Google Scholar] [CrossRef]
- Sahu, P.; Chhabra, P.; Mehendale, A.M. A Comprehensive Review on Non-Alcoholic Fatty Liver Disease. Cureus 2023, 15, e50159. [Google Scholar] [CrossRef] [PubMed]
- Habash, M.; Al-shakhshir, S.; Abusamak, M.; Mohammad, M.Y.; AbuSamak, M. The Association of Coffee Consumption Rate with Serum 25-Hydroxyvitamin D, Non-HDL Levels, and TC/HDL Ratio in Females with Vitamin D Deficiency. Women’s Health 2022, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.A.M.; Masroor, A.; Khorochkov, A.; Prieto, J.; Singh, K.B.; Nnadozie, M.C.; Abdal, M.; Shrestha, N.; Mohammed, L. The Role of Vitamins in Non-Alcoholic Fatty Liver Disease: A Systematic Review. Cureus 2021, 13, e16855. [Google Scholar] [CrossRef] [PubMed]
- Ivancovsky-Wajcman, D.; Fliss-Isakov, N.; Salomone, F.; Webb, M.; Shibolet, O.; Kariv, R.; Zelber-Sagi, S. Dietary Vitamin E and C Intake Is Inversely Associated with the Severity of Nonalcoholic Fatty Liver Disease. Digest. Liver Dis. 2019, 51, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, A.; Fiorotto, R.; Strazzabosco, M. Bile Acids and Their Receptors: Modulators and Therapeutic Targets in Liver Inflammation. Semin Immunopathol. 2022, 44, 547–564. [Google Scholar] [CrossRef]
- Li, J.; Cordero, P.; Nguyen, V.; Oben, J.A. The Role of Vitamins in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Integr. Med. Insights 2016, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Pickett-Blakely, O.; Young, K.; Carr, R.M. Micronutrients in Nonalcoholic Fatty Liver Disease Pathogenesis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Mao, T.; Sun, Y.; Xu, X.; He, K. Overview and Prospect of NAFLD: Significant Roles of Nutrients and Dietary Patterns in Its Progression or Prevention. Hepatol. Commun. 2023, 7, e0234. [Google Scholar] [CrossRef]
- Golabi, P.; Owrangi, S.; Younossi, Z.M. Global Perspective on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis—Prevalence, Clinical Impact, Economic Implications and Management Strategies. Aliment. Pharmacol. Ther. 2024, 59, S1–S9. [Google Scholar] [CrossRef]
- Miao, L.; Targher, G.; Byrne, C.D.; Cao, Y.-Y.; Zheng, M.-H. Current Status and Future Trends of the Global Burden of MASLD. Trends Endocrinol. Metab. 2024, 35, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Hua, R.; Peng, K.; Yin, Y.; Zeng, C.; Guo, Y.; Wang, Y.; Li, L.; Li, X.; Qiu, Y.; et al. High-Starchy Carbohydrate Diet Aggravates NAFLD by Increasing Fatty Acids Influx Mediated by NOX2. Food Sci. Hum. Wellness 2023, 12, 1081–1101. [Google Scholar] [CrossRef]
- Lujan, P.V.; Esmel, E.V.; Meseguer, E.S. Overview of Non-Alcoholic Fatty Liver Disease (NAFLD) and the Role of Sugary Food Consumption and Other Dietary Components in Its Development. Nutrients 2021, 13, 1442. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A Multisociety Delphi Consensus Statement on New Fatty Liver Disease Nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Bhopale, K.K.; Srinivasan, M.P. Therapeutics for Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD). Livers 2023, 3, 597–617. [Google Scholar] [CrossRef]
- Sangro, P.; de la Torre Aláez, M.; Sangro, B.; D’Avola, D. Metabolic Dysfunction–Associated Fatty Liver Disease (MAFLD): An Update of the Recent Advances in Pharmacological Treatment. J. Physiol. Biochem. 2023, 79, 869–879. [Google Scholar] [CrossRef]
- Boccatonda, A.; Andreetto, L.; D’Ardes, D.; Cocco, G.; Rossi, I.; Vicari, S.; Schiavone, C.; Cipollone, F.; Guagnano, M.T. From NAFLD to MAFLD: Definition, Pathophysiological Basis and Cardiovascular Implications. Biomedicines 2023, 11, 883. [Google Scholar] [CrossRef]
- Wang, S.-W.; Hsieh, T.-H.; Cheng, Y.-M.; Wang, C.-C.; Kao, J.-H. Liver and Atherosclerotic Risks of Patients with Cryptogenic Steatotic Liver Disease. Hepatol. Int. 2024, 18, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Sanz, M.J.; Real, J.T.; Marques, P.; Capuozzo, M.; Eldjoudi, D.A.; Gualillo, O. Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets? Biology 2022, 11, 1237. [Google Scholar] [CrossRef]
- Yang, K.; Song, M. New Insights into the Pathogenesis of Metabolic-Associated Fatty Liver Disease (MAFLD): Gut–Liver–Heart Crosstalk. Nutrients 2023, 15, 3970. [Google Scholar] [CrossRef]
- Kobayashi, T.; Iwaki, M.; Nakajima, A.; Nogami, A.; Yoneda, M. Current Research on the Pathogenesis of NAFLD/NASH and the Gut–Liver Axis: Gut Microbiota, Dysbiosis, and Leaky-Gut Syndrome. Int. J. Mol. Sci. 2022, 23, 11689. [Google Scholar] [CrossRef]
- Juanola, O.; Martínez-López, S.; Francés, R.; Gómez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef]
- Petrescu, M.; Vlaicu, S.I.; Ciumărnean, L.; Milaciu, M.V.; Mărginean, C.; Florea, M.; Vesa, Ș.C.; Popa, M. Chronic Inflammation—A Link between Nonalcoholic Fatty Liver Disease (NAFLD) and Dysfunctional Adipose Tissue. Medicina 2022, 58, 641. [Google Scholar] [CrossRef]
- Wang, B.; Du, M. Increasing Adipocyte Number and Reducing Adipocyte Size: The Role of Retinoids in Adipose Tissue Development and Metabolism. Crit. Rev. Food Sci. Nutr. 2023. ahead-of-print. [Google Scholar] [CrossRef]
- Nan, Y.; Su, H.; Lian, X.; Wu, J.; Liu, S.; Chen, P.; Liu, S. Pathogenesis of Liver Fibrosis and Its TCM Therapeutic Perspectives. Evid.-Based Complement. Altern. Med. 2022, 2022, 5325431. [Google Scholar] [CrossRef]
- Kumar, S.; Duan, Q.; Wu, R.; Harris, E.N.; Su, Q. Pathophysiological Communication between Hepatocytes and Non-Parenchymal Cells in Liver Injury from NAFLD to Liver Fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113869. [Google Scholar] [CrossRef]
- Sen, P.; Govaere, O.; Sinioja, T.; McGlinchey, A.; Geng, D.; Ratziu, V.; Bugianesi, E.; Schattenberg, J.M.; Vidal-Puig, A.; Allison, M.; et al. Quantitative Modeling of Human Liver Reveals Dysregulation of Glycosphingolipid Pathways in Nonalcoholic Fatty Liver Disease. iScience 2022, 25, 104949. [Google Scholar] [CrossRef]
- Panzarini, E.; Leporatti, S.; Tenuzzo, B.A.; Quarta, A.; Hanafy, N.A.N.; Giannelli, G.; Moliterni, C.; Vardanyan, D.; Sbarigia, C.; Fidaleo, M.; et al. Therapeutic Effect of Polymeric Nanomicelles Formulation of LY2157299-Galunisertib on CCl4-Induced Liver Fibrosis in Rats. J. Pers. Med. 2022, 12, 1812. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; He, W.; Dong, H.; Guo, Y.; Yuan, G.; Shi, X.; Wang, D.; Lu, F. Role of Liver Sinusoidal Endothelial Cell in Metabolic Dysfunction-Associated Fatty Liver Disease. Cell Commun. Signal. 2024, 22, 346. [Google Scholar] [CrossRef]
- Saito, M.; Okamatsu-Ogura, Y. Thermogenic Brown Fat in Humans: Implications in Energy Homeostasis, Obesity and Metabolic Disorders. World J. Men’s Health 2023, 41, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Zhao, Y.; Liu, P.; Cai, D.; Zhang, X.; Gao, L. Regulatory Mechanisms of Beta-Carotene and BCMO1 in Adipose Tissues: A Gene Enrichment-Based Bioinformatics Analysis. Hum. Exp. Toxicol. 2022, 41, 09603271211072871. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, J.; Zhu, D.; Jiang, X.; Wei, L.; Wang, W.; Chen, Y.Q. Adipose Tissue Plays a Major Role in Retinoic Acid-Mediated Metabolic Homoeostasis. Adipocyte 2022, 11, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, X.; Deng, H.; Huang, H.; Liu, Y.; Zhong, Z.; Shen, L.; Cao, S.; Ma, X.; Zhou, Z.; et al. Integrated Transcriptome and Metabolomics to Reveal the Mechanism of Adipose Mesenchymal Stem Cells in Treating Liver Fibrosis. Int. J. Mol. Sci. 2023, 24, 16086. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Lin, H.; Liu, C.; Yan, Y.; Ke, Y.; Chen, Y. Higher Serum Vitamin A Is Associated with a Worsened Progression of Non-Alcoholic Fatty Liver Disease in Adults: A Prospective Study. Food Funct. 2022, 13, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Linder, K.; Willmann, C.; Kantartzis, K.; Machann, J.; Schick, F.; Graf, M.; Kümmerle, S.; Häring, H.-U.; Fritsche, A.; Stefan, N.; et al. Dietary Niacin Intake Predicts the Decrease of Liver Fat Content During a Lifestyle Intervention. Sci. Rep. 2019, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lara, K.A.; Rodríguez-Millán, E.; Sebastián, D.; Blanco-Soto, R.; Camacho, M.; Nan, M.N.; Diarte-Añazco, E.M.G.; Mato, E.; Lope-Piedrafita, S.; Roglans, N.; et al. Nicotinamide Protects Against Diet-Induced Body Weight Gain, Increases Energy Expenditure, and Induces White Adipose Tissue Beiging. Mol. Nutr. Food Res. 2021, 65, e2100111. [Google Scholar] [CrossRef] [PubMed]
- Nejabati, H.R.; Ghaffari-Novin, M.; Fathi-Maroufi, N.; Faridvand, Y.; Holmberg, H.-C.; Hansson, O.; Nikanfar, S.; Nouri, M. N1-Methylnicotinamide: Is It Time to Consider It as a Dietary Supplement for Athletes? Curr. Pharm. Des. 2022, 28, 800–805. [Google Scholar] [CrossRef]
- Wei, X.; Tan, Y.; Huang, J.; Dong, X.; Feng, W.; Liu, T.; Yang, Z.; Yang, G.; Luo, X. N1-Methylnicotinamide Impairs Gestational Glucose Tolerance in Mice. J. Mol. Endocrinol. 2024, 72, e230126. [Google Scholar] [CrossRef]
- Kim, M.-B.; Pham, T.X.; vanLuling, M.; Kostour, V.; Kang, H.; Corvino, O.; Jang, H.; Odell, W.; Bae, M.; Park, Y.-K.; et al. Nicotinamide Riboside Supplementation Exerts an Anti–Obesity Effect and Prevents Inflammation and Fibrosis in White Adipose Tissue of Female Diet-Induced Obesity Mice. J. Nutr. Biochem. 2022, 107, 109058. [Google Scholar] [CrossRef]
- Zhao, H.; Tian, Y.; Zuo, Y.; Zhang, X.; Gao, Y.; Wang, P.; Sun, L.; Zhang, H.; Liang, H. Nicotinamide Riboside Ameliorates High-Fructose-Induced Lipid Metabolism Disorder in Mice via Improving FGF21 Resistance in the Liver and White Adipose Tissue. Food Funct. 2022, 13, 12400–12411. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, H.; Ye, R.; Yan, C.; Lin, J.; Huang, Y.; Jiang, X.; Yuan, S.; Chen, L.; Jiang, R.; et al. Pantothenate Protects against Obesity via Brown Adipose Tissue Activation. Am. J. Physiol.-Endocrinol. Metab. 2022, 323, E69–E79. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, M.; Kawabeta, K.; Uemura, M.; Koba, K.; Sawamura, H.; Watanabe, T. Dietary High-Dose Biotin Intake Activates Fat Oxidation and Hepatic Carnitine Palmitoyltransferase in Rat. J. Nutr. Sci. Vitaminol. 2022, 68, 250–259. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, J.; Liu, X.; Liu, R.; Wang, Y.; Huang, X.; Li, Y.; Liu, R.; Yang, X. Dietary Folic Acid Addition Reduces Abdominal Fat Deposition Mediated by Alterations in Gut Microbiota and SCFA Production in Broilers. Anim. Nutr. 2023, 12, 54–62. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhao, L.; Meng, C.; Zhao, X.; Liu, Y.; Shi, R.; Han, X.; Wang, T.; Li, J. Prophylactic and Therapeutic Effects of Different Doses of Vitamin C on High-Fat-Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. Biomed. Pharmacother. 2020, 131, 110792. [Google Scholar] [CrossRef]
- Lee, B.; An, H.J.; Kim, D.H.; Lee, M.-K.; Jeong, H.H.; Chung, K.W.; Go, Y.; Seo, A.Y.; Kim, I.Y.; Seong, J.K.; et al. SMP30-Mediated Synthesis of Vitamin C Activates the Liver PPARα/FGF21 Axis to Regulate Thermogenesis in Mice. Exp. Mol. Med. 2022, 54, 2036–2046. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, C.; Chen, X.; Sun, Y.; Xiong, M.; Fan, Y.; Petersen, R.B.; Chen, H.; Huang, K.; Zheng, L. Vitamin C Inhibits the Metabolic Changes Induced by Tet1 Insufficiency Under High Fat Diet Stress. Mol. Nutr. Food Res. 2021, 65, e2100417. [Google Scholar] [CrossRef]
- Elkhwanky, M.; Kummu, O.; Piltonen, T.T.; Laru, J.; Morin-Papunen, L.; Mutikainen, M.; Tavi, P.; Hakkola, J. Obesity Represses CYP2R1, the Vitamin D 25-Hydroxylase, in the Liver and Extrahepatic Tissues. JBMR Plus 2020, 4, e10397. [Google Scholar] [CrossRef]
- Szymczak-Pajor, I.; Miazek, K.; Selmi, A.; Balcerczyk, A.; Śliwińska, A. The Action of Vitamin D in Adipose Tissue: Is There the Link between Vitamin D Deficiency and Adipose Tissue-Related Metabolic Disorders? Int. J. Mol. Sci. 2022, 23, 956. [Google Scholar] [CrossRef]
- Chang, E. Effects of Vitamin D Supplementation on Adipose Tissue Inflammation and NF-ΚB/AMPK Activation in Obese Mice Fed a High-Fat Diet. Int. J. Mol. Sci. 2022, 23, 10915. [Google Scholar] [CrossRef] [PubMed]
- Marziou, A.; Philouze, C.; Couturier, C.; Astier, J.; Obert, P.; Landrier, J.-F.; Riva, C. Vitamin D Supplementation Improves Adipose Tissue Inflammation and Reduces Hepatic Steatosis in Obese C57BL/6J Mice. Nutrients 2020, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- MARZIOU, A.; AUBERT, B.; COUTURIER, C.; ASTIER, J.; PHILOUZE, C.; OBERT, P.; LANDRIER, J.-F.; RIVA, C. Combined Beneficial Effect of Voluntary Physical Exercise and Vitamin D Supplementation in Diet-Induced Obese C57BL/6J Mice. Med. Sci. Sports Exerc. 2021, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Kolieb, E.; Maher, S.A.; Shalaby, M.N.; Alsuhaibani, A.M.; Alharthi, A.; Hassan, W.A.; El-Sayed, K. Vitamin D and Swimming Exercise Prevent Obesity in Rats under a High-Fat Diet via Targeting FATP4 and TLR4 in the Liver and Adipose Tissue. Int. J. Environ. Res. Public Health 2022, 19, 13740. [Google Scholar] [CrossRef] [PubMed]
- Jodar, E.; Campusano, C.; de Jongh, R.T.; Holick, M.F. Calcifediol: A Review of Its Pharmacological Characteristics and Clinical Use in Correcting Vitamin D Deficiency. Eur. J. Nutr. 2023, 62, 1579–1597. [Google Scholar] [CrossRef] [PubMed]
- Juretić, N.; Sepúlveda, R.; D’Espessailles, A.; Vera, D.B.; Cadagan, C.; de Miguel, M.; González-Mañán, D.; Tapia, G. Dietary Alpha- and Gamma-Tocopherol (1:5 Ratio) Supplementation Attenuates Adipose Tissue Expansion, Hepatic Steatosis, and Expression of Inflammatory Markers in a High-Fat-Diet–Fed Murine Model. Nutrition 2021, 85, 111139. [Google Scholar] [CrossRef]
- Ali, M.A.; El-Tahan, R.A.; Kamel, M.A.; Matar, N.A.; Mahmoud, S.A. The Diabetogenic Effects of Chronic Supplementation of Vitamin C or E in Rats: Interplay between Liver and Adipose Tissues Transcriptional Machinery of Lipid Metabolism. Life Sci. 2022, 306, 120812. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, W.; Xiao, H.; Zhang, S.; Gao, C.; Piao, H.; Liu, L.; Li, S. Vitamin K2 Protects Mice against Non-Alcoholic Fatty Liver Disease Induced by High-Fat Diet. Sci. Rep. 2024, 14, 3075. [Google Scholar] [CrossRef]
- Maio, G.D.; Alessio, N.; Demirsoy, I.H.; Peluso, G.; Perrotta, S.; Monda, M.; Bernardo, G.D. Evaluation of Browning Agents on the White Adipogenesis of Bone Marrow Mesenchymal Stromal Cells: A Contribution to Fighting Obesity. Cells 2021, 10, 403. [Google Scholar] [CrossRef]
- Alser, M.; Naja, K.; Elrayess, M.A. Mechanisms of Body Fat Distribution and Gluteal-Femoral Fat Protection against Metabolic Disorders. Front. Nutr. 2024, 11, 1368966. [Google Scholar] [CrossRef]
- Chait, A.; Hartigh, L.J. den Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Yang, C.-F.; Liu, W.-W.; Wang, H.-Q.; Zhang, J.-L.; Li, K.; Diao, Z.-Y.; Yue, Q.-L.; Yan, G.-J.; Li, C.-J.; Sun, H.-X. Gonadal White Adipose Tissue Is Important for Gametogenesis in Mice through Maintenance of Local Metabolic and Immune Niches. J. Biol. Chem. 2022, 298, 101818. [Google Scholar] [CrossRef]
- Börgeson, E.; Boucher, J.; Hagberg, C.E. Of Mice and Men: Pinpointing Species Differences in Adipose Tissue Biology. Front. Cell Dev. Biol. 2022, 10, 1003118. [Google Scholar] [CrossRef]
- Zhang, H.; Ding, Y.; Zeng, Q.; Wang, D.; Liu, G.; Hussain, Z.; Xiao, B.; Liu, W.; Deng, T. Characteristics of Mesenteric Adipose Tissue Attached to Different Intestinal Segments and Their Roles in Immune Regulation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2022, 322, G310–G326. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial Adipose Tissue in Contemporary Cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Rubio-Contreras, D.; Gómez-Rosado, J.C.; Capitán-Morales, L.C.; Hmadcha, A.; Soria, B.; Lachaud, C.C. Human Omental Mesothelial Cells Impart an Immunomodulatory Landscape Impeding B- and T-Cell Activation. Int. J. Mol. Sci. 2022, 23, 5924. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Milanez, M.I.O.; Nishi, E.E.; Sato, A.Y.S.; Carvalho, P.M.; Nogueira, F.N.; Campos, R.R.; Oyama, L.M.; Bergamaschi, C.T. Retroperitoneal Adipose Tissue Denervation Improves Cardiometabolic and Autonomic Dysfunction in a High Fat Diet Model. Life Sci. 2021, 283, 119841. [Google Scholar] [CrossRef] [PubMed]
- Hachemi, I.; U-Din, M. Brown Adipose Tissue: Activation and Metabolism in Humans. Endocrinol. Metab. 2023, 38, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Gallerand, A.; Ivanov, S. Immune Cell Involvement in Brown Adipose Tissue Functions. Discov. Immunol. 2022, 1, kyac007. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gu, C.; Zhang, Z.; Arianti, R.; Swaminathan, A.; Tran, K.; Battist, A.; Kristóf, E.; Ruan, H.-B. Supraclavicular Brown Adipocytes Originate from Tbx1+ Myoprogenitors. PLOS Biol. 2023, 21, e3002413. [Google Scholar] [CrossRef]
- U-Din, M.; Rebelos, E.; Saari, T.; Niemi, T.; Kuellmer, K.; Eskola, O.; Fromme, T.; Rajander, J.; Taittonen, M.; Klingenspor, M.; et al. Thermogenic Capacity of Human Supraclavicular Brown Fat and Cold-Stimulated Brain Glucose Metabolism. Metabolites 2023, 13, 387. [Google Scholar] [CrossRef]
- Cresswell, E.; Basty, N.; Pasdar, N.A.; Karpe, F.; Pinnick, K.E. The Value of Neck Adipose Tissue as a Predictor for Metabolic Risk in Health and Type 2 Diabetes. Biochem. Pharmacol. 2024, 223, 116171. [Google Scholar] [CrossRef]
- Hammoud, S.H.; AlZaim, I.; Al-Dhaheri, Y.; Eid, A.H.; El-Yazbi, A.F. Perirenal Adipose Tissue Inflammation: Novel Insights Linking Metabolic Dysfunction to Renal Diseases. Front. Endocrinol. 2021, 12, 707126. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Ibáñez, C.F.; Xie, M. Perirenal Adipose Tissue Contains a Subpopulation of Cold-Inducible Adipocytes Derived from Brown-to-White Conversion. eLife 2024, 13, RP93151. [Google Scholar] [CrossRef]
- Fuentevilla-Álvarez, G.; Huesca-Gómez, C.; Paz-Torres, Y.E.; González-Moyotl, N.; Soto, M.E.; García-Valdivia, J.A.; Reyna-Sámano; Martínez-Rosas, M.; Meza-Toledo, S.E.; Gamboa, R. Evaluation of the Participation of ABCA1 Transporter in Epicardial and Mediastinal Adipose Tissue from Patients with Coronary Artery Disease. Arch. Endocrinol. Metab. 2023, 68, e230188. [Google Scholar] [CrossRef] [PubMed]
- Marttila, J.; Sipola, P.; Juutilainen, A.; Sillanmäki, S.; Hedman, M.; Kuusisto, J. Central Obesity Is Associated with Increased Left Ventricular Maximal Wall Thickness and Intrathoracic Adipose Tissue Measured with Cardiac Magnetic Resonance. High Blood Press. Cardiovasc. Prev. 2024, 31, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Mladenović, D.; Vesković, M.; Šutulović, N.; Hrnčić, D.; Stanojlović, O.; Radić, L.; Macut, J.B.; Macut, D. Adipose-Derived Extracellular Vesicles—A Novel Cross-Talk Mechanism in Insulin Resistance, Non-Alcoholic Fatty Liver Disease, and Polycystic Ovary Syndrome. Endocrine 2024, 85, 18–34. [Google Scholar] [CrossRef]
- Lee, E.; Korf, H.; Vidal-Puig, A. An Adipocentric Perspective on the Development and Progression of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2023, 78, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Qiao, Y.; Liu, S.; Yang, S.; Cong, S.; Wang, S.; Yu, D.; Wang, W.; Chai, X. Frontiers and Hotspots of Adipose Tissue and NAFLD: A Bibliometric Analysis from 2002 to 2022. Front. Physiol. 2023, 14, 1278952. [Google Scholar] [CrossRef]
- Myint, M.; Oppedisano, F.; Giorgi, V.D.; Kim, B.-M.; Marincola, F.M.; Alter, H.J.; Nesci, S. Inflammatory Signaling in NASH Driven by Hepatocyte Mitochondrial Dysfunctions. J. Transl. Med. 2023, 21, 757. [Google Scholar] [CrossRef]
- Darci-Maher, N.; Alvarez, M.; Arasu, U.T.; Selvarajan, I.; Lee, S.H.T.; Pan, D.Z.; Miao, Z.; Das, S.S.; Kaminska, D.; Örd, T.; et al. Cross-Tissue Omics Analysis Discovers Ten Adipose Genes Encoding Secreted Proteins in Obesity-Related Non-Alcoholic Fatty Liver Disease. eBioMedicine 2023, 92, 104620. [Google Scholar] [CrossRef]
- Hosokawa, Y.; Hosooka, T.; Imamori, M.; Yamaguchi, K.; Itoh, Y.; Ogawa, W. Adipose Tissue Insulin Resistance Exacerbates Liver Inflammation and Fibrosis in a Diet-Induced NASH Model. Hepatol. Commun. 2023, 7, e0161. [Google Scholar] [CrossRef]
- Lopez-Yus, M.; Lorente-Cebrian, S.; del Moral-Bergos, R.; Hörndler, C.; Garcia-Sobreviela, M.P.; Casamayor, C.; Sanz-Paris, A.; Bernal-Monterde, V.; Arbones-Mainar, J.M. Identification of Novel Targets in Adipose Tissue Involved in Non-alcoholic Fatty Liver Disease Progression. FASEB J. 2022, 36, e22429. [Google Scholar] [CrossRef]
- Eyck, A.V.; Kwanten, W.J.; Peleman, C.; Makhout, S.; Laere, S.V.; Maele, K.V.D.; Hoorenbeeck, K.V.; Man, J.D.; Winter, B.Y.D.; Francque, S.; et al. The Role of Adipose Tissue and Subsequent Liver Tissue Hypoxia in Obesity and Early Stage Metabolic Dysfunction Associated Steatotic Liver Disease. Int. J. Obes. 2023, 36, e22429. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, M.-F.; Jiang, S.; Wu, J.; Liu, J.; Yuan, X.-W.; Shen, D.; Zhang, J.-Z.; Zhou, N.; He, J.; et al. Liver Governs Adipose Remodelling via Extracellular Vesicles in Response to Lipid Overload. Nat. Commun. 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Liu, J. Friend or Foe for Obesity: How Hepatokines Remodel Adipose Tissues and Translational Perspective. Genes Dis. 2023, 10, 825–847. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Rong, X.; Xu, A.; Guo, J. Liver-Adipose Tissue Crosstalk: A Key Player in the Pathogenesis of Glucolipid Metabolic Disease. Chin. J. Integr. Med. 2017, 23, 410–414. [Google Scholar] [CrossRef]
- Yang, Z.; Kubant, R.; Cho, C.E.; Kranenburg, E.; Beaudry, J.; Bottiglieri, T.; Anderson, G.H. Micronutrients in High-Fat Diet Modify Insulin Resistance and Its Regulatory Genes in Adult Male Mice. Mol. Nutr. Food Res. 2023, 67, e2300199. [Google Scholar] [CrossRef]
- Vucic, V.; Ristic-Medic, D.; Arsic, A.; Petrovic, S.; Paunovic, M.; Vasiljevic, N.; Ilich, J.Z. Nutrition and Physical Activity as Modulators of Osteosarcopenic Adiposity: A Scoping Review and Recommendations for Future Research. Nutrients 2023, 15, 1619. [Google Scholar] [CrossRef] [PubMed]
- Marley, A.; Smith, S.C.; Ahmed, R.; Nightingale, P.; Cooper, S.C. Vitamin A Deficiency: Experience from a Tertiary Referral UK Hospital; Not Just a Low- and Middle-Income Country Issue. Public Health Nutr. 2021, 24, 6466–6471. [Google Scholar] [CrossRef]
- von Lintig, J.; Moon, J.; Lee, J.; Ramkumar, S. Carotenoid Metabolism at the Intestinal Barrier. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2020, 1865, 158580. [Google Scholar] [CrossRef]
- Paik, J.; Vogel, S.; Quadro, L.; Piantedosi, R.; Gottesman, M.; Lai, K.; Hamberger, L.; de Morais Vieira, M.; Blaner, W.S. Vitamin A: Overlapping Delivery Pathways to Tissues from the Circulation. J. Nutr. 2004, 134, S276–S280. [Google Scholar] [CrossRef] [PubMed]
- Haaker, M.W.; Vaandrager, A.B.; Helms, J.B. Retinoids in Health and Disease: A Role for Hepatic Stellate Cells in Affecting Retinoid Levels. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2020, 1865, 158674. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Weiskirchen, S.; Weiskirchen, R. Vitamin A: Too Good to Be Bad? Front. Pharmacol. 2023, 14, 1186336. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic Stellate Cell Lipid Droplets: A Specialized Lipid Droplet for Retinoid Storage. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Blaner, W.S. Retinol and Retinyl Esters: Biochemistry and Physiology Thematic Review Series: Fat-Soluble Vitamins: Vitamin A. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, J.S.; Lass, A.; Schupp, M. Biological Functions of RBP4 and Its Relevance for Human Diseases. Front. Physiol. 2021, 12, 659977. [Google Scholar] [CrossRef]
- Gilani, A.; Stoll, L.; Homan, E.A.; Lo, J.C. Adipose Signals Regulating Distal Organ Health and Disease. Diabetes 2024, 73, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Cero, C.; Lea, H.J.; Zhu, K.Y.; Shamsi, F.; Tseng, Y.-H.; Cypess, A.M. Β3-Adrenergic Receptors Regulate Human Brown/Beige Adipocyte Lipolysis and Thermogenesis. JCI Insight 2021, 6, e139160. [Google Scholar] [CrossRef] [PubMed]
- Queathem, E.D.; Welly, R.J.; Clart, L.M.; Rowles, C.C.; Timmons, H.; Fitzgerald, M.; Eichen, P.A.; Lubahn, D.B.; Vieira-Potter, V.J. White Adipose Tissue Depots Respond to Chronic Beta-3 Adrenergic Receptor Activation in a Sexually Dimorphic and Depot Divergent Manner. Cells 2021, 10, 3453. [Google Scholar] [CrossRef]
- Collins, S. β-Adrenergic Receptors and Adipose Tissue Metabolism: Evolution of an Old Story. Annu. Rev. Physiol. 2022, 84, 1–16. [Google Scholar] [CrossRef]
- Choi, M.; Yun, J.W. β-Carotene Induces UCP1-Independent Thermogenesis via ATP-Consuming Futile Cycles in 3T3-L1 White Adipocytes. Arch. Biochem. Biophys. 2023, 739, 109581. [Google Scholar] [CrossRef] [PubMed]
- Góes, É.; Cordeiro, A.; Bento, C.; Ramalho, A. Vitamin A Deficiency and Its Association with Visceral Adiposity in Women. Biomedicines 2023, 11, 991. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, D.; Chen, H.; Li, D.; Song, J.; Zhong, Y.; Xia, M. Retinol-Binding Protein 4 Induces Hepatic Mitochondrial Dysfunction and Promotes Hepatic Steatosis. J. Clin. Endocrinol. Metab. 2016, 101, 4338–4348. [Google Scholar] [CrossRef]
- Yao, J.-M.; Ying, H.-Z.; Zhang, H.-H.; Qiu, F.-S.; Wu, J.-Q.; Yu, C.-H. Exosomal RBP4 Potentiated Hepatic Lipid Accumulation and Inflammation in High-Fat-Diet-Fed Mice by Promoting M1 Polarization of Kupffer Cells. Free Radic. Biol. Med. 2023, 195, 58–73. [Google Scholar] [CrossRef]
- Kilicarslan, M.; de Weijer, B.A.; Sjödin, K.S.; Aryal, P.; Horst, K.W.t.; Cakir, H.; Romijn, J.A.; Ackermans, M.T.; Janssen, I.M.; Berends, F.J.; et al. RBP4 Increases Lipolysis in Human Adipocytes and Is Associated with Increased Lipolysis and Hepatic Insulin Resistance in Obese Women. FASEB J. 2020, 34, 6099–6110. [Google Scholar] [CrossRef]
- Flores-Cortez, Y.A.; Barragán-Bonilla, M.I.; Mendoza-Bello, J.M.; GonzÁlez-Calixto, C.; Flores-Alfaro, E.; Espinoza-Rojo, M. Interplay of Retinol Binding Protein 4 with Obesity and Associated Chronic Alterations. Mol. Med. Rep. 2022, 26, 244. [Google Scholar] [CrossRef]
- Park, Y.; Smith-Warner, S.A.; Zhang, X.; Park, Y.J.; Kim, H.; Park, H.; Lee, H.A.; Jung, S. Association between Use of Vitamin and Mineral Supplement and Non-Alcoholic Fatty Liver Disease in Hypertensive Adults. Sci. Rep. 2023, 13, 13670. [Google Scholar] [CrossRef]
- Mrowicka, M.; Mrowicki, J.; Dragan, G.; Majsterek, I. The Importance of Thiamine (Vitamin B1) in Humans. Biosci. Rep. 2023, 43, BSR20230374. [Google Scholar] [CrossRef] [PubMed]
- Hrubša, M.; Siatka, T.; Nejmanová, I.; Vopršalová, M.; Krčmová, L.K.; Matoušová, K.; Javorská, L.; Macáková, K.; Mercolini, L.; Remião, F.; et al. Biological Properties of Vitamins of the B-Complex, Part 1: Vitamins B1, B2, B3, and B5. Nutrients 2022, 14, 484. [Google Scholar] [CrossRef] [PubMed]
- Kalyesubula, M.; Mopuri, R.; Asiku, J.; Rosov, A.; Yosefi, S.; Edery, N.; Bocobza, S.; Moallem, U.; Dvir, H. High-Dose Vitamin B1 Therapy Prevents the Development of Experimental Fatty Liver Driven by Overnutrition. Dis. Model. Mech. 2021, 14, dmm048355. [Google Scholar] [CrossRef] [PubMed]
- Vinnai, B.Á.; Arianti, R.; Győry, F.; Bacso, Z.; Fésüs, L.; Kristóf, E. Extracellular Thiamine Concentration Influences Thermogenic Competency of Differentiating Neck Area-Derived Human Adipocytes. Front. Nutr. 2023, 10, 1207394. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Araújo, E.R.; Toscano, A.E.; Silva, P.B.P.; Junior, J.P.d.S.; Gouveia, H.J.C.B.; da Silva, M.M.; Souza, V.d.S.; Silva, S.R.d.F.; Manhães-de-Castro, R. Effects of Deficiency or Supplementation of Riboflavin on Energy Metabolism: A Systematic Review with Preclinical Studies. Nutr. Rev. 2024, 8, nuae041. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bian, X.; Wan, M.; Dong, W.; Gao, W.; Yao, Z.; Guo, C. Effects of Riboflavin Deficiency and High Dietary Fat on Hepatic Lipid Accumulation: A Synergetic Action in the Development of Non-Alcoholic Fatty Liver Disease. Nutr. Metab. 2024, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M.E. The Chemistry of the Vitamin B3 Metabolome. Biochem. Soc. Trans. 2018, 47, 131–147. [Google Scholar] [CrossRef]
- Pan, J.; Hu, Y.; Pang, N.; Yang, L. Association between Dietary Niacin Intake and Nonalcoholic Fatty Liver Disease: NHANES 2003–2018. Nutrients 2023, 15, 4128. [Google Scholar] [CrossRef]
- Ganji, S.H.; Kukes, G.D.; Lambrecht, N.; Kashyap, M.L.; Kamanna, V.S. Therapeutic Role of Niacin in the Prevention and Regression of Hepatic Steatosis in Rat Model of Nonalcoholic Fatty Liver Disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 306, G320–G327. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of Fenofibrate and Niacin on Intrahepatic Triglyceride Content, Very Low-Density Lipoprotein Kinetics, and Insulin Action in Obese Subjects with Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. The Absence of Adiponectin Alters Niacin’s Effects on Adipose Tissue Inflammation in Mice. Nutrients 2020, 12, 2427. [Google Scholar] [CrossRef]
- Miallot, R.; Millet, V.; Galland, F.; Naquet, P. The Vitamin B5/Coenzyme A Axis: A Target for Immunomodulation? Eur. J. Immunol. 2023, 53, e2350435. [Google Scholar] [CrossRef]
- Machado, M.V.; Kruger, L.; Jewell, M.L.; Michelotti, G.A.; Pereira, T.D.; Xie, G.; Moylan, C.A.; Diehl, A.M. Vitamin B5 and N-Acetylcysteine in Nonalcoholic Steatohepatitis: A Preclinical Study in a Dietary Mouse Model. Dig. Dis. Sci. 2016, 61, 137–148. [Google Scholar] [CrossRef]
- Chen, J.; Lu, R.S.; Diaz-Canestro, C.; Song, E.; Jia, X.; Liu, Y.; Wang, C.; Cheung, C.K.Y.; Panagiotou, G.; Xu, A. Distinct Changes in Serum Metabolites and Lipid Species in the Onset and Progression of NAFLD in Obese Chinese. Comput. Struct. Biotechnol. J. 2024, 23, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T. Disorders Affecting Vitamin B6 Metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef]
- Dewangan, S.; Bhatia, A.K. Chapter 5—Vitamins and metabolites. In Handbook of Biomolecules; Elsevier: Amsterdam, The Netherlands, 2023; pp. 119–131. [Google Scholar] [CrossRef]
- Wang, P.; Huang, J.; Xue, F.; Abuduaini, M.; Tao, Y.; Liu, H. Associations of Serum Vitamin B6 Status with the Risks of Cardiovascular, Cancer, and All-Cause Mortality in the Elderly. Front. Immunol. 2024, 15, 1354958. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kessoku, T.; Ozaki, A.; Iwaki, M.; Honda, Y.; Ogawa, Y.; Imajo, K.; Yoneda, M.; Saito, S.; Nakajima, A. Vitamin B6 Efficacy in the Treatment of Nonalcoholic Fatty Liver Disease: An Open-Label, Single-Arm, Single-Center Trial. J. Clin. Biochem. Nutr. 2021, 68, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Tripathi, D.; Vikram, N.K.; Madhusudan, K.S.; Pandey, R.M.; Bhatia, N. Association of Nutrient Intake with Non-Alcoholic Fatty Liver Disease and Liver Steatosis in Adult Indian Population—A Case Control Study. Hum. Nutr. Metab. 2023, 32, 200188. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, X.; Zhang, J.; Li, Q.; Qian, Z. Selenium and Vitamin B6 Cosupplementation Improves Dyslipidemia and Fatty Liver Syndrome by SIRT1/SREBP-1c Pathway in Hyperlipidemic Sprague-Dawley Rats Induced by High-Fat Diet. Nutr. Res. 2022, 106, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Alvarez, D.; Solórzano-Vargas, R.S.; Río, A.L.D. Biotin in Metabolism and Its Relationship to Human Disease. Arch. Méd. Res. 2002, 33, 439–447. [Google Scholar] [CrossRef]
- Riverón-Negrete, L.; Sicilia-Argumedo, G.; Álvarez-Delgado, C.; Coballase-Urrutia, E.; Alcántar-Fernández, J.; Fernandez-Mejia, C. Dietary Biotin Supplementation Modifies Hepatic Morphology without Changes in Liver Toxicity Markers. BioMed Res. Int. 2016, 2016, 7276463. [Google Scholar] [CrossRef]
- Voland, L.; Yuan, M.; Lecoutre, S.; Debédat, J.; Pelloux, V.; Pradeau, M.; Coles, E.; Merabtene, F.; Zhang, C.; Mardinoglu, A.; et al. Tissue Pleiotropic Effect of Biotin and Prebiotic Supplementation in Established Obesity. Am. J. Physiol.-Endocrinol. Metab. 2023, 325, E390–E405. [Google Scholar] [CrossRef]
- Quevedo-Ocampo, J.; Escobedo-Calvario, A.; Souza-Arroyo, V.; Miranda-Labra, R.U.; Bucio-Ortiz, L.; Gutiérrez-Ruiz, M.C.; Chávez-Rodríguez, L.; Gomez-Quiroz, L.E. Folate Metabolism in Hepatocellular Carcinoma. What Do We Know So Far? Technol. Cancer Res. Treat. 2022, 21, 15330338221144446. [Google Scholar] [CrossRef]
- Chen, H.-K.; Luo, J.; Li, X.-J.; Liao, W.-Z.; Hu, Y.-Q.; Guo, X.-G. Serum Folate Associated with Nonalcoholic Fatty Liver Disease and Advanced Hepatic Fibrosis. Sci. Rep. 2023, 13, 12933. [Google Scholar] [CrossRef]
- Yao, B.; Lu, X.; Xu, L.; Jiang, Y. Association of Serum Folate with Prevalence of Non-Alcoholic Fatty Liver Disease among Adults (NHANES 2011–2018). Front. Nutr. 2023, 10, 1141156. [Google Scholar] [CrossRef]
- Molaqanbari, M.R.; Zarringol, S.; Talari, H.R.; Taghizadeh, M.; Bahmani, F.; Mohtashamian, A.; Ebrahimzadeh, A.; Sharifi, N. Effects of Folic Acid Supplementation on Liver Enzymes, Lipid Profile, and Insulin Resistance in Patients with Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Adv. Biomed. Res. 2023, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Vazquez-Chantada, M.; Conde-Vancells, J.; Gonzalez-Lahera, A.; Mosen-Ansorena, D.; Blanco, F.J.; Clément, K.; Aron-Wisnewsky, J.; Tran, A.; Gual, P.; et al. Impaired Function of Solute Carrier Family 19 Leads to Low Folate Levels and Lipid Droplet Accumulation in Hepatocytes. Biomedicines 2023, 11, 337. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zheng, B.; Xu, S.; Zhao, Z.; Liu, W.; Wang, T.; Yuan, M.; Sun, X.; Tan, Y.; Xu, Q.; et al. Mitochondrial Folate Metabolism–Mediated α-Linolenic Acid Exhaustion Masks Liver Fibrosis Resolution. J. Biol. Chem. 2023, 299, 104909. [Google Scholar] [CrossRef] [PubMed]
- Glen, C.D.; McVeigh, L.E.; Voutounou, M.; Dubrova, Y.E. The Effects of Methyl-donor Deficiency on the Pattern of Gene Expression in Mice. Mol. Nutr. Food Res. 2015, 59, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic Steatohepatitis Is Associated with Altered Hepatic MicroRNA Expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Tryndyak, V.P.; Marrone, A.K.; Latendresse, J.R.; Muskhelishvili, L.; Beland, F.A.; Pogribny, I.P. MicroRNA Changes, Activation of Progenitor Cells and Severity of Liver Injury in Mice Induced by Choline and Folate Deficiency. J. Nutritional. Biochem. 2016, 28, 83–90. [Google Scholar] [CrossRef]
- Salman, M.; Kamel, M.A.; El-Nabi, S.E.H.; Ismail, A.H.A.; Ullah, S.; Al-Ghamdi, A.; Hathout, H.M.R.; El-Garawani, I.M. The Regulation of HBP1, SIRT1, and SREBP-1c Genes and the Related MicroRNAs in Non-Alcoholic Fatty Liver Rats: The Association with the Folic Acid Anti-Steatosis. PLoS ONE 2022, 17, e0265455. [Google Scholar] [CrossRef]
- Petrus, P.; Bialesova, L.; Checa, A.; Kerr, A.; Naz, S.; Bäckdahl, J.; Gracia, A.; Toft, S.; Dahlman-Wright, K.; Hedén, P.; et al. Adipocyte Expression of SLC19A1 Links DNA Hypermethylation to Adipose Tissue Inflammation and Insulin Resistance. J. Clin. Endocrinol. Metab. 2017, 103, 710–721. [Google Scholar] [CrossRef]
- Chan, C.-W.; Chan, P.-H.; Lin, B.-F. Folate Deficiency Increased Lipid Accumulation and Leptin Production of Adipocytes. Front. Nutr. 2022, 9, 852451. [Google Scholar] [CrossRef] [PubMed]
- Shane, B. Folate and Vitamin B12 Metabolism: Overview and Interaction with Riboflavin, Vitamin B6, and Polymorphisms. Food Nutr. Bull. 2008, 29, S5–S16. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Liang, B.; Su, Y.; Sun, S.; Xia, S.; Shao, J.; Zhang, Z.; Hong, M.; Zhang, F.; et al. Methionine Metabolism in Chronic Liver Diseases: An Update on Molecular Mechanism and Therapeutic Implication. Signal Transduct. Target. Ther. 2020, 5, 280. [Google Scholar] [CrossRef]
- Ge, Y.; Zadeh, M.; Mohamadzadeh, M. Vitamin B12 Regulates the Transcriptional, Metabolic, and Epigenetic Programing in Human Ileal Epithelial Cells. Nutrients 2022, 14, 2825. [Google Scholar] [CrossRef] [PubMed]
- Ashok, T.; Puttam, H.; Tarnate, V.C.A.; Jhaveri, S.; Avanthika, C.; Treviño, A.G.T.; Sandeep, S.L.; Ahmed, N.T.; Lakshmisai, S.S. Role of Vitamin B12 and Folate in Metabolic Syndrome. Cureus 2021, 13, e18521. [Google Scholar] [CrossRef] [PubMed]
- Guetterman, H.M.; Huey, S.L.; Knight, R.; Fox, A.M.; Mehta, S.; Finkelstein, J.L. Vitamin B-12 and the Gastrointestinal Microbiome: A Systematic Review. Adv. Nutr. 2022, 13, 530–558. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Schwartz, B. B Vitamins, Glucoronolactone and the Immune System: Bioavailability, Doses and Efficiency. Nutrients 2023, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.R.; Cavallucci, V.; Fidaleo, M. Altered Vitamin B12 Metabolism in the Central Nervous System Is Associated with the Modification of Ribosomal Gene Expression: New Insights from Comparative RNA Dataset Analysis. Funct. Integr. Genom. 2023, 23, 45. [Google Scholar] [CrossRef] [PubMed]
- Chiocchetti, A.; Prodam, F.; Dianzani, U. Homocysteine and Folate in Inflammatory Bowel Disease: Can Reducing Sulfur Reduce Suffering? Dig. Dis. Sci. 2018, 63, 3161–3163. [Google Scholar] [CrossRef]
- Harb, Z.; Deckert, V.; Bressenot, A.M.; Christov, C.; Guéant-Rodriguez, R.-M.; Raso, J.; Alberto, J.M.; de Barros, J.-P.P.; Umoret, R.; Peyrin-Biroulet, L.; et al. The Deficit in Folate and Vitamin B12 Triggers Liver Macrovesicular Steatosis and Inflammation in Rats with Dextran Sodium Sulfate-Induced Colitis. J. Nutr. Biochem. 2020, 84, 108415. [Google Scholar] [CrossRef]
- Halczuk, K.; Kaźmierczak-Barańska, J.; Karwowski, B.T.; Karmańska, A.; Cieślak, M. Vitamin B12—Multifaceted In Vivo Functions and In Vitro Applications. Nutrients 2023, 15, 2734. [Google Scholar] [CrossRef] [PubMed]
- Boachie, J.; Adaikalakoteswari, A.; Gázquez, A.; Zammit, V.; Larqué, E.; Saravanan, P. Vitamin B12 Induces Hepatic Fatty Infiltration through Altered Fatty Acid Metabolism. Cell. Physiol. Biochem. 2021, 55, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Koplay, M.; Gulcan, E.; Ozkan, F. Association between Serum Vitamin B12 Levels and the Degree of Steatosis in Patients with Nonalcoholic Fatty Liver Disease. J. Investig. Med. 2011, 59, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Patsiaoura, K.; Katsiki, E.; Zafeiriadou, E.; Zavos, C.; Deretzi, G.; Tsiaousi, E.; Slavakis, A. Serum Vitamin B12 and Folate Levels in Patients with Non-Alcoholic Fatty Liver Disease. Int. J. Food Sci. Nutr. 2012, 63, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Tayyem, R.F.; Al-Dayyat, H.M.; Rayyan, Y.M. Relationship between Lifestyle Factors and Nutritional Status and Non-Alcoholic Fatty Liver Disease among a Group of Adult Jordanians. Arab J. Gastroenterol. 2019, 20, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, Y.; Hu, Y.-Q. Bi-Directional Causal Effect between Vitamin B12 and Non-Alcoholic Fatty Liver Disease: Inferring from Large Population Data. Front. Nutr. 2023, 10, 1015046. [Google Scholar] [CrossRef]
- Talari, H.R.; Molaqanbari, M.R.; Mokfi, M.; Taghizadeh, M.; Bahmani, F.; Tabatabaei, S.M.H.; Sharifi, N. The Effects of Vitamin B12 Supplementation on Metabolic Profile of Patients with Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Sci. Rep. 2022, 12, 14047. [Google Scholar] [CrossRef]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.D.; Wu, X.; Still, C.D.; Chu, X.; Petrick, A.T.; Gerhard, G.S.; Conneely, K.N.; DiStefano, J.K. Differential DNA Methylation and Changing Cell-Type Proportions as Fibrotic Stage Progresses in NAFLD. Clin. Epigenetics 2021, 13, 152. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Edmison, J.; Marczewski, S.; Dasarathy, S.; Gruca, L.L.; Bennett, C.; Duenas, C.; Lopez, R. Methionine and Protein Metabolism in Non-Alcoholic Steatohepatitis: Evidence for Lower Rate of Transmethylation of Methionine. Clin. Sci. 2011, 121, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Singh, B.K.; Zhou, J.; Tikno, K.; Widjaja, A.; Sandireddy, R.; Arul, K.; Ghani, S.A.B.A.; Bee, G.G.B.; Wong, K.A.; et al. Vitamin B12 and Folate Decrease Inflammation and Fibrosis in NASH by Preventing Syntaxin 17 Homocysteinylation. J. Hepatol. 2022, 77, 1246–1255. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Wood, C.; Mina, T.H.; Webster, C.; Goljan, I.; Weldeselassie, Y.; Reynolds, R.M.; Saravanan, P. Vitamin B12 Deficiency and Altered One-Carbon Metabolites in Early Pregnancy Is Associated with Maternal Obesity and Dyslipidaemia. Sci. Rep. 2020, 10, 11066. [Google Scholar] [CrossRef] [PubMed]
- Aureli, A.; Recupero, R.; Mariani, M.; Manco, M.; Carlomagno, F.; Bocchini, S.; Nicodemo, M.; Marchili, M.R.; Cianfarani, S.; Cappa, M.; et al. Low Levels of Serum Total Vitamin B12 Are Associated with Worse Metabolic Phenotype in a Large Population of Children, Adolescents and Young Adults, from Underweight to Severe Obesity. Int. J. Mol. Sci. 2023, 24, 16588. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Cheng, H.; Gao, L.; Zhao, X.; Mi, J. Genetically Proxied Vitamin B12 and Homocysteine in Relation to Life Course Adiposity and Body Composition. Diabetes Metab. Syndr. Clin. Res. Rev. 2023, 17, 102883. [Google Scholar] [CrossRef] [PubMed]
- Mercantepe, F. Relationship of Vitamin B12 Levels With Different Degrees of Obesity and Diabetes Mellitus. Cureus 2023, 15, e47352. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yin, Z.; Han, L.; Zhang, M.; Li, H.; Yang, X.; Chen, Y.; Zhang, S.; Han, J.; Duan, Y. Ascorbic Acid Inhibits Transcriptional Activities of LXRα to Ameliorate Lipid Metabolism Disorder. J. Funct. Food 2022, 88, 104901. [Google Scholar] [CrossRef]
- Hussain, M.M.; Rava, P.; Walsh, M.; Rana, M.; Iqbal, J. Multiple Functions of Microsomal Triglyceride Transfer Protein. Nutr. Metab. 2012, 9, 14. [Google Scholar] [CrossRef]
- Nishikata, N.; Shikata, N.; Kimura, Y.; Noguchi, Y. Dietary Lipid-Dependent Regulation of de Novo Lipogenesis and Lipid Partitioning by Ketogenic Essential Amino Acids in Mice. Nutr. Diabetes 2011, 1, e5. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De Novo Lipogenesis in Health and Disease. Metabolis 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Tutunchi, H.; Saghafi-Asl, M.; Asghari-Jafarabadi, M.; Ostadrahimi, A. The Relationship between Severity of Liver Steatosis and Metabolic Parameters in a Sample of Iranian Adults. BMC Res. Notes 2020, 13, 218. [Google Scholar] [CrossRef]
- Hou, X.; Guan, Y.; Tang, Y.; Song, A.; Zhao, J.; Ren, L.; Chen, S.; Wei, L.; Ma, H.; Song, G. A Correlation Study of the Relationships between Nonalcoholic Fatty Liver Disease and Serum Triglyceride Concentration after an Oral Fat Tolerance Test. Lipids Health Dis. 2021, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Skat-Rørdam, J.; Pedersen, K.; Skovsted, G.F.; Gregersen, I.; Vangsgaard, S.; Ipsen, D.H.; Latta, M.; Lykkesfeldt, J.; Tveden-Nyborg, P. Vitamin C Deficiency May Delay Diet-Induced NASH Regression in the Guinea Pig. Antioxidants 2021, 11, 69. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Corey, K.E.; Lim, J.K. AGA Clinical Practice Update on Lifestyle Modification Using Diet and Exercise to Achieve Weight Loss in the Management of Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2021, 160, 912–918. [Google Scholar] [CrossRef]
- Keinicke, H.; Sun, G.; Mentzel, C.M.J.; Fredholm, M.; John, L.M.; Andersen, B.; Raun, K.; Kjærgaard, M. FGF21 Regulates Hepatic Metabolic Pathways to Improve Steatosis and Inflammation. Endocr. Connect. 2020, 1, 755–768. [Google Scholar] [CrossRef]
- Raptis, D.D.; Mantzoros, C.S.; Polyzos, S.A. Fibroblast Growth Factor-21 as a Potential Therapeutic Target of Nonalcoholic Fatty Liver Disease. Ther. Clin. Risk Manag. 2023, 19, 77–96. [Google Scholar] [CrossRef]
- Lee, H.J.; Shon, J.; Park, Y.J. Association of NAFLD with FGF21 Polygenic Hazard Score, and Its Interaction with Protein Intake Level in Korean Adults. Nutrients 2023, 15, 2385. [Google Scholar] [CrossRef]
- Pavlovic, V.; Ciric, M.; Petkovic, M.; Golubovic, M. Vitamin C and Epigenetics: A Short Physiological Overview. Open Med. 2023, 18, 20230688. [Google Scholar] [CrossRef] [PubMed]
- Holmer, M.; Hagström, H.; Chen, P.; Danielsson, O.; Aouadi, M.; Rydén, M.; Stål, P. Associations between Subcutaneous Adipocyte Hypertrophy and Nonalcoholic Fatty Liver Disease. Sci. Rep. 2022, 12, 20519. [Google Scholar] [CrossRef]
- Lim, J.; Heo, Y.; Choi, S.S. Investigation of Changes in DNA Methylation Associated with Alterations in Gene Expression Resulting in Differences between Lean and Obese Adipogenesis. Genomics 2023, 115, 110623. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Zhuo, Q.; Tseng, Y.; Wang, J.; Ma, Y.; Zhang, J.; Liu, J. TET1 Promotes Fatty Acid Oxidation and Inhibits NAFLD Progression by Hydroxymethylation of PPARα Promoter. Nutr. Metab. 2020, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Theys, C.; Lauwers, D.; Perez-Novo, C.; Berghe, W.V. PPARα in the Epigenetic Driver Seat of NAFLD: New Therapeutic Opportunities for Epigenetic Drugs? Biomedicines 2022, 10, 3041. [Google Scholar] [CrossRef]
- Vachher, M.; Bansal, S.; Kumar, B.; Yadav, S.; Burman, A. Deciphering the Role of Aberrant DNA Methylation in NAFLD and NASH. Heliyon 2022, 8, e11119. [Google Scholar] [CrossRef]
- Alswailmi, F.K.; Shah, S.I.A.; Nawaz, H.; Al-Mazaideh, G.M. Molecular Mechanisms of Vitamin D-Mediated Immunomodulation. Galen Méd. J. 2021, 10, 2097. [Google Scholar] [CrossRef]
- Nelson, J.E.; Roth, C.L.; Wilson, L.A.; Yates, K.P.; Aouizerat, B.; Morgan-Stevenson, V.; Whalen, E.; Hoofnagle, A.; Mason, M.; Gersuk, V.; et al. Vitamin D Deficiency Is Associated With Increased Risk of Non-Alcoholic Steatohepatitis in Adults With Non-Alcoholic Fatty Liver Disease: Possible Role for MAPK and NF-ΚB? Am. J. Gastroenterol. 2016, 111, 852–863. [Google Scholar] [CrossRef]
- Huang, X.; Yang, Y.; Jiang, Y.; Zhou, Z.; Zhang, J. Association between Vitamin D Deficiency and Lipid Profiles in Overweight and Obese Adults: A Systematic Review and Meta-Analysis. BMC Public Health 2023, 23, 1653. [Google Scholar] [CrossRef]
- Nimitphong, H.; Guo, W.; Holick, M.F.; Fried, S.K.; Lee, M. Vitamin D Inhibits Adipokine Production and Inflammatory Signaling Through the Vitamin D Receptor in Human Adipocytes. Obesity 2021, 29, 562–568. [Google Scholar] [CrossRef]
- Micu, E.S.; Amzolini, A.M.; Abu-Alhija, A.B.; Fortofoiu, M.C.; Vladu, I.M.; Clenciu, D.; Mitrea, A.; Mogoanta, S.S.; Crisan, A.E.; Predescu, O.I.; et al. Systemic and Adipose Tissue Inflammation in NASH: Correlations with Histopathological Aspects. Rom J. Morphol. Embryo 2021, 62, 509–515. [Google Scholar] [CrossRef]
- Martínez–Sánchez, C.; Bassegoda, O.; Deng, H.; Almodóvar, X.; Ibarzabal, A.; de Hollanda, A.; Torre, R.M.G.d.l.; Blaya, D.; Ariño, S.; Jiménez-Esquivel, N.; et al. Therapeutic Targeting of Adipose Tissue Macrophages Ameliorates Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. JHEP Rep. 2023, 5, 100830. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Tinahones, A.; Bekay, R.E.; Malagón, M.M.; Tinahones, F.J. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites 2020, 10, 179. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, R. Vitamin D3 Affects Browning of White Adipocytes by Regulating Autophagy via PI3K/Akt/MTOR/P53 Signaling in Vitro and in Vivo. Apoptosis 2022, 27, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Reda, D.; Elshopakey, G.E.; Albukhari, T.A.; Almehmadi, S.J.; Refaat, B.; Risha, E.F.; Mahgoub, H.A.; El-Boshy, M.E.; Abdelhamid, F.M. Vitamin D3 Alleviates Nonalcoholic Fatty Liver Disease in Rats by Inhibiting Hepatic Oxidative Stress and Inflammation via the SREBP-1-c/ PPARα-NF-ΚB/IR-S2 Signaling Pathway. Front. Pharmacol. 2023, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Zhou, Y.; Wang, W.; Scott, J.; Kim, K.; Sun, Z.; Guo, Q.; Lu, Y.; Gonzales, N.M.; Wu, H.; et al. Vitamin D Receptor Activation in Liver Macrophages Ameliorates Hepatic Inflammation, Steatosis, and Insulin Resistance in Mice. Hepatology 2020, 71, 1559–1574. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.A.; Ong, F.J.; Barra, N.G.; Blondin, D.P.; Gunn, E.; Oreskovich, S.M.; Szamosi, J.C.; Syed, S.A.; Hutchings, E.K.; Konyer, N.B.; et al. Lower Brown Adipose Tissue Activity Is Associated with Non-Alcoholic Fatty Liver Disease but Not Changes in the Gut Microbiota. Cell Rep. Med. 2021, 2, 100397. [Google Scholar] [CrossRef]
- Mukai, T.; Kusudo, T. Bidirectional Effect of Vitamin D on Brown Adipogenesis of C3H10T1/2 Fibroblast-like Cells. PeerJ 2023, 11, e14785. [Google Scholar] [CrossRef]
- Tourkochristou, E.; Mouzaki, A.; Triantos, C. Gene Polymorphisms and Biological Effects of Vitamin D Receptor on Nonalcoholic Fatty Liver Disease Development and Progression. Int. J. Mol. Sci. 2023, 24, 8288. [Google Scholar] [CrossRef]
- Gázquez, A.; Sánchez-Campillo, M.; Barranco, A.; Rueda, R.; Chan, J.P.; Kuchan, M.J.; Larqué, E. Calcifediol During Pregnancy Improves Maternal and Fetal Availability of Vitamin D Compared to Vitamin D3 in Rats and Modifies Fetal Metabolism. Front. Nutr. 2022, 9, 871632. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, Y.; Wang, Y.; Hong, H. Vitamin D/Vitamin D Receptor Pathway in Non-Alcoholic Fatty Liver Disease. Expert Opin. Ther. Targets 2023, 27, 1145–1157. [Google Scholar] [CrossRef]
- Tanoğlu, A.; Çağiltay, E.; Tanoğlu, E.G.; Gökhan, A.; Şirin, C.; Çavuşoğlu, T.; Yeşilbaş, R.S. Circulating Mir-200c and Mir-33a May Be Used as Biomarkers for Predicting High Fructose Corn Syrup-Induced Fatty Liver and Vitamin D Supplementation-Related Liver Changes. Turk. J. Med. Sci. 2022, 52, 1448–1457. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Carotti, S.; Bertoccini, L.; Baroni, M.G.; Vespasiani-Gentilucci, U.; Cavallo, M.-G.; Morini, S. Relationship between Adipose Tissue Dysfunction, Vitamin D Deficiency and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2017, 23, 3407–3417. [Google Scholar] [CrossRef]
- Karkeni, E.; Bonnet, L.; Marcotorchino, J.; Tourniaire, F.; Astier, J.; Ye, J.; Landrier, J.-F. Vitamin D Limits Inflammation-Linked MicroRNA Expression in Adipocytes in Vitro and in Vivo: A New Mechanism for the Regulation of Inflammation by Vitamin D. Epigenetics 2018, 13, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Karkeni, E.; Payet, T.; Astier, J.; Sicard, F.; Mounien, L.; Landrier, J.-F. Proposal of a Bioinformatics Approach to Predict Molecular Mechanisms Involved in Inflammatory Response: Case of ATRA and 1,25(OH)2D in Adipocytes. Epigenetics 2023, 18, 2201516. [Google Scholar] [CrossRef] [PubMed]
- Burz, S.D.; Monnoye, M.; Philippe, C.; Farin, W.; Ratziu, V.; Strozzi, F.; Paillarse, J.-M.; Chêne, L.; Blottière, H.M.; Gérard, P. Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms 2021, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Boughanem, H.; Ruiz-Limón, P.; Pilo, J.; Lisbona-Montañez, J.M.; Tinahones, F.J.; Indias, I.M.; Macías-González, M. Linking Serum Vitamin D Levels with Gut Microbiota after 1-Year Lifestyle Intervention with Mediterranean Diet in Patients with Obesity and Metabolic Syndrome: A Nested Cross-Sectional and Prospective Study. Gut Microbes 2023, 15, 2249150. [Google Scholar] [CrossRef]
- Sukik, A.; Alalwani, J.; Ganji, V. Vitamin D, Gut Microbiota, and Cardiometabolic Diseases—A Possible Three-Way Axis. Int. J. Mol. Sci. 2023, 24, 940. [Google Scholar] [CrossRef]
- Le, H.H.; Lee, M.-T.; Besler, K.R.; Johnson, E.L. Host Hepatic Metabolism Is Modulated by Gut Microbiota-Derived Sphingolipids. Cell Host Microbe 2022, 30, 798–808.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shang, X.; Jin, S.; Ma, Z.; Wang, H.; AO, N.; Yang, J.; Du, J. Vitamin D Ameliorates High-Fat-Diet-Induced Hepatic Injury via Inhibiting Pyroptosis and Alters Gut Microbiota in Rats. Arch. Biochem. Biophys. 2021, 705, 108894. [Google Scholar] [CrossRef]
- Du, T.; Xiang, L.; Zhang, J.; Yang, C.; Zhao, W.; Li, J.; Zhou, Y.; Ma, L. Vitamin D Improves Hepatic Steatosis in NAFLD via Regulation of Fatty Acid Uptake and β-Oxidation. Front. Endocrinol. 2023, 14, 1138078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Metabolism of Natural Forms of Vitamin E and Biological Actions of Vitamin E Metabolites. Free Radic. Biol. Med. 2022, 179, 375–387. [Google Scholar] [CrossRef]
- Liao, S.; Omage, S.O.; Börmel, L.; Kluge, S.; Schubert, M.; Wallert, M.; Lorkowski, S. Vitamin E and Metabolic Health: Relevance of Interactions with Other Micronutrients. Antioxidants 2022, 11, 1785. [Google Scholar] [CrossRef] [PubMed]
- Aksoz, E.; Korkut, O.; Aksit, D.; Gokbulut, C. Vitamin E (A-, β + Γ- and Δ-tocopherol) Levels in Plant Oils. Flavour Fragr. J. 2020, 35, 504–510. [Google Scholar] [CrossRef]
- Trugilho, L.; Alvarenga, L.; Cardozo, L.F.M.F.; Barboza, I.; Leite, M.; Fouque, D.; Mafra, D. Vitamin E and Conflicting Understandings in Noncommunicable Diseases: Is It Worth Supplementing? Clin. Nutr. ESPEN 2024, 59, 343–354. [Google Scholar] [CrossRef]
- Zingg, J. Vitamin E: Regulatory Role on Signal Transduction. Iubmb Life 2019, 71, 456–478. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Lipid Oxidation That Is, and Is Not, Inhibited by Vitamin E: Consideration about Physiological Functions of Vitamin E. Free Radic. Biol. Med. 2021, 176, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Head, B. Vitamin E: How Much Is Enough, Too Much and Why! Free. Radic. Biol. Med. 2021, 177, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.M.; Cansanção, K.; de Mello Perez, R.; Leite, N.C.; Padilha, P.; Ramalho, A.; Peres, W. Association between Serum and Dietary Antioxidant Micronutrients and Advanced Liver Fibrosis in Non-Alcoholic Fatty Liver Disease: An Observational Study. Peerj 2020, 8, e9838. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.A.M.; Abdelrahman, M.M.; Attia, H.M.A.-A.S.; Hafez, A.; Rashed, S.A.; Amin, Y.A.; Hemdan, S.B. Decreased Serum Zinc, Selenium, and Vitamin E as Possible Risk Factors of Hepatic Fibrosis in Non-Alcoholic Fatty Liver Disease. Nutr. Health 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Torquato, P.; Barola, C.; Russo, A.; Rychlicki, C.; Giusepponi, D.; Bellezza, G.; Sidoni, A.; Galarini, R.; Svegliati-Baroni, G.; et al. Nonalcoholic Fatty Liver Disease Impairs the Cytochrome P-450-Dependent Metabolism of α-Tocopherol (Vitamin E). J. Nutritional. Biochem. 2017, 47, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Violet, P.-C.; Ebenuwa, I.C.; Wang, Y.; Niyyati, M.; Padayatty, S.J.; Head, B.; Wilkins, K.; Chung, S.; Thakur, V.; Ulatowski, L.; et al. Vitamin E Sequestration by Liver Fat in Humans. JCI Insight 2019, 5, e133309. [Google Scholar] [CrossRef] [PubMed]
- Karedath, J.; Javed, H.; Talpur, F.A.; Lal, B.; Kumari, A.; Kivan, H.; Chunchu, V.A.; Hirani, S. Effect of Vitamin E on Clinical Outcomes in Patients With Non-Alcoholic Fatty Liver Disease: A Meta-Analysis. Cureus 2022, 14, e32764. [Google Scholar] [CrossRef]
- Ellatif, M.A.; El-Karib, A.O.; Dallak, M.; Eid, R.A.; Al-Ani, R.; Haidara, M.A. Vitamin E Protects Against Hepatocyte Ultrastructural Damage Induced by High Fat Diet in a Rat Model of Pre-Diabetes. Int. J. Morphol. 2018, 36, 1350–1355. [Google Scholar] [CrossRef]
- Podszun, M.C.; Alawad, A.S.; Lingala, S.; Morris, N.; Huang, W.-C.A.; Yang, S.; Schoenfeld, M.; Rolt, A.; Ouwerkerk, R.; Valdez, K.; et al. Vitamin E Treatment in NAFLD Patients Demonstrates That Oxidative Stress Drives Steatosis through Upregulation of De-Novo Lipogenesis. Redox Biol. 2020, 37, 101710. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liang, J.; Han, M.; Gao, Z. Polyphenols Synergistic Drugs to Ameliorate Non-Alcoholic Fatty Liver Disease via Signal Pathway and Gut Microbiota: A Review. J. Adv. Res. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Radu, F.; Potcovaru, C.-G.; Salmen, T.; Filip, P.V.; Pop, C.; Fierbințeanu-Braticievici, C. The Link between NAFLD and Metabolic Syndrome. Diagnostics 2023, 13, 614. [Google Scholar] [CrossRef] [PubMed]
- D’Espessailles, A.; Campos, V.; Juretić, N.; Tapia, G.S.; Pettinelli, P. Hepatic Retinaldehyde Dehydrogenases Are Modulated by Tocopherol Supplementation in Mice with Hepatic Steatosis. Nutrition 2022, 94, 111539. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial Oxidative Function in NAFLD: Friend or Foe? Mol. Metab. 2021, 50, 101134. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics That Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate Intake and Nonalcoholic Fatty Liver Disease: Fructose as a Weapon of Mass Destruction. Hepatobiliary Surg. Nutr. 2014, 4, 109–116. [Google Scholar] [CrossRef]
- Poolsri, W.; Phokrai, P.; Suwankulanan, S.; Phakdeeto, N.; Phunsomboon, P.; Pekthong, D.; Richert, L.; Pongcharoen, S.; Srisawang, P. Combination of Mitochondrial and Plasma Membrane Citrate Transporter Inhibitors Inhibits De Novo Lipogenesis Pathway and Triggers Apoptosis in Hepatocellular Carcinoma Cells. Biomed. Res. Int. 2018, 2018, 3683026. [Google Scholar] [CrossRef]
- Dai, L.; Mafra, D.; Shiels, P.G.; Hackeng, T.M.; Stenvinkel, P.; Schurgers, L.J. Vitamin K and Hallmarks of Ageing: Focus on Diet and Gut Microbiome. Nutrients 2023, 15, 2727. [Google Scholar] [CrossRef]
- Akbulut, A.C.; Pavlic, A.; Petsophonsakul, P.; Halder, M.; Maresz, K.; Kramann, R.; Schurgers, L. Vitamin K2 Needs an RDI Separate from Vitamin K1. Nutrients 2020, 12, 1852. [Google Scholar] [CrossRef] [PubMed]
- Mrštná, K.; Matoušová, K.; Krčmová, L.K.; Carazo, A.; Pourová, J.; Mladěnka, P.; Matysová, L.; Švec, F. Analysis of Vitamin K1 and Major K2 Variants in Rat/Human Serum and Lipoprotein Fractions by a Rapid, Simple, and Sensitive UHPLC-MS/MS Method. J. Chromatogr. A 2024, 1714, 464548. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Cui, W.; Cao, Q.; Gao, M.; Liu, H.; Su, G.; Gross, M.L.; Li, W. The Catalytic Mechanism of Vitamin K Epoxide Reduction in a Cellular Environment. J. Biol. Chem. 2021, 296, 100145. [Google Scholar] [CrossRef]
- Stępień, A.; Koziarska-Rościszewska, M.; Rysz, J.; Stępień, M. Biological Role of Vitamin K—With Particular Emphasis on Cardiovascular and Renal Aspects. Nutrients 2022, 14, 262. [Google Scholar] [CrossRef]
- dos Santos, E.A.; Giudici, K.V.; França, N.A.G.d.; Peters, B.S.E.; Fisberg, R.M.; Martini, L.A. Correlations among Vitamin K Intake, Body Fat, Lipid Profile and Glucose Homeostasis in Adults and the Elderly. Arch. Endocrinol. Metab. 2020, 64, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.-J.; Wen, Z.-M.; Huang, X.-M.; Feng, H.-J.; Lin, S.-S.; Liu, Y.-N.; Li, S.-C.; Zhang, Y.; Peng, W.-G.; Yang, J.-R.; et al. Better Performance of PIVKA-II for Detecting Hepatocellular Carcinoma in Patients with Chronic Liver Disease with Normal Total Bilirubin. World J. Gastroenterol. 2023, 29, 1359–1373. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Huang, J.; Li, H.; Gao, J. The Relationship between Vitamin K and Metabolic Dysfunction-Associated Fatty Liver Disease among the United States Population: National Health and Nutrition Examination Survey 2017–2018. Front. Nutr. 2023, 10, 1086477. [Google Scholar] [CrossRef]
- Bordoloi, J.; Ozah, D.; Bora, T.; Kalita, J.; Manna, P. Gamma-Glutamyl Carboxylated Gas6 Mediates the Beneficial Effect of Vitamin K on Lowering Hyperlipidemia via Regulating the AMPK/SREBP1/PPARα Signaling Cascade of Lipid Metabolism. J. Nutr. Biochem. 2019, 70, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Apostolo, D.; Ferreira, L.L.; Vincenzi, F.; Vercellino, N.; Minisini, R.; Latini, F.; Ferrari, B.; Burlone, M.E.; Pirisi, M.; Bellan, M. From MASH to HCC: The Role of Gas6/TAM Receptors. Front. Immunol. 2024, 15, 1332818. [Google Scholar] [CrossRef]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [CrossRef]
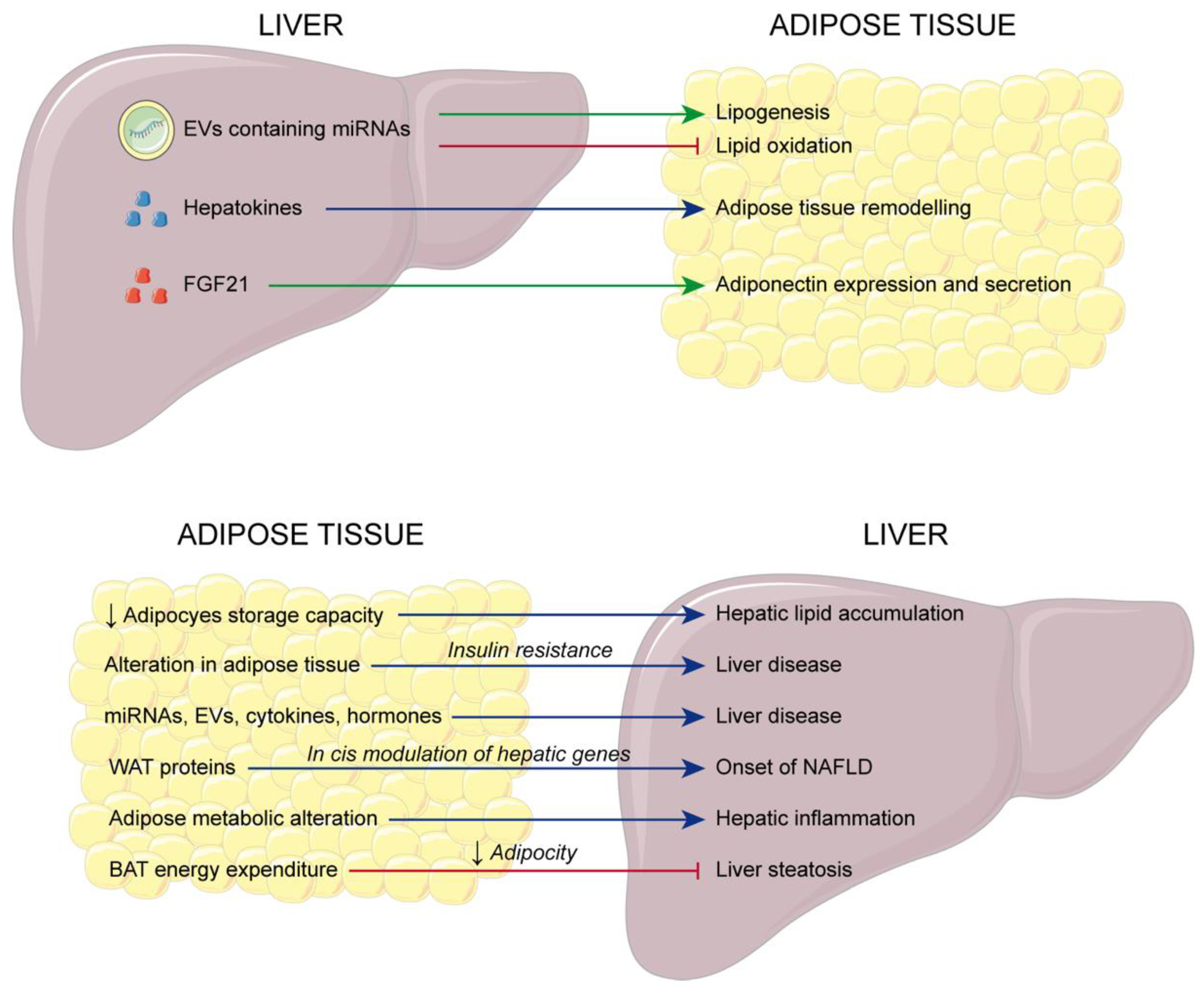
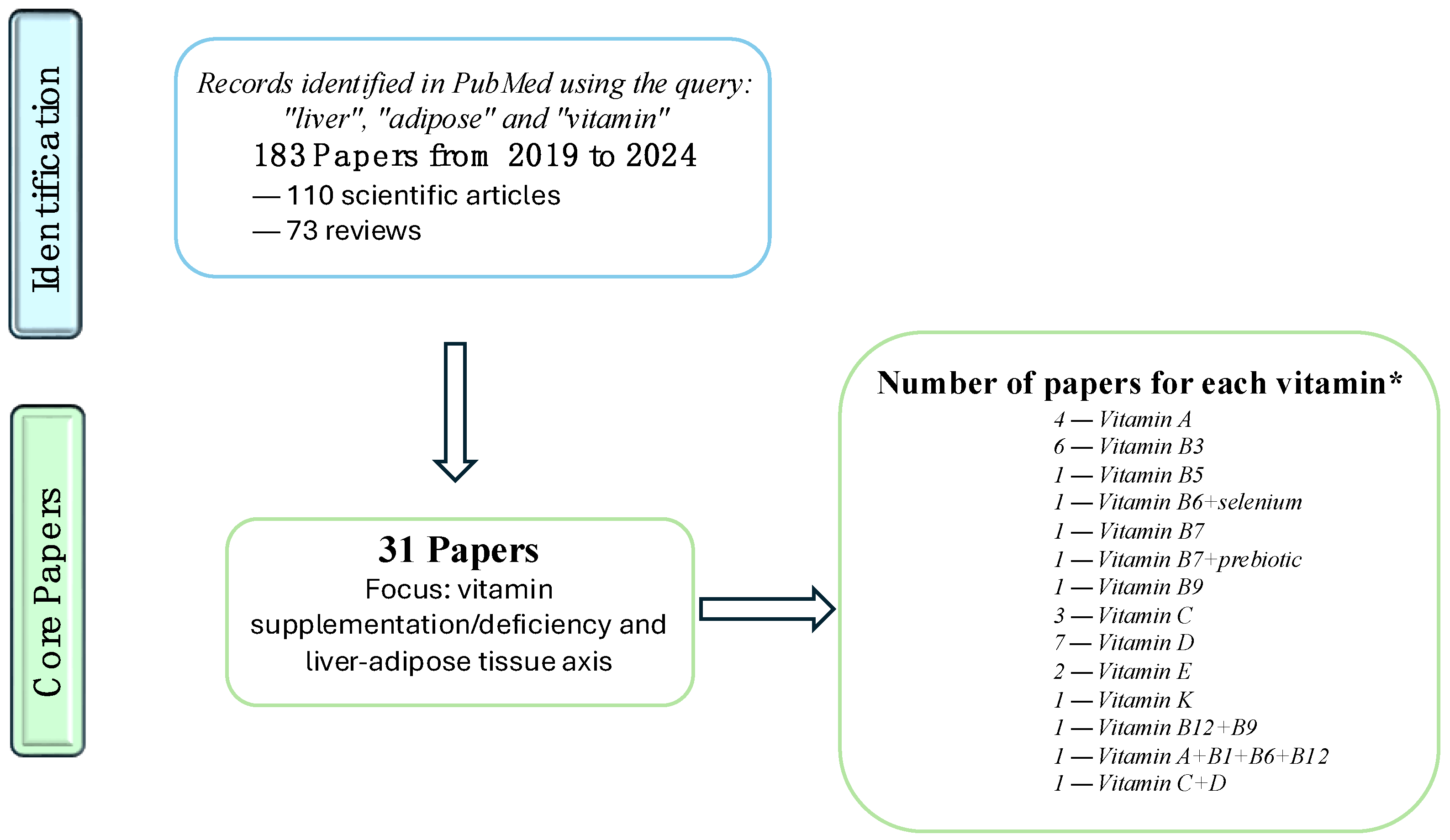
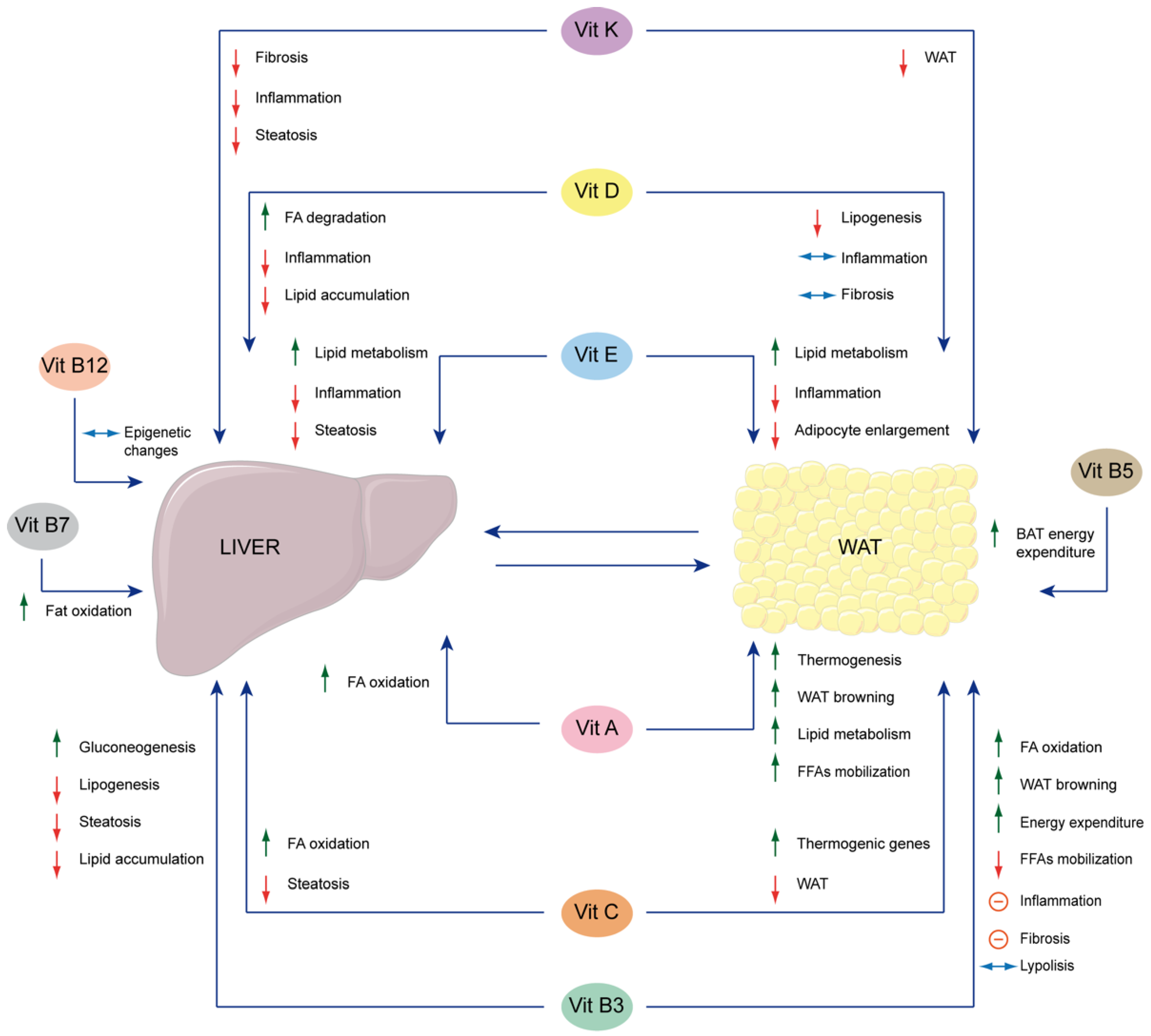
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tattoli, I.; Mathew, A.R.; Verrienti, A.; Pallotta, L.; Severi, C.; Andreola, F.; Cavallucci, V.; Giorgi, M.; Massimi, M.; Bencini, L.; et al. The Interplay between Liver and Adipose Tissue in the Onset of Liver Diseases: Exploring the Role of Vitamin Deficiency. Cells 2024, 13, 1631. https://doi.org/10.3390/cells13191631
Tattoli I, Mathew AR, Verrienti A, Pallotta L, Severi C, Andreola F, Cavallucci V, Giorgi M, Massimi M, Bencini L, et al. The Interplay between Liver and Adipose Tissue in the Onset of Liver Diseases: Exploring the Role of Vitamin Deficiency. Cells. 2024; 13(19):1631. https://doi.org/10.3390/cells13191631
Chicago/Turabian StyleTattoli, Ivan, Aimee Rachel Mathew, Antonella Verrienti, Lucia Pallotta, Carola Severi, Fausto Andreola, Virve Cavallucci, Mauro Giorgi, Mara Massimi, Lapo Bencini, and et al. 2024. "The Interplay between Liver and Adipose Tissue in the Onset of Liver Diseases: Exploring the Role of Vitamin Deficiency" Cells 13, no. 19: 1631. https://doi.org/10.3390/cells13191631