
Endothelial Myosin IIA Is Required for the Maintenance of Blood–Brain Barrier Integrity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Lines
2.3. FACS Sorting and Analysis of Brain ECs
2.4. Evans Blue Leakage
2.5. BBB Permeability Assay
2.6. Immunohistochemistry
2.7. Transmission Electron Microscopy (TEM)
2.8. Western Blot
2.9. Real-Time PCR (RT-PCR) Analysis
2.10. Seizure Model
2.11. Statistics
3. Results
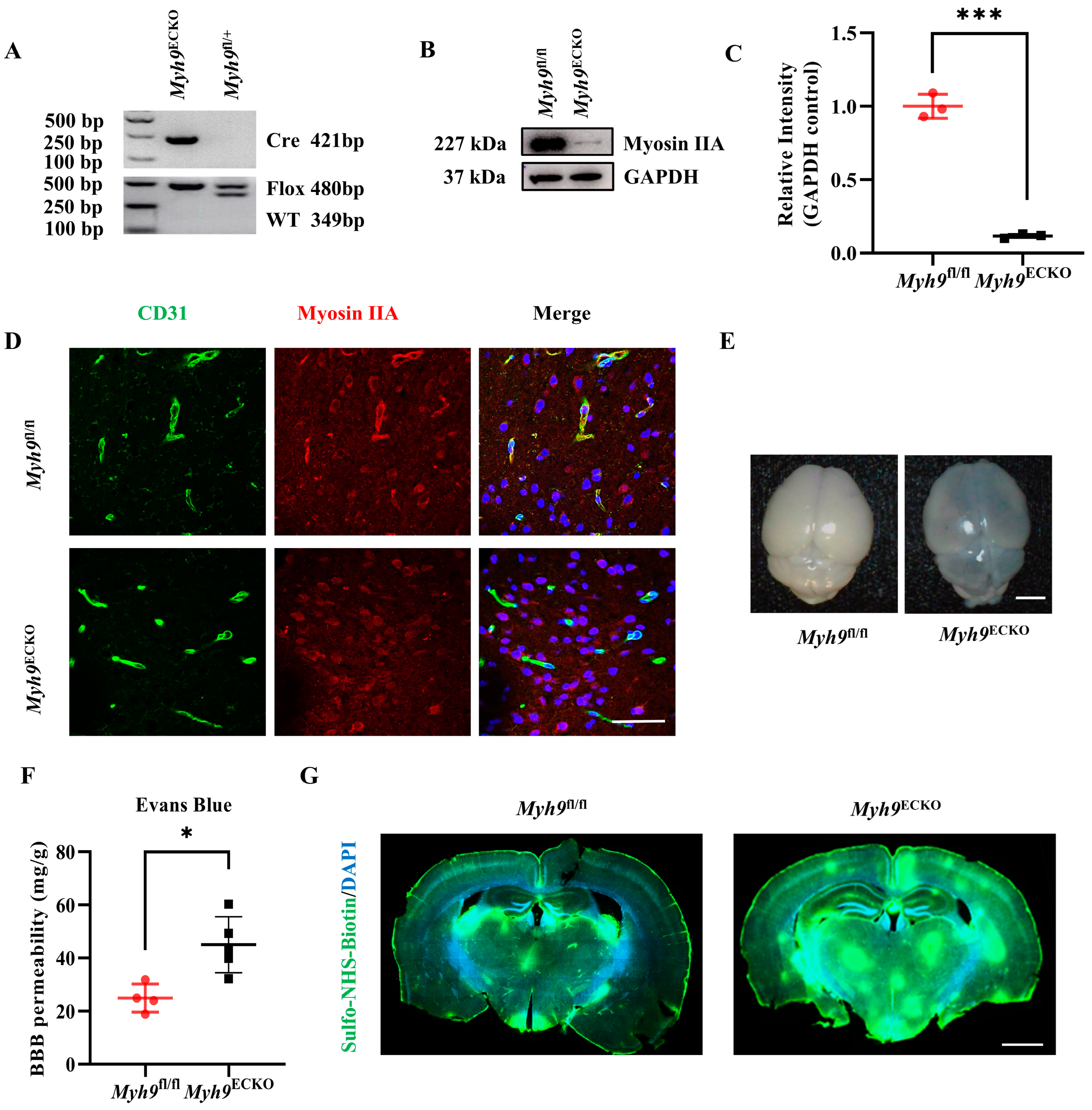
3.1. Cerebral Endothelial Deletion of Myosin IIA Impairs the Integrity of the BBB in Mice
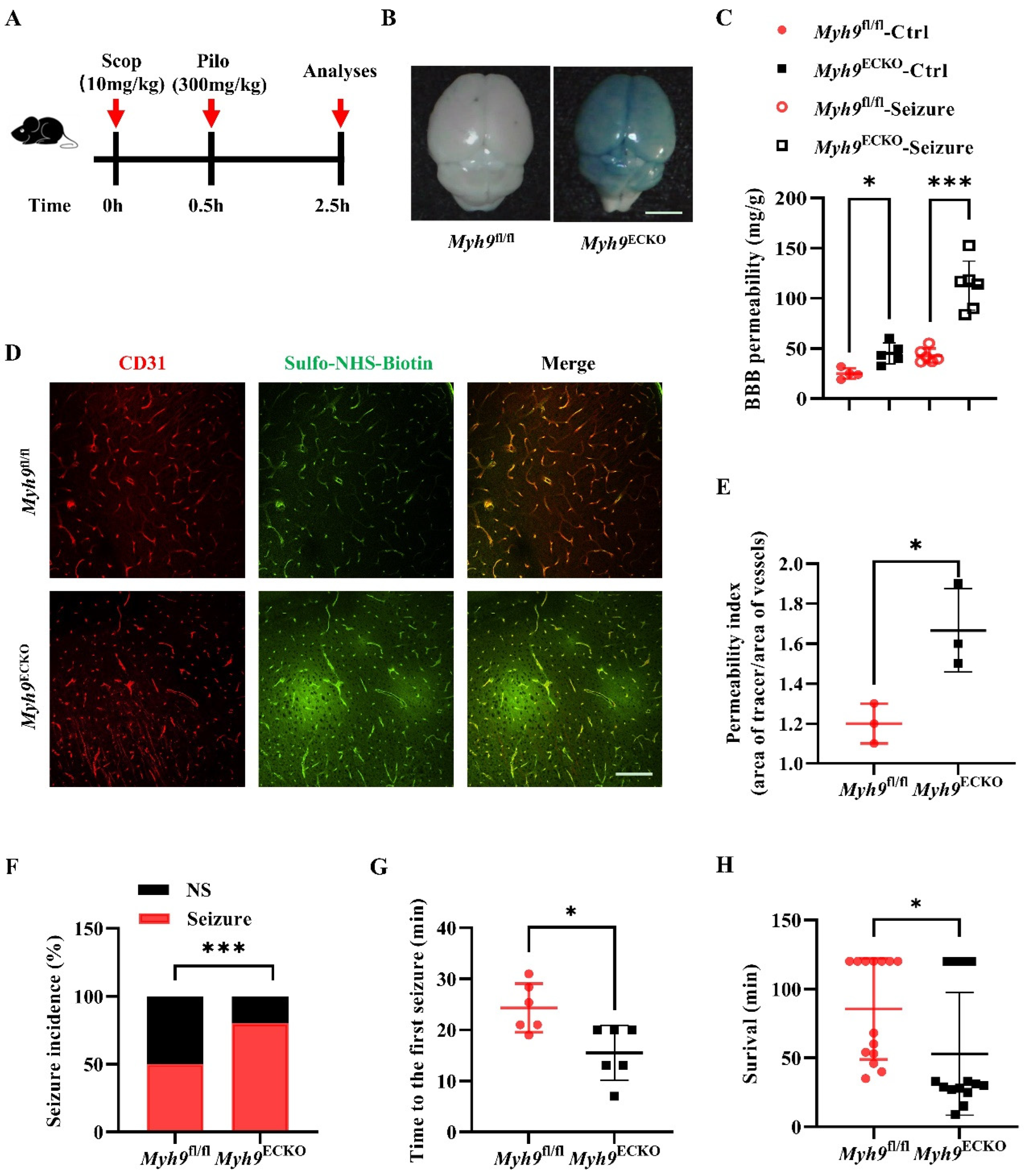
3.2. Cerebral Endothelial Deletion of Myosin IIA Increases Seizure Susceptibility and Seizure-Induced Mortality
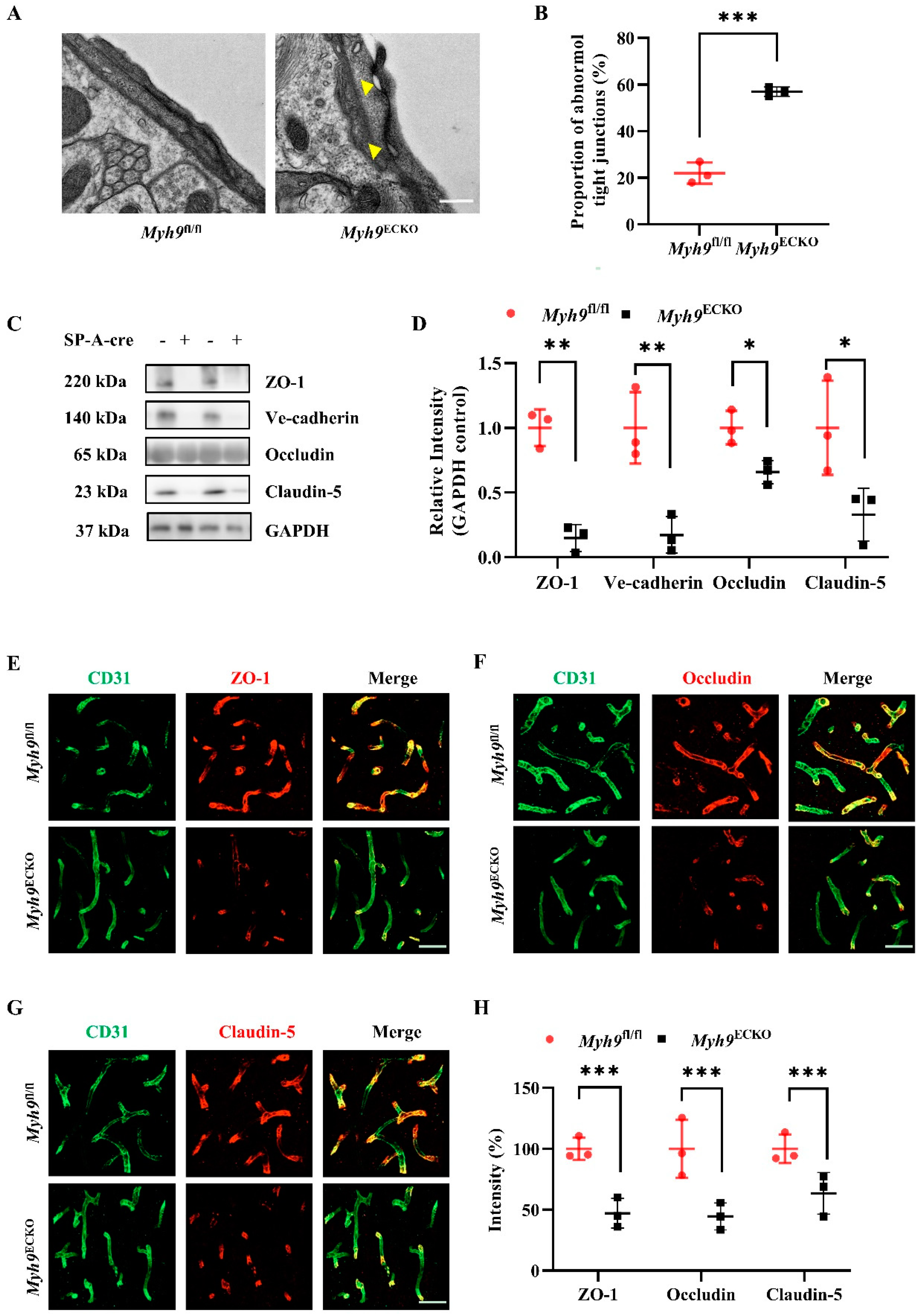
3.3. Cerebral Endothelial Deletion of Myosin IIA Disrupts Brain Endothelial Tight Junctions
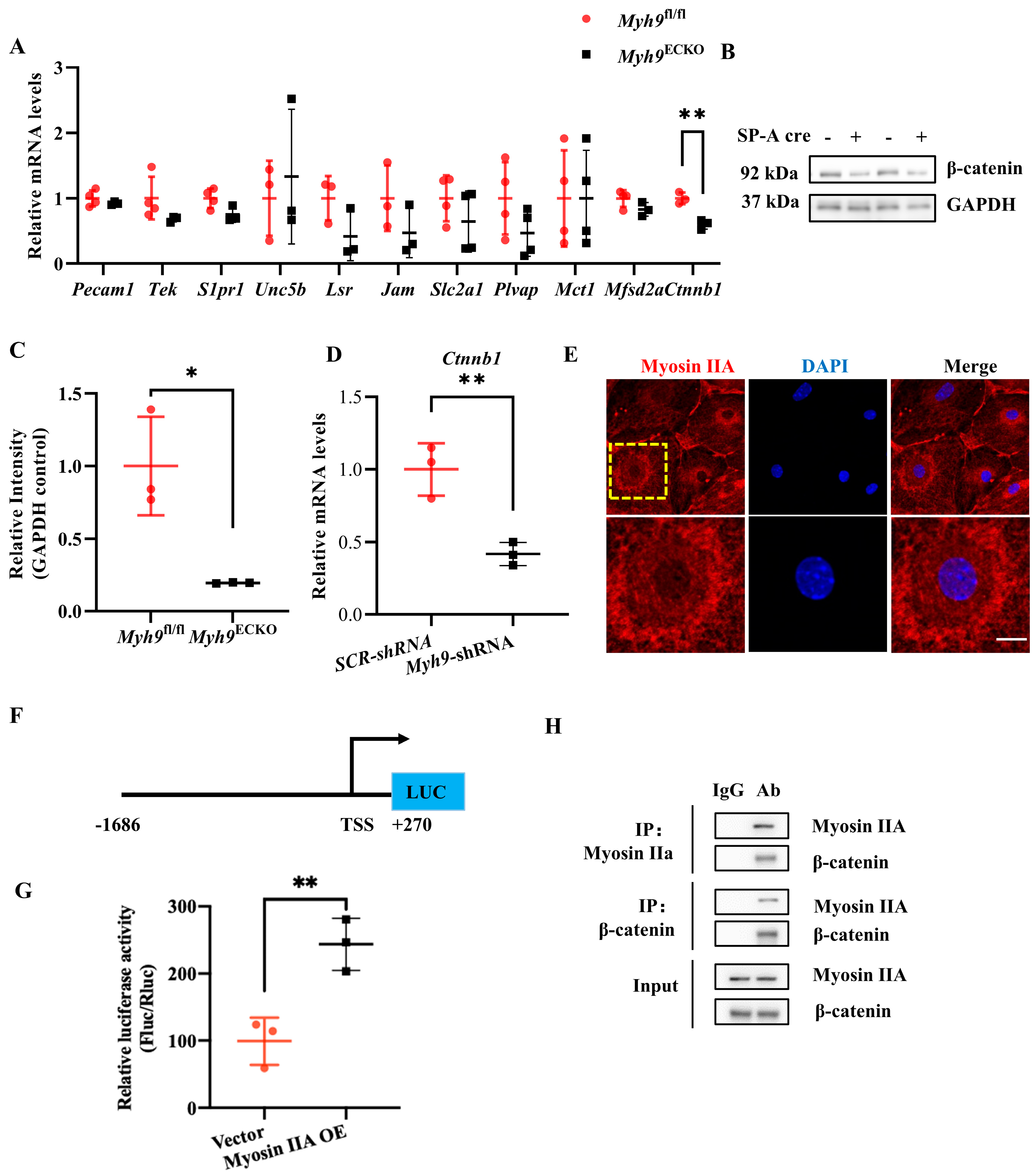
3.4. Myosin IIA Mediates the Transcription of the Ctnnb1 Gene and Interacts with Its Protein β-Catenin
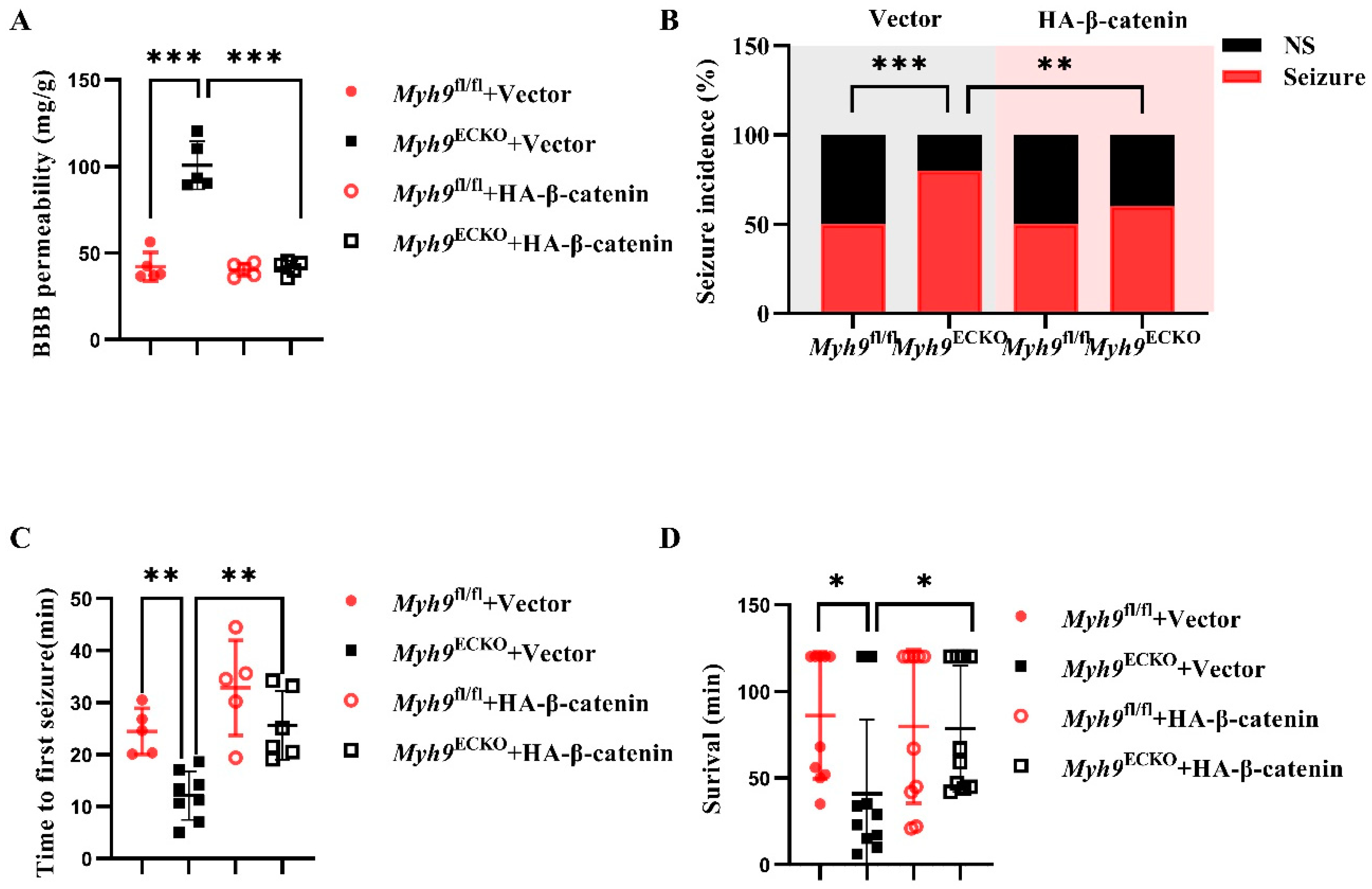
3.5. Overexpression of β-Catenin Ameliorates BBB Leakage in Myh9ECKO Mice
3.6. Overexpression of β-Catenin Ameliorates Epileptic Damage in Myh9ECKO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Gu, C. The Molecular Constituents of the Blood–Brain Barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow. Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouche, A.; Garcia-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef]
- Pathan, N.; Shende, P. Tailoring of P-glycoprotein for effective transportation of actives across blood-brain-barrier. J. Control Release 2021, 335, 398–407. [Google Scholar] [CrossRef]
- Nilles, K.L.; Williams, E.I.; Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Blood-Brain Barrier Transporters: Opportunities for Therapeutic Development in Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 1898. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.S.; Munji, R.N.; Chan, T.C.; Quirk, C.R.; Weiner, G.A.; Weger, B.D.; Rossi, M.J.; Elmsaouri, S.; Malfavon, M.; Deng, A.; et al. Neuronal Activity Regulates Blood-Brain Barrier Efflux Transport through Endothelial Circadian Genes. Neuron 2020, 108, 937–952.e937. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Boswell, C.A.; Jeet, S.; Beach, T.G.; Hoyte, K.; Luk, W.; Shihadeh, V.; Ulufatu, S.; Foreman, O.; Lu, Y.; et al. Lack of Widespread BBB Disruption in Alzheimer’s Disease Models: Focus on Therapeutic Antibodies. Neuron 2015, 88, 289–297. [Google Scholar] [CrossRef]
- Zhan, R.; Meng, X.; Tian, D.; Xu, J.; Cui, H.; Yang, J.; Xu, Y.; Shi, M.; Xue, J.; Yu, W.; et al. NAD(+) rescues aging-induced blood-brain barrier damage via the CX43-PARP1 axis. Neuron 2023, 111, 3634–3649.e7. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Marchi, N.; Angelov, L.; Masaryk, T.; Fazio, V.; Granata, T.; Hernandez, N.; Hallene, K.; Diglaw, T.; Franic, L.; Najm, I.; et al. Seizure-Promoting Effect of Blood–Brain Barrier Disruption. Epilepsia 2007, 48, 732–742. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef]
- van Vliet, E.A.; da Costa Araujo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534. [Google Scholar] [CrossRef]
- Martin, M.; Vermeiren, S.; Bostaille, N.; Eubelen, M.; Spitzer, D.; Vermeersch, M.; Profaci, C.P.; Pozuelo, E.; Toussay, X.; Raman-Nair, J.; et al. Engineered Wnt ligands enable blood-brain barrier repair in neurological disorders. Science 2022, 375, eabm4459. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell. Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, P.; Yang, Z.; Huang, X.; Wei, G.; Sun, Y.; Kang, X.; Hu, X.; Deng, Q.; Chen, L.; et al. Zyxin regulates endothelial von Willebrand factor secretion by reorganizing actin filaments around exocytic granules. Nat. Commun. 2017, 8, 14639. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Sun, Y.; Deng, Y.; Wei, G.; Liu, J.; Jin, S.; Dong, C.; Kang, X.; Huo, Y.; Zhang, J.; et al. Defective VWF secretion due to expression of MYH9-RD E1841K mutant in endothelial cells disrupts hemostasis. Blood Adv. 2022, 6, 4537–4552. [Google Scholar] [CrossRef]
- Li, P.; Wei, G.; Cao, Y.; Deng, Q.; Han, X.; Huang, X.; Huo, Y.; He, Y.; Chen, L.; Luo, J. Myosin IIa is critical for cAMP-mediated endothelial secretion of von Willebrand factor. Blood 2018, 131, 686–698. [Google Scholar] [CrossRef]
- Wang, A.; Ma, X.; Conti, M.A.; Liu, C.; Kawamoto, S.; Adelstein, R.S. Nonmuscle myosin II isoform and domain specificity during early mouse development. Proc. Natl. Acad. Sci. USA 2010, 107, 14645–14650. [Google Scholar] [CrossRef]
- Conti, M.A.; Even-Ram, S.; Liu, C.; Yamada, K.M.; Adelstein, R.S. Defects in Cell Adhesion and the Visceral Endoderm following Ablation of Nonmuscle Myosin Heavy Chain II-A in Mice. J. Biol. Chem. 2004, 279, 41263–41266. [Google Scholar] [CrossRef]
- Gong, S.; Cao, G.; Li, F.; Chen, Z.; Pan, X.; Ma, H.; Zhang, Y.; Yu, B.; Kou, J. Endothelial Conditional Knockdown of NMMHC IIA (Nonmuscle Myosin Heavy Chain IIA) Attenuates Blood-Brain Barrier Damage During Ischemia-Reperfusion Injury. Stroke 2021, 52, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Elegheert, J.; Behiels, E.; Bishop, B.; Scott, S.; Woolley, R.E.; Griffiths, S.C.; Byrne, E.F.X.; Chang, V.T.; Stuart, D.I.; Jones, E.Y.; et al. Lentiviral transduction of mammalian cells for fast, scalable and high-level production of soluble and membrane proteins. Nat. Protoc. 2018, 13, 2991–3017. [Google Scholar] [CrossRef]
- Petrovskaya, A.V.; Tverskoi, A.M.; Barykin, E.P.; Varshavskaya, K.B.; Dalina, A.A.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. Distinct Effects of Beta-Amyloid, Its Isomerized and Phosphorylated Forms on the Redox Status and Mitochondrial Functioning of the Blood-Brain Barrier Endothelium. Int. J. Mol. Sci. 2022, 24, 183. [Google Scholar] [CrossRef]
- Leow, D.M.; Cheah, I.K.; Fong, Z.W.; Halliwell, B.; Ong, W.Y. Protective Effect of Ergothioneine against 7-Ketocholesterol-Induced Mitochondrial Damage in hCMEC/D3 Human Brain Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 5498. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Reschke, C.R.; Reddy, A.; Mäe, M.A.; Connolly, R.; Behan, C.; O’Keeffe, E.; Bolger, I.; Hudson, N.; et al. Microvascular stabilization via blood-brain barrier regulation prevents seizure activity. Nat. Commun. 2022, 13, 2003. [Google Scholar] [CrossRef] [PubMed]
- Kalailingam, P.; Wang, K.Q.; Toh, X.R.; Nguyen, T.Q.; Chandrakanthan, M.; Hasan, Z.; Habib, C.; Schif, A.; Radio, F.C.; Dallapiccola, B.; et al. Deficiency of MFSD7c results in microcephaly-associated vasculopathy in Fowler syndrome. J. Clin. Investig. 2020, 130, 4081–4093. [Google Scholar] [CrossRef]
- Meng, F.; Shi, L.; Cheng, X.; Hou, N.; Wang, Y.; Teng, Y.; Meng, A.; Yang, X. Surfactant protein A promoter directs the expression of Cre recombinase in brain microvascular endothelial cells of transgenic mice. Matrix Biol. 2007, 26, 54–57. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, Y.; Song, X.; Ning, H.; Zhang, Y.; Teng, Y.; Wang, J.; Yang, X. Brain endothelial PTEN/AKT/NEDD4-2/MFSD2A axis regulates blood-brain barrier permeability. Cell Rep. 2021, 36, 109327. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lan, Y.; He, W.; Chen, D.; Wang, J.; Zhou, F.; Wang, Y.; Sun, H.; Chen, X.; Xu, C.; et al. Mouse embryonic head as a site for hematopoietic stem cell development. Cell Stem Cell 2012, 11, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lan, Y.; Wang, Y.; Wang, J.; Yang, G.; Meng, F.; Han, H.; Meng, A.; Wang, Y.; Yang, X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell 2011, 20, 291–302. [Google Scholar] [CrossRef]
- Wang, J.; Cui, Y.; Yu, Z.; Wang, W.; Cheng, X.; Ji, W.; Guo, S.; Zhou, Q.; Wu, N.; Chen, Y.; et al. Brain Endothelial Cells Maintain Lactate Homeostasis and Control Adult Hippocampal Neurogenesis. Cell Stem Cell 2019, 25, 754–767.e759. [Google Scholar] [CrossRef]
- Suidan, G.L.; Brill, A.; De Meyer, S.F.; Voorhees, J.R.; Cifuni, S.M.; Cabral, J.E.; Wagner, D.D. Endothelial Von Willebrand factor promotes blood-brain barrier flexibility and provides protection from hypoxia and seizures in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2112–2120. [Google Scholar] [CrossRef]
- Liu, X.X.; Yang, L.; Shao, L.X.; He, Y.; Wu, G.; Bao, Y.H.; Lu, N.N.; Gong, D.M.; Lu, Y.P.; Cui, T.T.; et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 2020, 217, e20180992. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Hoettels, B.A.; Meegan, J.E.; Wertz, T.S.; Cha, B.J.; Yang, X.; Oxford, J.T.; Wu, M.H.; Yuan, S.Y. AKT2 maintains brain endothelial claudin-5 expression and selective activation of IR/AKT2/FOXO1-signaling reverses barrier dysfunction. J. Cereb. Blood Flow. Metab. 2020, 40, 374–391. [Google Scholar] [CrossRef]
- Chen, T.; Dai, Y.; Hu, C.; Lin, Z.; Wang, S.; Yang, J.; Zeng, L.; Li, S.; Li, W. Cellular and molecular mechanisms of the blood-brain barrier dysfunction in neurodegenerative diseases. Fluids Barriers CNS 2024, 21, 60. [Google Scholar] [CrossRef]
- Wang, D.; Kranz-Eble, P.; De Vivo, D.C. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum. Mutat. 2000, 16, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Vazquez-Liebanas, E.; Nahar, K.; Bertuzzi, G.; Keller, A.; Betsholtz, C.; Mae, M.A. Adult-induced genetic ablation distinguishes PDGFB roles in blood-brain barrier maintenance and development. J. Cereb. Blood Flow. Metab. 2022, 42, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; Machado, R.F.; Göthert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.-Y. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood–Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation 2016, 133, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Fang, C.; Huang, X.; Feng, Z.; Yao, Y.; Wang, Y.; Chang, J. Endothelial β-Catenin Deficiency Causes Blood-Brain Barrier Breakdown via Enhancing the Paracellular and Transcellular Permeability. Front. Mol. Neurosci. 2022, 15, 895429. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Cui, Y.; Wang, Y.; Zhang, Y.; He, Q.; Yu, Z.; Xu, C.; Ning, H.; Han, Y.; Cai, Y.; et al. Genome Editing with AAV-BR1-CRISPR in Postnatal Mouse Brain Endothelial Cells. Int. J. Biol. Sci. 2022, 18, 652–660. [Google Scholar] [CrossRef]
- Ye, G.; Yang, Q.; Lei, X.; Zhu, X.; Li, F.; He, J.; Chen, H.; Ling, R.; Zhang, H.; Lin, T.; et al. Nuclear MYH9-induced CTNNB1 transcription, targeted by staurosporin, promotes gastric cancer cell anoikis resistance and metastasis. Theranostics 2020, 10, 7545–7560. [Google Scholar] [CrossRef]
- Ren, J.; Huang, Y.; Ren, Y.; Tu, T.; Qiu, B.; Ai, D.; Bi, Z.; Bai, X.; Li, F.; Li, J.L.; et al. Somatic variants of MAP3K3 are sufficient to cause cerebral and spinal cord cavernous malformations. Brain 2023, 146, 3634–3647. [Google Scholar] [CrossRef] [PubMed]
- Krolak, T.; Chan, K.Y.; Kaplan, L.; Huang, Q.; Wu, J.; Zheng, Q.; Kozareva, V.; Beddow, T.; Tobey, I.G.; Pacouret, S.; et al. A High-Efficiency AAV for Endothelial Cell Transduction Throughout the Central Nervous System. Nat. Cardiovasc. Res. 2022, 1, 389–400. [Google Scholar] [CrossRef]
- Ma, X.; Adelstein, R.S. The role of vertebrate nonmuscle Myosin II in development and human disease. Bioarchitecture 2014, 4, 88–102. [Google Scholar] [CrossRef]
- Chen, S.; Li, L.; Peng, C.; Bian, C.; Ocak, P.E.; Zhang, J.H.; Yang, Y.; Zhou, D.; Chen, G.; Luo, Y. Targeting Oxidative Stress and Inflammatory Response for Blood-Brain Barrier Protection in Intracerebral Hemorrhage. Antioxid. Redox Signal 2022, 37, 115–134. [Google Scholar] [CrossRef]
- Zolkiewski, M.; Redowicz, M.J.; Korn, E.D.; Ginsburg, A. Thermally induced unfolding of Acanthamoeba myosin II and skeletal muscle myosin: Nucleotide effects. Arch. Biochem. Biophys. 1995, 318, 207–214. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Milán, J.A.; Mulet, M.; Serra, A.; Gallart-Palau, X. Trioxidized cysteine and aging: A molecular binomial that extends far beyond classical proteinopathic paradigms. Aging 2024, 16, 11484–11490. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Milan, J.A.; Fernandez-Rhodes, M.; Guo, X.; Mulet, M.; Ngan, S.C.; Iyappan, R.; Katoueezadeh, M.; Sze, S.K.; Serra, A.; Gallart-Palau, X. Trioxidized cysteine in the aging proteome mimics the structural dynamics and interactome of phosphorylated serine. Aging Cell 2024, 23, e14062. [Google Scholar] [CrossRef]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef]
- Bejot, Y.; Barnay, J.L.; Chavent, A.; Daubail, B.; Jacquin, A.; Kazemi, A.; Ricolfi, F.; Giroud, M. Subarachnoid Hemorrhage Revealing Moyamoya Syndrome in a Patient With May-Hegglin Anomaly. Neurologist 2017, 22, 204–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Fann, C.S.; Liu, C.M.; Chen, W.J.; Wu, J.Y.; Hung, S.I.; Chen, C.H.; Jou, Y.S.; Liu, S.K.; Hwang, T.J.; et al. RASD2, MYH9, and CACNG2 genes at chromosome 22q12 associated with the subgroup of schizophrenia with non-deficit in sustained attention and executive function. Biol. Psychiatry. 2008, 64, 789–796. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Chen, J.; Luo, Y.; Hui, H.; Cai, T.; Huang, H.; Yang, F.; Feng, J.; Zhang, J.; Yan, X. CD146 coordinates brain endothelial cell-pericyte communication for blood-brain barrier development. Proc. Natl. Acad. Sci. USA 2017, 114, 7622–7631. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef]
- Boye, K.; Geraldo, L.H.; Furtado, J.; Pibouin-Fragner, L.; Poulet, M.; Kim, D.; Nelson, B.; Xu, Y.; Jacob, L.; Maissa, N.; et al. Endothelial Unc5B controls blood-brain barrier integrity. Nat. Commun. 2022, 13, 1169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Tischfield, M.; Williams, J.; Smallwood, P.M.; Rattner, A.; Taketo, M.M.; Nathans, J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Investig. 2014, 124, 3825–3846. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, G.; Davis, T.P. Physiology and pathophysiology of the blood-brain barrier: P-glycoprotein and occludin trafficking as therapeutic targets to optimize central nervous system drug delivery. J. Investig. Med. 2012, 60, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, G.; Willis, C.L.; Staatz, W.D.; Nametz, N.; Quigley, C.A.; Hom, S.; Lochhead, J.J.; Davis, T.P. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J. Neurochem. 2009, 110, 58–71. [Google Scholar] [CrossRef]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012, 26, 1875–1883. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Qiao, Z.; Zhou, C.; Pei, Y.; Xu, H.; Kang, X.; Luo, J. Endothelial Myosin IIA Is Required for the Maintenance of Blood–Brain Barrier Integrity. Cells 2024, 13, 1635. https://doi.org/10.3390/cells13191635
Deng Y, Qiao Z, Zhou C, Pei Y, Xu H, Kang X, Luo J. Endothelial Myosin IIA Is Required for the Maintenance of Blood–Brain Barrier Integrity. Cells. 2024; 13(19):1635. https://doi.org/10.3390/cells13191635
Chicago/Turabian StyleDeng, Yanan, Ziqi Qiao, Changping Zhou, Yujun Pei, Han Xu, Xuya Kang, and Jincai Luo. 2024. "Endothelial Myosin IIA Is Required for the Maintenance of Blood–Brain Barrier Integrity" Cells 13, no. 19: 1635. https://doi.org/10.3390/cells13191635
APA StyleDeng, Y., Qiao, Z., Zhou, C., Pei, Y., Xu, H., Kang, X., & Luo, J. (2024). Endothelial Myosin IIA Is Required for the Maintenance of Blood–Brain Barrier Integrity. Cells, 13(19), 1635. https://doi.org/10.3390/cells13191635