
Bile Acids-Based Therapies for Primary Sclerosing Cholangitis: Current Landscape and Future Developments

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Clinical Variants of PSC
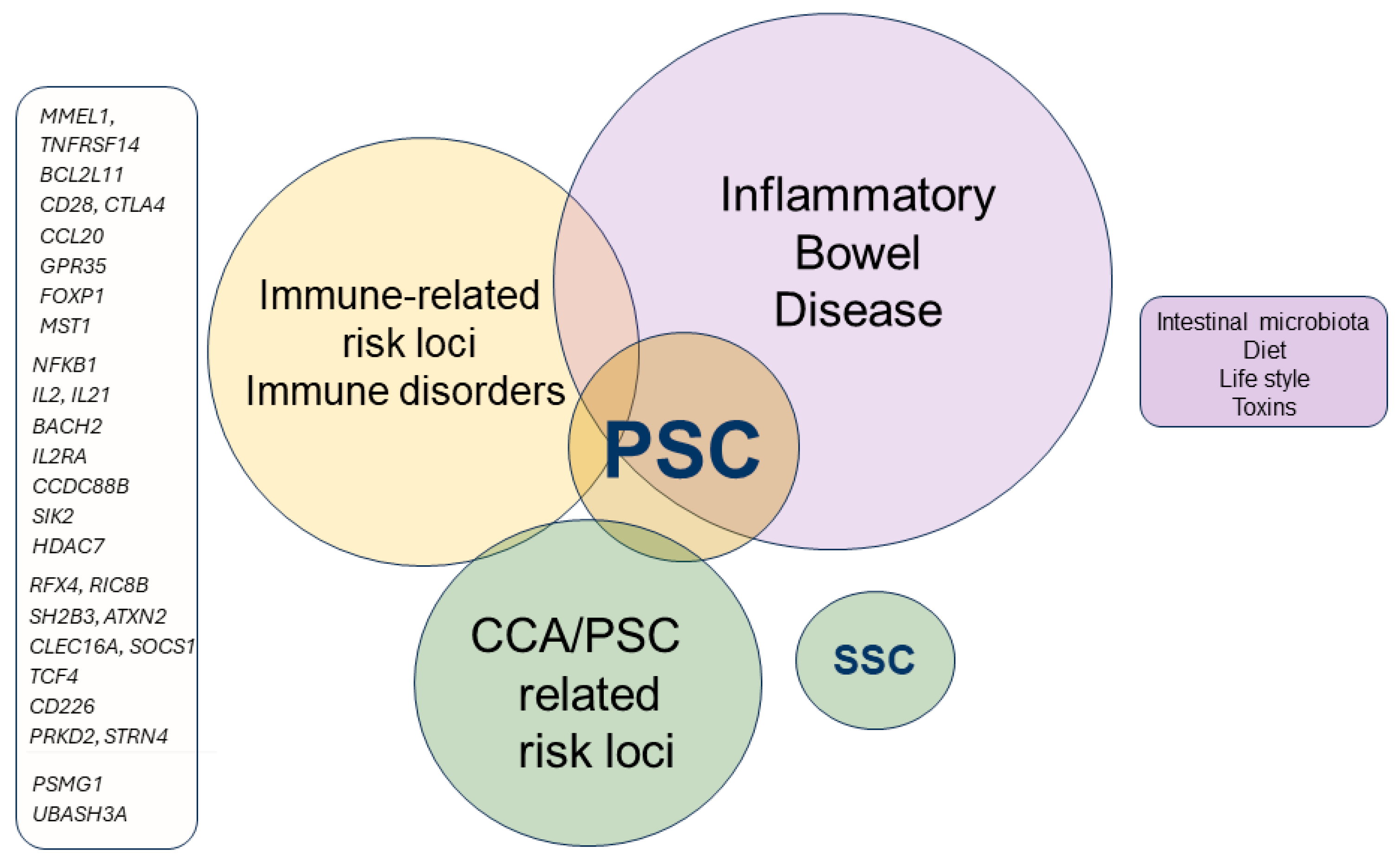
3. Genetic and Immunopathogenesis of PSC
4. Cholangiocytes Biology in PSC
4.1. Senescence in PSC
4.2. Biliary Fibrosis
5. Bile Acids Metabolism
5.1. Bile Acid-Regulated Receptors (BARRs)
5.1.1. Membrane Receptors
S1P2R
Leukemia Inhibitory Factor Receptor (LIFR)
5.1.2. Nuclear Receptors
5.2. Bile Acids in PSC
6. Intestinal Microbiota in PSC
7. Assessing Therapeutic Efficacy in PSC
7.1. Biochemical Biomarkers
7.2. Radiology
7.3. Staging of Liver Fibrosis
8. Treatment of PSC
8.1. UDCA in PSC
8.2. UDCA Derivatives
8.3. FXR Agonists in PSC
8.4. FGF-19
8.5. GPBAR1 in PSC
8.6. ASBT/IBAT Inhibitors
8.7. PPARs
8.8. Antifibrotic Agents
8.9. Immunosuppression
8.10. Microbiota-Based Therapies in PSC
8.11. Antibiotic Therapy
8.12. Fecal Microbiota Transplantation (FMT)
8.13. Treating Pruritus
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.M.; Wang, J.; Williamson, K.D.; Leeflang, M.M.; Boonstra, K.; Weersma, R.K.; Beuers, U.; Chapman, R.W.; Geskus, R.B.; Ponsioen, C.Y. A novel prognostic model for transplant-free survival in primary sclerosing cholangitis. Gut 2018, 67, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Bowlus, C.L.; Arrivé, L.; Bergquist, A.; Deneau, M.; Forman, L.; Ilyas, S.I.; Lunsford, K.E.; Martinez, M.; Sapisochin, G.; Shroff, R.; et al. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2023, 77, 659–702. [Google Scholar] [CrossRef]
- Vesterhus, M.; Hov, J.R.; Holm, A.; Schrumpf, E.; Nygård, S.; Godang, K.; Andersen, I.M.; Naess, S.; Thorburn, D.; Saffioti, F.; et al. Enhanced liver fibrosis score predicts transplant-free survival in primary sclerosing cholangitis. Hepatology 2015, 62, 188–197. [Google Scholar] [CrossRef]
- Williamson, K.D.; Chapman, R.W. Primary sclerosing cholangitis: A clinical update. Br. Med. Bull. 2015, 114, 53–64. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Scalera, I.; Slaney, E.; Laing, R.W.; Gunson, B.; Hirschfield, G.M.; Schlegel, A.; Ferguson, J.; Muiesan, P. Clinical outcomes of donation after circulatory death liver transplantation in primary sclerosing cholangitis. J. Hepatol. 2017, 67, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Weismüller, T.J.; Trivedi, P.J.; Bergquist, A.; Imam, M.; Lenzen, H.; Ponsioen, C.Y.; Holm, K.; Gotthardt, D.; Färkkilä, M.A.; Marschall, H.-U.; et al. Patient Age, Sex, and Inflammatory Bowel Disease Phenotype Associate with Course of Primary Sclerosing Cholangitis. Gastroenterology 2017, 152, 1975–1984.e8. [Google Scholar] [CrossRef]
- de Vries, E.; Bolier, R.; Goet, J.; Parés, A.; Verbeek, J.; de Vree, M.; Drenth, J.; van Erpecum, K.; van Nieuwkerk, K.; van der Heide, F.; et al. Fibrates for Itch (FITCH) in Fibrosing Cholangiopathies: A Double-Blind, Randomized, Placebo-Controlled Trial. Gastroenterology 2021, 160, 734–743.e6. [Google Scholar] [CrossRef] [PubMed]
- Prokopič, M.; Beuers, U. Management of primary sclerosing cholangitis and its complications: An algorithmic approach. Hepatol. Int. 2021, 15, 6–20. [Google Scholar] [CrossRef]
- Hov, J.-R.; Boberg, K.-M.; Karlsen, T.-H. Autoantibodies in primary sclerosing cholangitis. World J. Gastroenterol. 2008, 14, 3781–3791. [Google Scholar] [CrossRef]
- Ismail, S.; Kylänpää, L.; Mustonen, H.; Halttunen, J.; Lindström, O.; Jokelainen, K.; Udd, M.; Färkkilä, M. Risk factors for complications of ERCP in primary sclerosing cholangitis. Endoscopy 2012, 44, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Ponsioen, C.Y.; Assis, D.N.; Boberg, K.M.; Bowlus, C.L.; Deneau, M.; Thorburn, D.; Aabakken, L.; Färkkilä, M.; Petersen, B.; Rupp, C.; et al. Defining Primary Sclerosing Cholangitis: Results From an International Primary Sclerosing Cholangitis Study Group Consensus Process. Gastroenterology 2021, 161, 1764–1775.e5. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Bowlus, C.L. Primary sclerosing cholangitis: A review and update. Liver Res. 2017, 1, 221–230. [Google Scholar] [CrossRef]
- Eaton, J.E.; Talwalkar, J.A.; Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology 2013, 145, 521–536. [Google Scholar] [CrossRef]
- Rai, R.P.; Liu, Y.; Iyer, S.S.; Liu, S.; Gupta, B.; Desai, C.; Kumar, P.; Smith, T.; Singhi, A.D.; Nusrat, A.; et al. Blocking integrin α4β7-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J. Hepatol. 2020, 73, 1013–1022. [Google Scholar] [CrossRef]
- Gupta, B.; Rai, R.P.; Pal, P.B.; Rossmiller, D.; Chaudhary, S.; Chiaro, A.; Seaman, S.; Singhi, A.D.; Liu, S.; Monga, S.P.; et al. Selective Targeting of α4β7/MAdCAM-1 Axis Suppresses Fibrosis Progression by Reducing Proinflammatory T Cell Recruitment to the Liver. Cells 2024, 13, 756. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.K.; Karlsen, T.H.; Folseraas, T. Cholangiocytes in the pathogenesis of primary sclerosing cholangitis and development of cholangiocarcinoma. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.-G.; Juran, B.D.; Mucha, S.; Folseraas, T.; Jostins, L.; Melum, E.; Kumasaka, N.; Atkinson, E.J.; Schlicht, E.M.; Liu, J.Z.; et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat. Genet. 2017, 49, 269–273. [Google Scholar] [CrossRef]
- Folseraas, T.; Melum, E.; Rausch, P.; Juran, B.D.; Ellinghaus, E.; Shiryaev, A.; Laerdahl, J.K.; Ellinghaus, D.; Schramm, C.; Weismüller, T.J.; et al. Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. J. Hepatol. 2012, 57, 366–375. [Google Scholar] [CrossRef]
- Grimsrud, M.M.; Forster, M.; Goeppert, B.; Hemmrich-Stanisak, G.; Sax, I.; Grzyb, K.; Braadland, P.R.; Charbel, A.; Metzger, C.; Albrecht, T.; et al. Whole-exome sequencing reveals novel cancer genes and actionable targets in biliary tract cancers in primary sclerosing cholangitis. Hepatol. Commun. 2024, 8, e0461. [Google Scholar] [CrossRef]
- Melum, E.; Franke, A.; Schramm, C.; Weismüller, T.J.; Gotthardt, D.N.; Offner, F.A.; Juran, B.D.; Laerdahl, J.K.; Labi, V.; Björnsson, E.; et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat. Genet. 2011, 43, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Hov, J.R.; Folseraas, T.; Ellinghaus, E.; Rushbrook, S.M.; Doncheva, N.T.; Andreassen, O.A.; Weersma, R.K.; Weismüller, T.J.; Eksteen, B.; et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013, 45, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and structural features of cholangiocytes in health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Shin, K.; Fogg, V.C.; Margolis, B. Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol. 2006, 22, 207–235. [Google Scholar] [CrossRef] [PubMed]
- Bogert, P.T.; LaRusso, N.F. Cholangiocyte biology. Curr. Opin. Gastroenterol. 2007, 23, 299–305. [Google Scholar] [CrossRef]
- Pinto, C.; Giordano, D.M.; Maroni, L.; Marzioni, M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1270–1278. [Google Scholar] [CrossRef]
- Marzioni, M.; Glaser, S.S.; Francis, H.; Phinizy, J.L.; LeSage, G.; Alpini, G. Functional heterogeneity of cholangiocytes. Semin. Liver Dis. 2002, 22, 227–240. [Google Scholar] [CrossRef]
- Benedetti, A.; Bassotti, C.; Rapino, K.; Marucci, L.; Jezequel, A.M. A morphometric study of the epithelium lining the rat intrahepatic biliary tree. J. Hepatol. 1996, 24, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Francis, H.; Demorrow, S.; Lesage, G.; Fava, G.; Marzioni, M.; Venter, J.; Alpini, G. Heterogeneity of the intrahepatic biliary epithelium. World J. Gastroenterol. 2006, 12, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Masyuk, A.I.; Masyuk, T.V.; O’Hara, S.P.; LaRusso, N.F. Physiology of cholangiocytes. Compr. Physiol. 2013, 3, 541–565. [Google Scholar] [CrossRef] [PubMed]
- Raven, A.; Lu, W.-Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef]
- Ogawa, M.; Jiang, J.-X.; Xia, S.; Yang, D.; Ding, A.; Laselva, O.; Hernandez, M.; Cui, C.; Higuchi, Y.; Suemizu, H.; et al. Generation of functional ciliated cholangiocytes from human pluripotent stem cells. Nat. Commun. 2021, 12, 6504. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Masyuk, T.V.; Splinter, P.L.; Huang, B.Q.; Stroope, A.J.; LaRusso, N.F. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology 2006, 131, 911–920. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Masyuk, T.V.; LaRusso, N.F. Cholangiocyte primary cilia in liver health and disease. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 2007–2012. [Google Scholar] [CrossRef]
- Wiegreffe, S.; Löhrer, D.; Wirtz, M.; Wiemuth, D. The bile acid-sensitive ion channel (BASIC) mediates bile acid-dependent currents in bile duct epithelial cells. Pflügers Arch.-Eur. J. Physiol. 2021, 473, 1841–1850. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004, 127, 1565–1577. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. The Cholangiopathies. Mayo Clin. Proc. 2015, 90, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Pham, L.; Glaser, S.; Francis, H.; Alpini, G. Pathophysiological Roles of Ductular Reaction in Liver Inflammation and Hepatic Fibrogenesis. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 803–805. [Google Scholar] [CrossRef] [PubMed]
- Syal, G.; Fausther, M.; Dranoff, J.A. Advances in cholangiocyte immunobiology. Am. J. Physiol. Liver Physiol. 2012, 303, G1077–G1086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhong, X.; Shen, H.; Sheng, L.; Liangpunsakul, S.; Lok, A.S.; Omary, M.B.; Wang, S.; Rui, L. Biliary NIK promotes ductular reaction and liver injury and fibrosis in mice. Nat. Commun. 2022, 13, 5111. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bravo, B.; Rodrigo-Torres, D.; Ariño, S.; Coll, M.; Pose, E.; Blaya, D.; Graupera, I.; Perea, L.; Vallverdú, J.; Rubio-Tomás, T.; et al. Ductular Reaction Cells Display an Inflammatory Profile and Recruit Neutrophils in Alcoholic Hepatitis. Hepatology 2019, 69, 2180–2195. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef]
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Trussoni, C.E.; O’Hara, S.P.; Splinter, P.L.; Heimbach, J.K.; LaRusso, N.F. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab. Investig. 2014, 94, 1126–1133. [Google Scholar] [CrossRef]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; LaRusso, N.F. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar] [CrossRef]
- Trussoni, C.E.; O’Hara, S.P.; LaRusso, N.F. Cellular senescence in the cholangiopathies: A driver of immunopathology and a novel therapeutic target. Semin. Immunopathol. 2022, 44, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ikeda, H.; Yamaguchi, J.; Nakada, S.; Nakanuma, Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology 2008, 48, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Jalan-Sakrikar, N.; Anwar, A.; Yaqoob, U.; Gan, C.; Lagnado, A.B.; Wixom, A.Q.; Jurk, D.; Huebert, R.C. Telomere dysfunction promotes cholangiocyte senescence and biliary fibrosis in primary sclerosing cholangitis. JCI Insight 2023, 8, e170320. [Google Scholar] [CrossRef]
- Murillo Perez, C.F.; Fisher, H.; Hiu, S.; Kareithi, D.; Adekunle, F.; Mayne, T.; Malecha, E.; Ness, E.; van der Meer, A.J.; Lammers, W.J.; et al. Greater Transplant-Free Survival in Patients Receiving Obeticholic Acid for Primary Biliary Cholangitis in a Clinical Trial Setting Compared to Real-World External Controls. Gastroenterology 2022, 163, 1630–1642.e3. [Google Scholar] [CrossRef]
- Friedman, S.L.; Pinzani, M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology 2022, 75, 473–488. [Google Scholar] [CrossRef]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sévigny, J.; Wells, R.G. Transforming growth factor-β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef]
- Kunzmann, L.K.; Schoknecht, T.; Poch, T.; Henze, L.; Stein, S.; Kriz, M.; Grewe, I.; Preti, M.; Hartl, J.; Pannicke, N.; et al. Monocytes as Potential Mediators of Pathogen-Induced T-Helper 17 Differentiation in Patients with Primary Sclerosing Cholangitis (PSC). Hepatology 2020, 72, 1310–1326. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.T. Disorders of bile acid synthesis. J. Inherit. Metab. Dis. 2011, 34, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.H.; Walker, M.E.; Stewart, A.K.; O’Flaherty, S.; Gentry, E.C.; Patel, S.; Beaty, V.V.; Allen, G.; Pan, M.; Simpson, J.B.; et al. Bile salt hydrolases shape the bile acid landscape and restrict Clostridioides difficile growth in the murine gut. Nat. Microbiol. 2023, 8, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Rimal, B.; Collins, S.L.; Tanes, C.E.; Rocha, E.R.; Granda, M.A.; Solanki, S.; Hoque, N.J.; Gentry, E.C.; Koo, I.; Reilly, E.R.; et al. Bile salt hydrolase catalyses formation of amine-conjugated bile acids. Nature 2024, 626, 859–863. [Google Scholar] [CrossRef]
- Fu, T.; Huan, T.; Rahman, G.; Zhi, H.; Xu, Z.; Oh, T.G.; Guo, J.; Coulter, S.; Tripathi, A.; Martino, C.; et al. Paired microbiome and metabolome analyses associate bile acid changes with colorectal cancer progression. Cell Rep. 2023, 42, 112997. [Google Scholar] [CrossRef]
- Mohanty, I.; Mannochio-Russo, H.; Schweer, J.V.; El Abiead, Y.; Bittremieux, W.; Xing, S.; Schmid, R.; Zuffa, S.; Vasquez, F.; Muti, V.B.; et al. The underappreciated diversity of bile acid modifications. Cell 2024, 187, 1801–1818.e20. [Google Scholar] [CrossRef]
- Fiorucci, S.; Marchianò, S.; Urbani, G.; Di Giorgio, C.; Distrutti, E.; Zampella, A.; Biagioli, M. Immunology of bile acids regulated receptors. Prog. Lipid Res. 2024, 95, 101291. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Reich, M.; Spomer, L.; Klindt, C.; Fuchs, K.; Stindt, J.; Deutschmann, K.; Höhne, J.; Liaskou, E.; Hov, J.R.; Karlsen, T.H.; et al. Downregulation of TGR5 (GPBAR1) in biliary epithelial cells contributes to the pathogenesis of sclerosing cholangitis. J. Hepatol. 2021, 75, 634–646. [Google Scholar] [CrossRef]
- Deutschmann, K.; Reich, M.; Klindt, C.; Dröge, C.; Spomer, L.; Häussinger, D.; Keitel, V. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; Renga, B.; D’Amore, C.; Sepe, V.; Finamore, C.; De Marino, S.; Carino, A.; Cipriani, S.; Monti, M.C.C.; Zampella, A.; et al. Exploitation of cholane scaffold for the discovery of potent and selective farnesoid X receptor (FXR) and G-protein coupled bile acid receptor 1 (GP-BAR1) ligands. J. Med. Chem. 2014, 57, 8477–8495. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Renga, B.; Festa, C.; D’Amore, C.; Masullo, D.; Cipriani, S.; Di Leva, F.S.S.; Monti, M.C.C.; Novellino, E.; Limongelli, V.; et al. Modification on ursodeoxycholic acid (UDCA) scaffold. discovery of bile acid derivatives as selective agonists of cell-surface G-protein coupled bile acid receptor 1 (GP-BAR1). J. Med. Chem. 2014, 57, 7687–7701. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Renga, B.; D’Amore, C.; Simonetti, M.; De Tursi, A.A.A.; Carino, A.; Monti, M.C.C.; Sepe, V.; Zampella, A.; Fiorucci, S. Impaired Itching Perception in Murine Models of Cholestasis Is Supported by Dysregulation of GPBAR1 Signaling. PLoS ONE 2015, 10, e0129866. [Google Scholar] [CrossRef] [PubMed]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Bellini, R.; Lupia, A.; Massa, C.; Bordoni, M.; Marchianò, S.; Rosselli, R.; Sepe, V.; Rapacciuolo, P.; Moraca, F.; et al. Discovery of BAR502, as potent steroidal antagonist of leukemia inhibitory factor receptor for the treatment of pancreatic adenocarcinoma. Front. Oncol. 2023, 13, 1140730. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Morretta, E.; Lupia, A.; Bellini, R.; Massa, C.; Urbani, G.; Bordoni, M.; Marchianò, S.; Lachi, G.; Rapacciuolo, P.; et al. Bile acids serve as endogenous antagonists of the Leukemia inhibitory factor (LIF) receptor in oncogenesis. Biochem. Pharmacol. 2024, 223, 116134. [Google Scholar] [CrossRef]
- Xu, G.; Song, M. Recent advances in the mechanisms underlying the beneficial effects of bariatric and metabolic surgery. Surg. Obes. Relat. Dis. 2020, 17, 231–238. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pruzanski, M.; Morelli, A.; Pellicciari, R. A farnesoid x receptor-small heterodimer partner regulatory cascade modulates tissue metalloproteinase inhibitor-1 and matrix metalloprotease expression in hepatic stellate cells and promotes resolution of liver fibrosis. J. Pharmacol. Exp. Ther. 2005, 314, 584–595. [Google Scholar] [CrossRef]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Cariello, M.; Piglionica, M.; Gadaleta, R.M.; Moschetta, A. The Enterokine Fibroblast Growth Factor 15/19 in Bile Acid Metabolism. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2019; Volume 256, pp. 73–93. [Google Scholar] [CrossRef]
- Boyer, J.L.; Trauner, M.; Mennone, A.; Soroka, C.J.; Cai, S.Y.; Moustafa, T.; Zollner, G.; Lee, J.Y.; Ballatori, N. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1124–G1130. [Google Scholar] [CrossRef]
- Song, K.H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009, 49, 297–305. [Google Scholar] [CrossRef]
- Yang, H.; Liu, T.; Wang, J.; Li, T.W.; Fan, W.; Peng, H.; Krishnan, A.; Gores, G.J.; Mato, J.M.; Lu, S.C. Deregulated methionine adenosyltransferase α1, c-Myc, and Maf proteins together promote cholangiocarcinoma growth in mice and humans. Hepatology 2016, 64, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Milkiewicz, M.; Klak, M.; Kempinska-Podhorodecka, A.; Wiechowska-Kozlowska, A.; Urasinska, E.; Blatkiewicz, M.; Wunsch, E.; Elias, E.; Milkiewicz, P. Impaired Hepatic Adaptation to Chronic Cholestasis induced by Primary Sclerosing Cholangitis. Sci. Rep. 2016, 6, 39573. [Google Scholar] [CrossRef]
- Renga, B.; Migliorati, M.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. Farnesoid X receptor suppresses constitutive androstane receptor activity at the multidrug resistance protein-4 promoter. Biochim. Biophys. Acta 2011, 1809, 157–165. [Google Scholar] [CrossRef]
- Stedman, C.; Liddle, C.; Coulter, S.; Sonoda, J.; Alvarez, J.G.; Evans, R.M.; Downes, M. Benefit of farnesoid X receptor inhibition in obstructive cholestasis. Proc. Natl. Acad. Sci. USA 2006, 103, 11323–11328. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; D’Auria, M.V.V.; Sepe, V.; Chini, M.G.G.; Monti, M.C.C.; Bifulco, G.; Zampella, A.; et al. Discovery that theonellasterol a marine sponge sterol is a highly selective FXR antagonist that protects against liver injury in cholestasis. PLoS ONE 2012, 7, e30443. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Wu, X.; Ge, H.; Lemon, B.; Weiszmann, J.; Gupte, J.; Hawkins, N.; Li, X.; Tang, J.; Lindberg, R.; Li, Y. Selective activation of FGFR4 by an FGF19 variant does not improve glucose metabolism in ob/ob mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14379–14384. [Google Scholar] [CrossRef] [PubMed]
- Al-Dury, S.; Wahlström, A.; Panzitt, K.; Thorell, A.; Ståhlman, M.; Trauner, M.; Fickert, P.; Bäckhed, F.; Fändriks, L.; Wagner, M.; et al. Obeticholic acid may increase the risk of gallstone formation in susceptible patients. J. Hepatol. 2019, 71, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed]
- Chini, M.G.G.M.G.; Jones, C.R.R.C.R.; Zampella, A.; D’Auria, M.V.M.V.V.; Renga, B.; Fiorucci, S.; Butts, C.P.P.C.P.; Bifulco, G. Quantitative NMR-derived interproton distances combined with quantum mechanical calculations of 13C chemical shifts in the stereochemical determination of conicasterol F, a nuclear receptor ligand from Theonella swinhoei. J. Org. Chem. 2012, 77, 1489–1496. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Mencarelli, A.; Migliorati, M.; Barbanti, M.; Cipriani, S.; Palladino, G.; Distrutti, E.; Renga, B.; Fiorucci, S. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem. Pharmacol. 2010, 80, 1700–1707. [Google Scholar] [CrossRef]
- Yoshihara, A.; Kawasaki, H.; Masuno, H.; Takada, K.; Numoto, N.; Ito, N.; Hirata, N.; Kanda, Y.; Ishizawa, M.; Makishima, M.; et al. Lithocholic Acid Amides as Potent Vitamin D Receptor Agonists. Biomolecules 2022, 12, 130. [Google Scholar] [CrossRef]
- Biagioli, M.; Marchianò, S.; Carino, A.; Di Giorgio, C.; Santucci, L.; Distrutti, E.; Fiorucci, S. Bile Acids Activated Receptors in Inflammatory Bowel Disease. Cells 2021, 10, 1281. [Google Scholar] [CrossRef]
- Solt, L.A.; Kumar, N.; Nuhant, P.; Wang, Y.; Lauer, J.L.; Liu, J.; Istrate, M.A.; Kamenecka, T.M.; Roush, W.R.; Vidović, D.; et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011, 472, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, I.; Allaband, C.; Mannochio-Russo, H.; El Abiead, Y.; Hagey, L.R.; Knight, R.; Dorrestein, P.C. The changing metabolic landscape of bile acids—Keys to metabolism and immune regulation. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 493–516. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Withers, D.R.; Hepworth, M.R.; Wang, X.; Mackley, E.C.; Halford, E.E.; Dutton, E.E.; Marriott, C.L.; Brucklacher-Waldert, V.; Veldhoen, M.; Kelsen, J.; et al. Transient inhibition of ROR-γt therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat. Med. 2016, 22, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Kanai, T.; Sujino, T.; Nemoto, Y.; Kanai, Y.; Mikami, Y.; Hayashi, A.; Matsumoto, A.; Takaishi, H.; Ogata, H.; et al. T-helper 17 and interleukin-17-producing lymphoid tissue inducer-like cells make different contributions to colitis in mice. Gastroenterology 2012, 143, 1288–1297. [Google Scholar] [CrossRef]
- Braadland, P.R.; Schneider, K.M.; Bergquist, A.; Molinaro, A.; Lövgren-Sandblom, A.; Henricsson, M.; Karlsen, T.H.; Vesterhus, M.; Trautwein, C.; Hov, J.R.; et al. Suppression of bile acid synthesis as a tipping point in the disease course of primary sclerosing cholangitis. JHEP Rep. Innov. Hepatol. 2022, 4, 100561. [Google Scholar] [CrossRef]
- Özdirik, B.; Schnabl, B. Microbial Players in Primary Sclerosing Cholangitis: Current Evidence and Concepts. Cell. Mol. Gastroenterol. Hepatol. 2024, 17, 423–438. [Google Scholar] [CrossRef]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef]
- Kummen, M.; Thingholm, L.B.; Rühlemann, M.C.; Holm, K.; Hansen, S.H.; Moitinho-Silva, L.; Liwinski, T.; Zenouzi, R.; Storm-Larsen, C.; Midttun, Ø.; et al. Altered Gut Microbial Metabolism of Essential Nutrients in Primary Sclerosing Cholangitis. Gastroenterology 2021, 160, 1784–1798.e0. [Google Scholar] [CrossRef]
- Awoniyi, M.; Wang, J.; Ngo, B.; Meadows, V.; Tam, J.; Viswanathan, A.; Lai, Y.; Montgomery, S.; Farmer, M.; Kummen, M.; et al. Protective and aggressive bacterial subsets and metabolites modify hepatobiliary inflammation and fibrosis in a murine model of PSC. Gut 2023, 72, 671–685. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised clinical trial: Vancomycin or metronidazole in patients with primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef]
- Goode, E.C.; Clark, A.B.; Mells, G.F.; Srivastava, B.; Spiess, K.; Gelson, W.T.H.; Trivedi, P.J.; Lynch, K.D.; Castren, E.; Vesterhus, M.N.; et al. Factors Associated with Outcomes of Patients With Primary Sclerosing Cholangitis and Development and Validation of a Risk Scoring System. Hepatology 2019, 69, 2120–2135. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.E.; Vesterhus, M.; McCauley, B.M.; Atkinson, E.J.; Schlicht, E.M.; Juran, B.D.; Gossard, A.A.; LaRusso, N.F.; Gores, G.J.; Karlsen, T.H.; et al. Primary Sclerosing Cholangitis Risk Estimate Tool (PREsTo) Predicts Outcomes of the Disease: A Derivation and Validation Study Using Machine Learning. Hepatology 2020, 71, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Cazzagon, N.; El Mouhadi, S.; Trivedi, P.J.; Dohan, A.; Kemgang, A.; Ben Belkacem, K.; Housset, C.; Chretien, Y.; Corpechot, C.; et al. Simple Magnetic Resonance Scores Associate with Outcomes of Patients with Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 2785–2792.e3. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Eaton, J.; Ringe, K.I.; Venkatesh, S.; Yamamura, J. Recommendations on the use of magnetic resonance imaging in PSC-A position statement from the International PSC Study Group. Hepatology 2017, 66, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, A.; Imeen Ringe, K.; Bengtsson, J.; Baubeta, E.; Forsman, C.; Korsavidou-Hult, N.; Rorsman, F.; Nilsson, E.; Kartalis, N.; Bergquist, A. Development of a prognostic MRCP-score (DiStrict) for individuals with large-duct primary sclerosing cholangitis. JHEP Rep. Innov. Hepatol. 2022, 4, 100595. [Google Scholar] [CrossRef]
- Muir, A.J.; Levy, C.; Janssen, H.L.A.; Montano-Loza, A.J.; Shiffman, M.L.; Caldwell, S.; Luketic, V.; Ding, D.; Jia, C.; McColgan, B.J.; et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results with Insights on the Natural History of the Disease. Hepatology 2019, 69, 684–698. [Google Scholar] [CrossRef]
- Dillman, J.R.; Trout, A.T.; Taylor, A.E.; Khendek, L.; Kasten, J.L.; Sheridan, R.M.; Sharma, D.; Karns, R.A.; Castro-Rojas, C.; Zhang, B.; et al. Association Between MR Elastography Liver Stiffness and Histologic Liver Fibrosis in Children and Young Adults with Autoimmune Liver Disease. AJR. Am. J. Roentgenol. 2024, 223, e2431108. [Google Scholar] [CrossRef]
- Vesterhus, M.; Karlsen, T.H. Emerging therapies in primary sclerosing cholangitis: Pathophysiological basis and clinical opportunities. J. Gastroenterol. 2020, 55, 588–614. [Google Scholar] [CrossRef]
- Jansen, P.L.M. New therapies target the toxic consequences of cholestatic liver disease. Expert. Rev. Gastroenterol. Hepatol. 2018, 12, 277–285. [Google Scholar] [CrossRef]
- Lindor, K.D. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N. Engl. J. Med. 1997, 336, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Boberg, K.M.; de Muckadell, O.S.; Lindgren, S.; Hultcrantz, R.; Folvik, G.; Bell, H.; Gangsøy-Kristiansen, M.; Matre, J.; Rydning, A.; et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: A 5-year multicenter, randomized, controlled study. Gastroenterology 2005, 129, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.C.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.R.; Mack, C.; Abdou, R.; Amin, M.; Amir, A.; Auth, M.; Bazerbachi, F.; Marie Broderick, A.; Chan, A.; DiGuglielmo, M.; et al. Gamma Glutamyltransferase Reduction Is Associated with Favorable Outcomes in Pediatric Primary Sclerosing Cholangitis. Hepatol. Commun. 2018, 2, 1369–1378. [Google Scholar] [CrossRef]
- Fickert, P.; Hirschfield, G.M.; Denk, G.; Marschall, H.-U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef]
- Halilbasic, E.; Steinacher, D.; Trauner, M. Nor-Ursodeoxycholic Acid as a Novel Therapeutic Approach for Cholestatic and Metabolic Liver Diseases. Dig. Dis. 2017, 35, 288–292. [Google Scholar] [CrossRef]
- Steinacher, D.; Claudel, T.; Trauner, M. Therapeutic Mechanisms of Bile Acids and Nor-Ursodeoxycholic Acid in Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2017, 35, 282–287. [Google Scholar] [CrossRef]
- Zhu, C.; Boucheron, N.; Müller, A.C.; Májek, P.; Claudel, T.; Halilbasic, E.; Baazim, H.; Lercher, A.; Viczenczova, C.; Hainberger, D.; et al. 24-Norursodeoxycholic acid reshapes immunometabolism in CD8(+) T cells and alleviates hepatic inflammation. J. Hepatol. 2021, 75, 1164–1176. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. The EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Parés, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Manns, M.; Hirschfield, G. New Treatment Paradigms in Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2023, 21, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K.V.; Milkiewicz, P.; Janczewska, E.; Malova, E.S.; Sanni, J.; et al. Farnesoid X receptor agonist tropifexor attenuates cholestasis in a randomised trial in patients with primary biliary cholangitis. JHEP Rep. Innov. Hepatol. 2022, 4, 100544. [Google Scholar] [CrossRef]
- Sepe, V.; Distrutti, E.; Fiorucci, S.; Zampella, A. Farnesoid X receptor modulators (2011–2014): A patent review. Expert. Opin. Ther. Pat. 2015, 25, 885–896. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Moschetta, A. Metabolic Messengers: Fibroblast growth factor 15/19. Nat. Metab. 2019, 1, 588–594. [Google Scholar] [CrossRef]
- Kim, Y.C.; Byun, S.; Seok, S.; Guo, G.; Xu, H.E.; Kemper, B.; Kemper, J.K. Small Heterodimer Partner and Fibroblast Growth Factor 19 Inhibit Expression of NPC1L1 in Mouse Intestine and Cholesterol Absorption. Gastroenterology 2019, 156, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ge, H.; Gupte, J.; Weiszmann, J.; Shimamoto, G.; Stevens, J.; Hawkins, N.; Lemon, B.; Shen, W.; Xu, J.; et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J. Biol. Chem. 2007, 282, 29069–29072. [Google Scholar] [CrossRef]
- Kunst, R.F.; Bolt, I.; van Dasselaar, R.D.J.; Nijmeijer, B.A.; Beuers, U.; Oude Elferink, R.P.J.; van de Graaf, S.F.J. Combined inhibition of bile salt synthesis and intestinal uptake reduces cholestatic liver damage and colonic bile salts in mice. JHEP Rep. Innov. Hepatol. 2024, 6, 100917. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Chazouillères, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lin, Z.; Ji, X.; Luo, X.; Zhang, Z.; Sun, M.; Chen, X.; Zhang, B.; Liang, H.; Liu, D.; et al. FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J. Hepatol. 2023, 79, 109–125. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Bellini, R.; Lupia, A.; Massa, C.; Urbani, G.; Bordoni, M.; Marchianò, S.; Rosselli, R.; De Gregorio, R.; Rapacciuolo, P.; et al. The leukemia inhibitory factor regulates fibroblast growth factor receptor 4 transcription in gastric cancer. Cell. Oncol. 2023, 47, 695–710. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Ling, L.; Beuers, U.; DePaoli, A.M.; Lieu, H.D.; Harrison, S.A.; Hirschfield, G.M. Potent suppression of hydrophobic bile acids by aldafermin, an FGF19 analogue, across metabolic and cholestatic liver diseases. JHEP Rep. Innov. Hepatol. 2021, 3, 100255. [Google Scholar] [CrossRef]
- Reich, M.; Deutschmann, K.; Sommerfeld, A.; Klindt, C.; Kluge, S.; Kubitz, R.; Ullmer, C.; Knoefel, W.T.; Herebian, D.; Mayatepek, E.; et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut 2016, 65, 487–501. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.M.G.G.M.G.M.G.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef]
- Hov, J.R.; Keitel, V.; Laerdahl, J.K.; Spomer, L.; Ellinghaus, E.; ElSharawy, A.; Melum, E.; Boberg, K.M.; Manke, T.; Balschun, T.; et al. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLoS ONE 2010, 5, e12403. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Eksteen, B.; Cheung, A.C.; Thorburn, D.; Moylan, C.A.; Pockros, P.J.; Forman, L.M.; Dorenbaum, A.; Hirschfield, G.M.; Kennedy, C.; et al. Safety, tolerability, and efficacy of maralixibat in adults with primary sclerosing cholangitis: Open-label pilot study. Hepatol. Commun. 2023, 7, e0153. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, G.M.; Alsuwayt, B.; Auclair, A.M.; Boyer, J.L.; Assis, D.N.; Ghonem, N.S. Fenofibrate Downregulates NF-κB Signaling to Inhibit Pro-inflammatory Cytokine Secretion in Human THP-1 Macrophages and During Primary Biliary Cholangitis. Inflammation 2022, 45, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef]
- Eksteen, B.; Bowlus, C.L.; Montano-Loza, A.J.; Lefebvre, E.; Fischer, L.; Vig, P.; Martins, E.B.; Ahmad, J.; Yimam, K.K.; Pockros, P.J.; et al. Efficacy and Safety of Cenicriviroc in Patients With Primary Sclerosing Cholangitis: PERSEUS Study. Hepatol. Commun. 2021, 5, 478–490. [Google Scholar] [CrossRef]
- Carey, E.J.; Eaton, J.; Clayton, M.; Gossard, A.; Iqbal, S.; Ullah, H.; Zhang, N.; Butterfield, R.; Lindor, K.D. A pilot study of vidofludimus calcium for treatment of primary sclerosing cholangitis. Hepatol. Commun. 2022, 6, 1589–1597. [Google Scholar] [CrossRef]
- Voskens, C.; Stoica, D.; Rosenberg, M.; Vitali, F.; Zundler, S.; Ganslmayer, M.; Knott, H.; Wiesinger, M.; Wunder, J.; Kummer, M.; et al. Autologous regulatory T-cell transfer in refractory ulcerative colitis with concomitant primary sclerosing cholangitis. Gut 2023, 72, 49–53. [Google Scholar] [CrossRef]
- Bogatic, D.; Bryant, R.V.; Lynch, K.D.; Costello, S.P. Systematic review: Microbial manipulation as therapy for primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2023, 57, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Rahimpour, S.; Nasiri-Toosi, M.; Khalili, H.; Ebrahimi-Daryani, N.; Nouri-Taromlou, M.K.; Azizi, Z. A Triple Blinded, Randomized, Placebo-Controlled Clinical Trial to Evaluate the Efficacy and Safety of Oral Vancomycin in Primary Sclerosing Cholangitis: A Pilot Study. J. Gastrointest. Liver Dis. 2016, 25, 457–464. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients With Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Meixiong, J.; Vasavda, C.; Green, D.; Zheng, Q.; Qi, L.; Kwatra, S.G.; Hamilton, J.P.; Snyder, S.H.; Dong, X. Identification of a bilirubin receptor that may mediate a component of cholestatic itch. eLife 2019, 8, e44116. [Google Scholar] [CrossRef] [PubMed]
- Meixiong, J.; Vasavda, C.; Snyder, S.H.; Dong, X. MRGPRX4 is a G protein-coupled receptor activated by bile acids that may contribute to cholestatic pruritus. Proc. Natl. Acad. Sci. USA 2019, 116, 10525–10530. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Harrington, A.M.; Lieu, T.; Garcia-Caraballo, S.; Maddern, J.; Schober, G.; O’Donnell, T.; Grundy, L.; Lumsden, A.L.; Miller, P.; et al. Activation of pruritogenic TGR5, MrgprA3, and MrgprC11 on colon-innervating afferents induces visceral hypersensitivity. JCI Insight 2019, 4, e131712. [Google Scholar] [CrossRef]
- Carino, A.; Biagioli, M.; Marchianò, S.; Fiorucci, C.; Zampella, A.; Monti, M.C.C.; Scarpelli, P.; Ricci, P.; Distrutti, E.; Fiorucci, S. Ursodeoxycholic acid is a GPBAR1 agonist and resets liver/intestinal FXR signaling in a model of diet-induced dysbiosis and NASH. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Bowlus, C.L.; Levy, C.; Akarca, U.S.; Alvares-da-Silva, M.R.; Andreone, P.; Arrese, M.; Corpechot, C.; Francque, S.M.; Heneghan, M.A.; et al. Efficacy and Safety of Elafibranor in Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 795–805. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Bowlus, C.L.; Mayo, M.J.; Kremer, A.E.; Vierling, J.M.; Kowdley, K.V.; Levy, C.; Villamil, A.; Ladrón de Guevara Cetina, A.L.; Janczewska, E.; et al. A Phase 3 Trial of Seladelpar in Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 783–794. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Main Histopathology and Genetic Characteristics |
|---|---|
| Classical, large-duct PSC | Is the prototypical form of PSC, involves large extrahepatic bile ducts, and is more prevalent in males, with a ratio of 3:2. Age of onset is the fourth decade. Associated with IBD, generally pancolitis, in 60–70% of cases. Progression to transplant ranges from 12 to 20 years. Associated with HLA haplotypes: HLA-A*01; HLA-B*08 or HLA-DRB1*03031. |
| Non-IBD PSC | Equally distributed among males and females. Onset at older age. Same HLA haplotypes as PSC-IBD. Better outcome. |
| Small-bile-duct PSC | This form accounts for 10% of PSC. This phenotype lacks typical magnetic resonance cholangiopancreatography (MRCP) characteristics. IBD is required for diagnosis. Patients might be considered PBC-AMA. If IBD is not present, the HLA haplotypes differ from the large-duct PSC. |
| Overlap syndrome: PSC-autoimmune hepatitis | A variable percentage of PSC patients show features of auto-immune hepatitis, showing pANCA, ANA and SMA antibodies. Typically shows elevated levels of AST and ALT (up to ten folds) at onset. |
| Pediatric PSC | PSC in children shares most of the features of large-duct PSC, including association with IBD. Association with autoimmune hepatitis occurs in 10% of these patients. |
| Non-Caucasian PSC | PSC occurs in the African-American population with the same prevalence as the Caucasian population. The prevalent haplotype is HLA-B*08. Less frequent association with IBD. |
| Secondary sclerosing cholangitis | Infections, IgG4-associated SSC, HIV infection, sarcoidosis, biliary stones, traumatic, ischemic. Cholangiocarcinoma, pancreatic and ampullary cancer. Papillary stenosis. Chronic pancreatitis. Infestation (ascaris, liver fluke). |
| Study ID | Status | Drug-Target | CT Phase | Primary and Secondary End Points and Results |
|---|---|---|---|---|
| NCT04480840 | Active, not recruiting | PLN-74809, αvβ6 and αvβ1 inhibitor | 2 | Evaluation of safety, tolerability, and PK of PLN-74809 in participants with PSC and suspected liver fibrosis |
| NCT05642468 | Recruiting | A3907, ASBT inhibitor | 2 | Evaluation of safety and tolerability of A3907 in PSC patients |
| NCT04663308 | Recruiting | Volixibat, ASBT inhibitor | 2 | Evaluation of Volixibat use in PSC-associated itching treatment Secondary Evaluation of Volixibat impact on PSC disease progression |
| NCT05896137 | Recruiting | CS0159, FXR inhibitor | 2 | Evaluation of safety, tolerability, and efficacy of CS0159 in patients with PSC |
| NCT05082779 | Completed | CS0159, FXR inhibitor | 1 | Primary Evaluation of safety, tolerability, and PK and PD profiles of CS0159 in SAD and MAD studies Secondary Assessment of Food Effect (FE) on single-dose PK profile of CS0159 from SAD study in healthy subjects |
| NCT05295680 | Recruiting | Hymecromone, antineoplastic agent/hyaluronic acid synthesis inhibitor | 2 | Primary Evaluation of the efficacy of hymecromone plus standard of care (SOC) compared with SOC alone in the treatment of adolescents and adults with primary sclerosing cholangitis (PSC) Secondary Evaluation of changes in ALP from baseline to 6 months post-treatment following treatment with hymecromone plus SOC compared with SOC Evaluation of changes in PSC biomarkers: fibrotic effect (FibroScan), inflammation (serum Hyaluronan, HA), T-cell count |
| NCT05866809 | Completed | HK-660S, anti-inflammatory/anti-fibrotic agent | 2 | Evaluation of improvement of bile duct strictures via MRCP and ALP levels assessment following the administration of HK-660S in patients with PSC |
| NCT03722576 | Completed with results | Vidofludimus | 2 | Primary Evaluation of safety, tolerability, and efficacy of daily dosing with VC over a 6-month period via assessment of ALP and liver biochemistry (3 and 6 months). Secondary Assessment of IL-17 and IFNγ (6 weeks and 6 months) Results - Combined reduction in AST and ALT: 27.3% for Abnormal AST and ALT: 80% - Abnormal total bilirubin: 30%; abnormal direct bilirubin: 40% |
| NCT05627362 | Recruiting | Elafibranor, PPARα/δ agonist | 2 | Primary Evaluation of safety and side effects of Elafibranor in participants with PSC Secondary Evaluation of drug’s effects on blood tests and other tests related to PSC disease activity |
| NCT04024813 | Completed | Seladelpar, PPARδ agonist | 2 | Evaluation efficacy, safety, and tolerability of effects of Seladelpar compared to placebo in patients with PSC |
| NCT04309773 | Recruiting | Benzafibrate, PPARα agonist | 3 | Primary Evaluation of effects of Benzafibrate treatment compared to placebo on efficacy and safety in patients with PSC despite standard UDCA therapy Secondary Effects of Benzafibrate treatment compared to placebo on persistent cholestasis in patients with PSC despite standard UDCA therapy |
| NCT06026865 | Recruiting | S-adenosyl-methionine (SAMe), | N.A. | Primary Investigation of clinical effects (liver biochemistry, health-related quality of life, liver stiffness) of SAMe in patients with PSC Secondary Investigation of underlying mechanisms of hepatoprotection (plasma concentrations of SOD2, FGF-19, TNF-α, IL-6, and others) of SAMe in patients with PSC |
| NCT05525520 | Recruiting | EP547, MrgprX4 antagonist | 2 | Evaluation of the effects of EP547 in subjects with cholestatic pruritus due to PBC or PSC |
| NCT04060147 | Terminated with results | Cilofexor, FXR agonist | 1 | Evaluation of safety and tolerability of escalating doses of CILO in participants with PSC and compensated cirrhosis Results - Treatment-emergent adverse events (TEAEs): 81.8% - Treatment-emergent serious adverse events (SAEs): 0% |
| NCT03890120 | Terminated with results | Cilofexor, FXR agonist | 3 | Cilofexor reduce the risk of fibrosis progression among non-cirrhotic adults with PSC Results - Liver fibrosis progression in blinded phase, cilofexor vs. placebo: 30.8% vs. 32.8% - Severe adverse event (SAEs) in blinded phase, cilofexor vs. placebo: 19.1% vs. 18.7% - Changes in serum ALT concentration in blinded phase, cilofdexor vs. placebo (LSM): −13 vs. −3 - Changes in serum fasting total BAs concentration in blinded phase, cilofexor vs placebo (LSM): 7.2 vs. 9.8 - Liver fibrosis improvement in blinded phase, cilofexor vs. placebo: 25.6% vs. 17.2% |
| NCT05912387 | Recruiting | RosuvastatinHMG-CoA reductase inhibitor | Early 1 | Evaluation of bile acids profile (total BAs, SBAs:PBAs ratio, conjugated:deconjugated BAs ratio, and other) and microbiome impact of Rosuvastatin in PSC patients |
| NCT04133792 | Recruiting | Simvastatin | 3 | Assessment of effect of PSC prognosis according to long-term intake of Simvastatin (death, liver transplantation, cholangiocarcinoma, esophageal varices bleeding) |
| NCT05876182 | Recruiting | Vancomyci, | 2 | Evaluation of safety and efficacy of two doses of oral Vancomycin in subjects between 15 and 70 years old with PSC |
| NCT03710122 | Recruiting | Vancomycin | 2/3 | Primary Evaluation of safety and efficacy of Vancomycin for therapy in adult patients with PSC via assessment of serum ALP levels, liver fibrosis, and pro-inflammatory cytokines (TGF-ß, IL-4 and others) Secondary Determination of changes in the intestinal microbiota in relation to the use of Vancomycin and its correlation with changes in serum ALP and liver fibrosis |
| NCT06037577 | Completed | CM-101, anti-CCl24 monoclonal antibody | 1 | Evaluation of safety, tolerability, PKs, and PDs of single escalating subcutaneous doses of CM-101 in healthy male subjects |
| NCT04595825 | Active, not recruiting | CM-101, anti-CCl24 monoclonal antibody | 2 | Evaluation of safety, tolerability, and activity of the anti-human CCL24 monoclonal antibody CM-101 in adult subjects with PSC |
| NCT06553768 | Not yet recruiting | Maralixibat, IBAT inhibitor | 3 | Primary Evaluation of the mean change in the ItchRO(Obs) severity score from baseline to average of week 13 to week 20 Secondary Evaluation of the mean change in total sBA levels from baseline to average of week 12 and week 20 |
| NCT06286709 | Recruiting | Fecal Microbiota Transplantation | 2 | Primary Evaluation of reduction in serum ALP measured at 48 weeks following the first dose of FMT or FMT placebo Secondary Evaluation of PSC-PRO, SF-36, 5D-Itch, and SIBDQ questionnaires at screening (week-2) and weeks 8, 12, 24, 36, and 48; evaluation of VCTE at screening and week 48; evaluation of ELF at weeks 1, 5, 12, and 48; evaluation of AST, ALT, bilirubin, gGT, and albumin and assessment of UK-PSC score and Amsterdam-Oxford PSC score at screening and weeks 1, 3, 5, 8, 12, 24, 36, and 48; quantitative assessment of SES-CD and fecal calprotectin at screening and weeks 1, 5, 12, and 48; evaluation of occurrence of adverse events measured by CTCAE v5.0 |
| NCT06197308 | Recruiting | Vancomycin | Early 1 | Evaluation of safety and feasibility of microbiota transplant therapy (MTT) by determination of the frequency of serious adverse events or other adverse events and determination of the proportion of subjects taking 100% of the MTT per protocol, respectively |
| NCT06095986 | Not yet recruiting | Aramchol meglumine, Stearoyl-CoA desaturase 1 inhibitor | 2 | Primary Evaluation of changes in ALP levels from baseline to week 48 Secondary Evaluation of changes in Nakanuma stage classification, ELF test, MRCP, Gadoxetate clearance test, 5D-Itch scale, PROMIS-19 questionnaire, and Mayo IBD symptom severity score, as well as rMRS, UK-PSC, and PREsTo, from baseline to 48 weeks |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorucci, S.; Urbani, G.; Di Giorgio, C.; Biagioli, M.; Distrutti, E. Bile Acids-Based Therapies for Primary Sclerosing Cholangitis: Current Landscape and Future Developments. Cells 2024, 13, 1650. https://doi.org/10.3390/cells13191650
Fiorucci S, Urbani G, Di Giorgio C, Biagioli M, Distrutti E. Bile Acids-Based Therapies for Primary Sclerosing Cholangitis: Current Landscape and Future Developments. Cells. 2024; 13(19):1650. https://doi.org/10.3390/cells13191650
Chicago/Turabian StyleFiorucci, Stefano, Ginevra Urbani, Cristina Di Giorgio, Michele Biagioli, and Eleonora Distrutti. 2024. "Bile Acids-Based Therapies for Primary Sclerosing Cholangitis: Current Landscape and Future Developments" Cells 13, no. 19: 1650. https://doi.org/10.3390/cells13191650