


The Neolignan Honokiol and Its Synthetic Derivative Honokiol Hexafluoro Reduce Neuroinflammation and Cellular Senescence in Microglia Cells

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. BV2 Cell Culture
2.2. Cell Treatment
2.3. Sulforhodamine B (SRB) Assay
2.4. Senescence-Associated Heterochromatin Foci Analysis (SAHFs)
2.5. Senescence-Associated ß-Galactosidase Activity Assay
2.6. Senescence-Associated β-Galactosidase Staining
2.7. Protein Lysates from Cells
2.8. Western Blot (WB)
2.9. Real-Time PCR (RT-PCR)
2.10. Total ROS Production
2.11. Cell Morphology Analysis
2.12. Statistical Analysis
3. Results
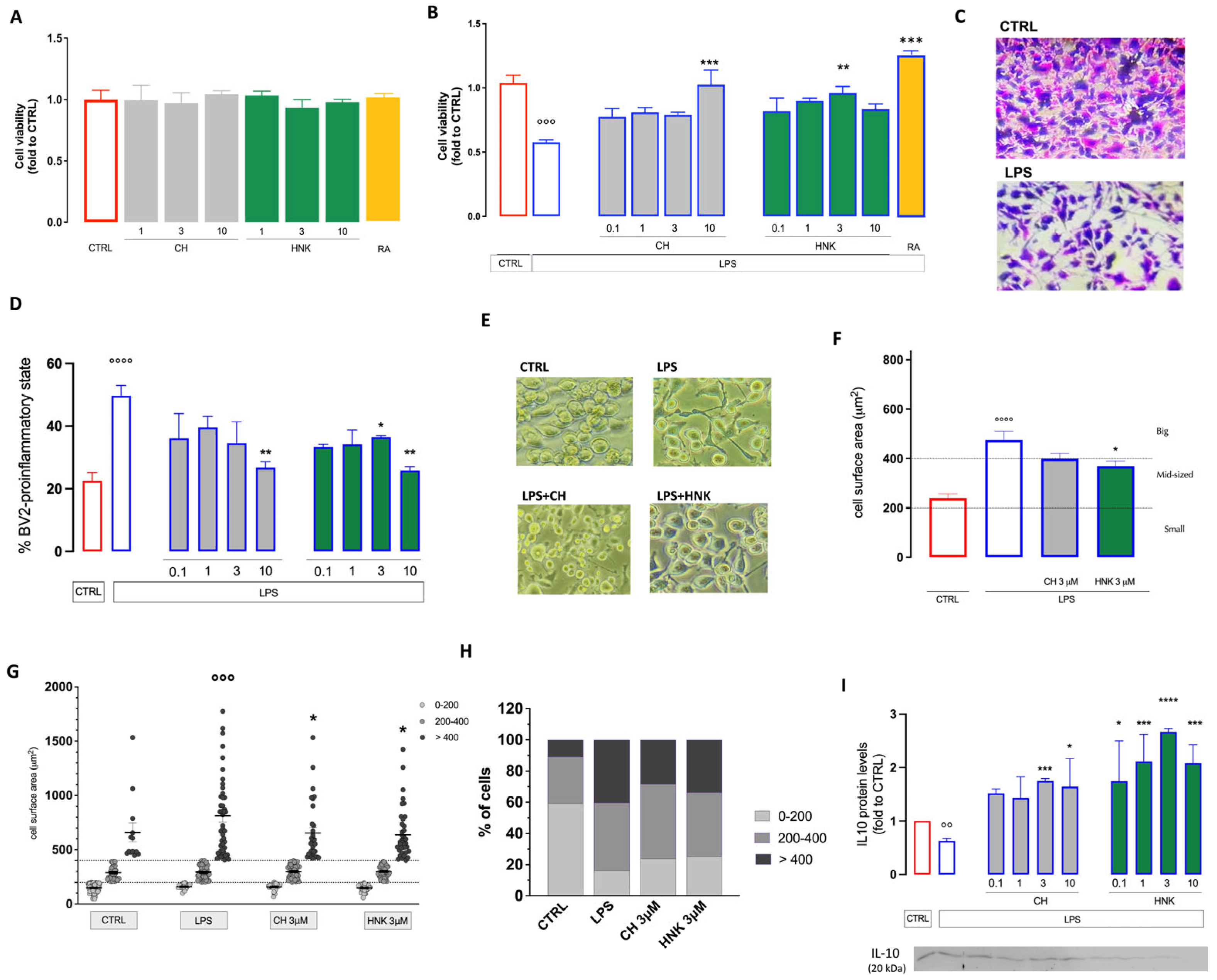
3.1. Effect of HNK and CH in an In Vitro Model of Neuroinflammation
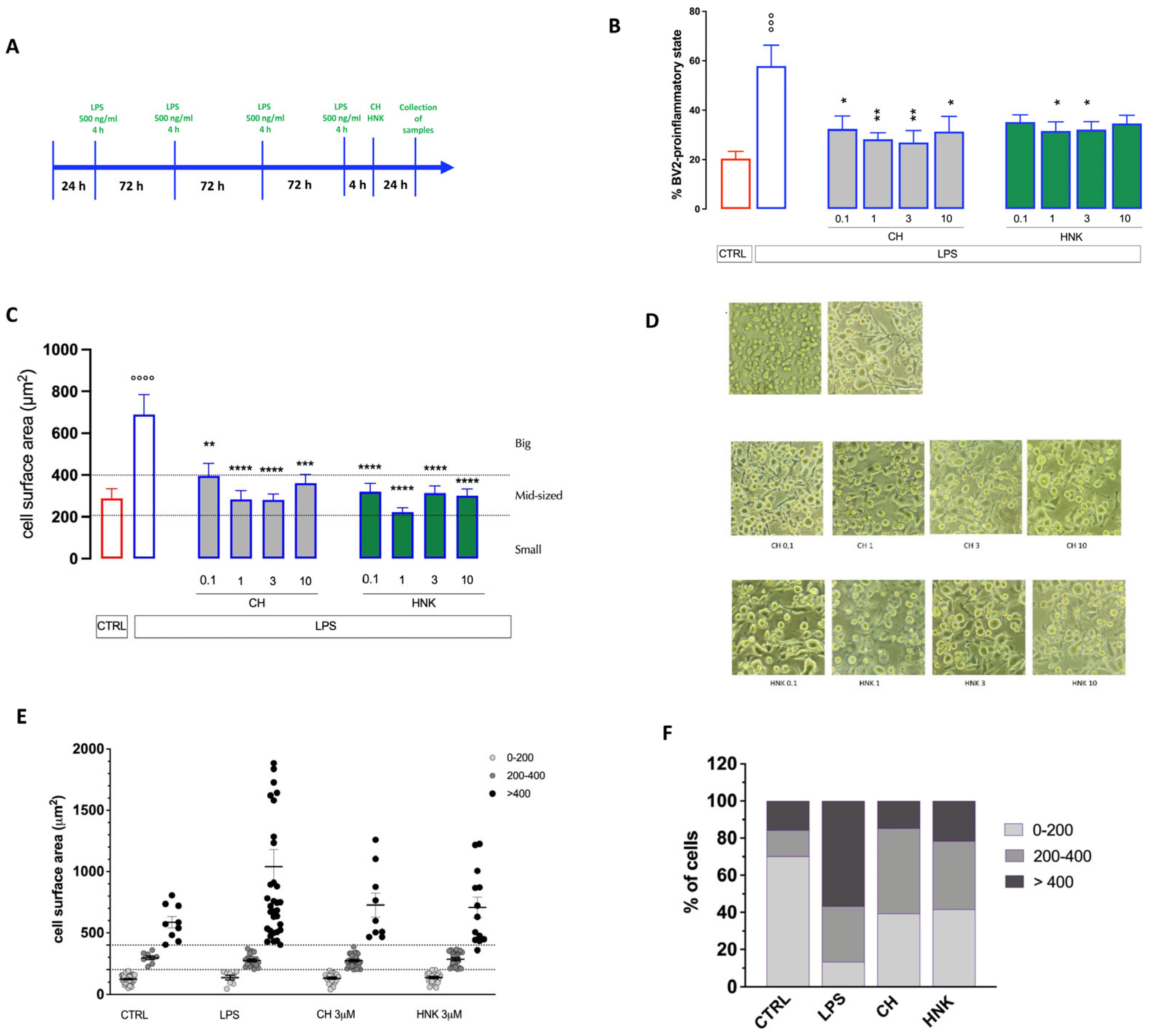
3.2. Effect of HNK and CH in an In Vitro Model of Cellular Senescence
3.2.1. Effect of HNK and CH on Altered Cell Morphology of Senescent BV Cells
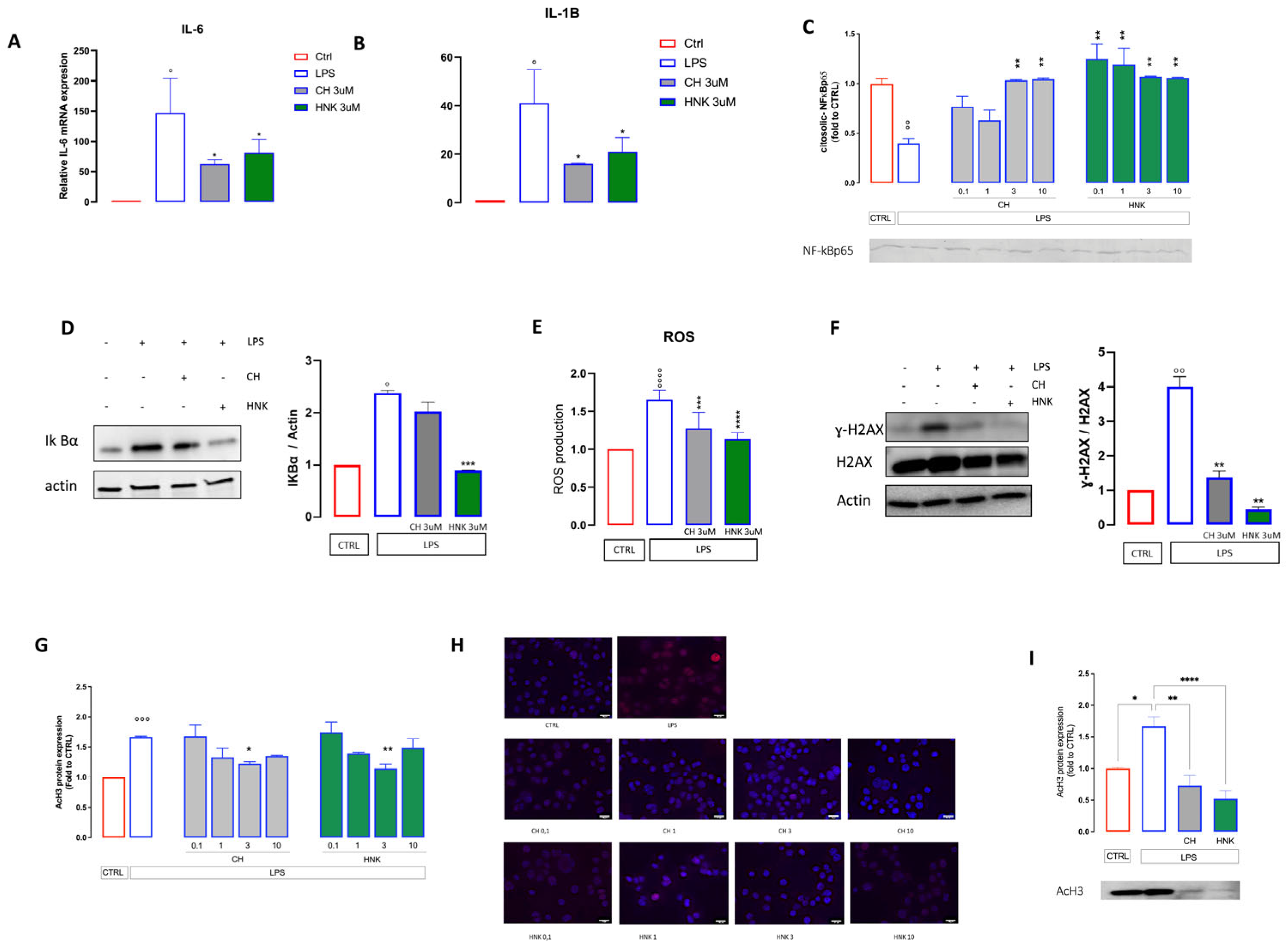
3.2.2. Effect of CH and HNK on Senescence-Associated Markers
3.2.3. Anti-SASP Activity of CH and HNK
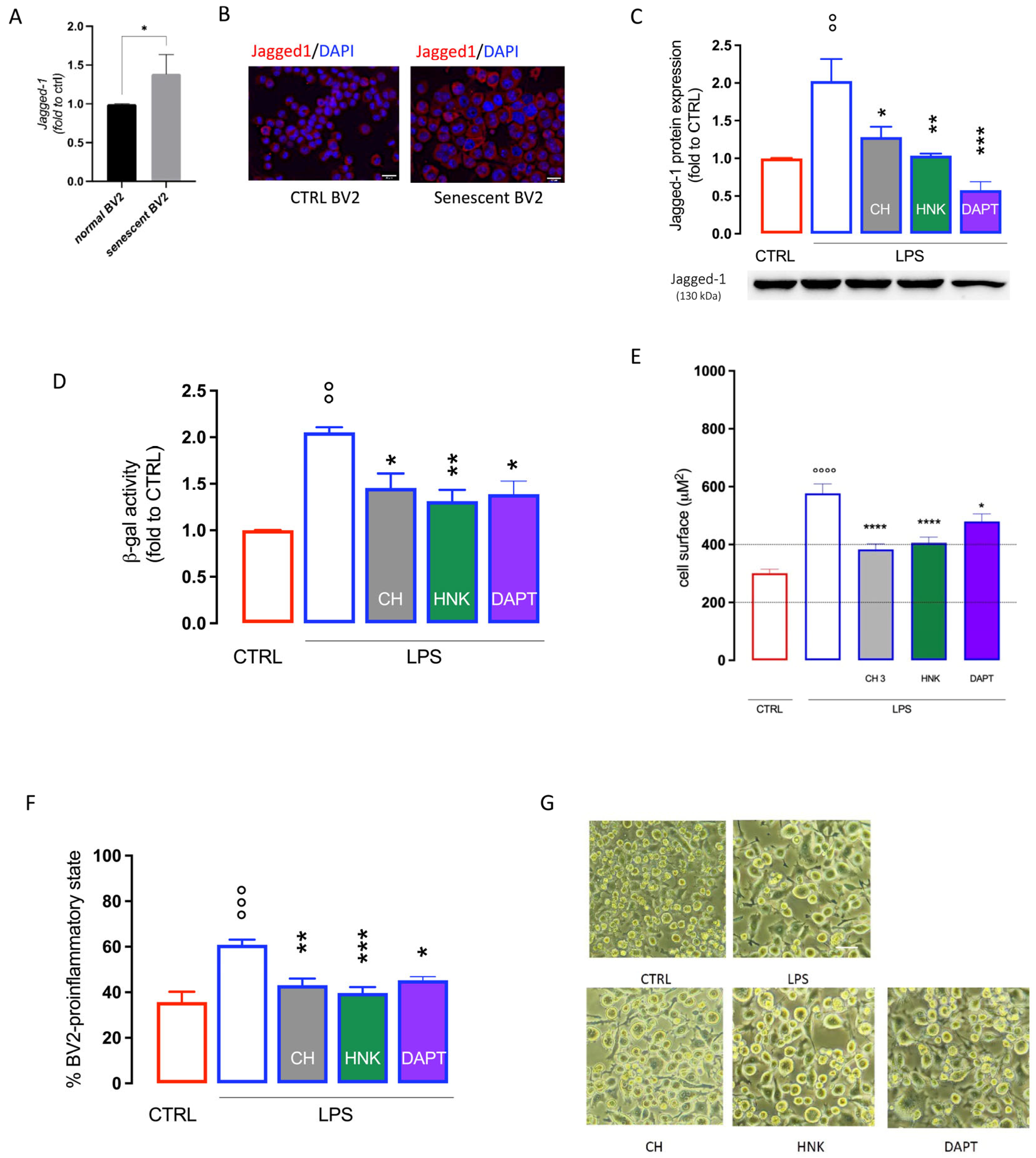
3.3. Effect of CH and HNK on Jagged-1 Expression in BV2 Senescent Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular Senescence: A Key Therapeutic Target in Aging and Diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef] [PubMed]
- Holloway, K.; Neherin, K.; Dam, K.U.; Zhang, H. Cellular Senescence and Neurodegeneration. Hum. Genet. 2023, 142, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.R.; Rani, L.; Subba, R.; Mondal, A.C. Cellular Senescence in the Aging Brain: A Promising Target for Neurodegenerative Diseases. Mech. Ageing Dev. 2022, 204, 111675. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Ginhoux, F.; Prinz, M. Origin of Microglia: Current Concepts and Past Controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Neumann, P.; Lenz, D.E.; Streit, W.J.; Bechmann, I. Is Microglial Dystrophy a Form of Cellular Senescence? An Analysis of Senescence Markers in the Aged Human Brain. Glia 2023, 71, 377–390. [Google Scholar] [CrossRef]
- Nelke, C.; Schroeter, C.B.; Pawlitzki, M.; Meuth, S.G.; Ruck, T. Cellular Senescence in Neuroinflammatory Disease: New Therapies for Old Cells? Trends Mol. Med. 2022, 28, 850–863. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for Targeting Senescent Cells in Human Disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Lucas, V.; Cavadas, C.; Aveleira, C.A. Cellular Senescence: From Mechanisms to Current Biomarkers and Senotherapies. Pharmacol. Rev. 2023, 75, 675–713. [Google Scholar] [CrossRef] [PubMed]
- Talarek, S.; Listos, J.; Barreca, D.; Tellone, E.; Sureda, A.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Neuroprotective Effects of Honokiol: From Chemistry to Medicine. Biofactors 2017, 43, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Mottaghi, S.; Abbaszadeh, H. Natural Lignans Honokiol and Magnolol as Potential Anticarcinogenic and Anticancer Agents. A Comprehensive Mechanistic Review. Nutr. Cancer 2021, 74, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sung, B. NF-κB in Cancer: A Matter of Life and Death. Cancer Discov. 2011, 1, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The Complexity of NF-κB Signaling in Inflammation and Cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Leo, A.; Pranzini, E.; Pietrovito, L.; Pardella, E.; Parri, M.; Cirri, P.; Bruno, G.; Calvani, M.; Peppicelli, S.; Torre, E.; et al. Claisened Hexafluoro Inhibits Metastatic Spreading of Amoeboid Melanoma Cells. Cancers 2021, 13, 3551. [Google Scholar] [CrossRef]
- Cohen, J.; Huang, S.; Koczwara, K.E.; Woods, K.T.; Ho, V.; Woodman, K.G.; Arbiser, J.L.; Daman, K.; Lek, M.; Emerson, C.P.; et al. Flavones Provide Resistance to DUX4-Induced Toxicity via an MTor-Independent Mechanism. Cell Death Dis. 2023, 14, 749. [Google Scholar] [CrossRef]
- Rauf, A.; Olatunde, A.; Imran, M.; Alhumaydhi, F.A.; Aljohani, A.S.M.; Khan, S.A.; Uddin, M.S.; Mitra, S.; Emran, T.B.; Khayrullin, M.; et al. Honokiol: A Review of Its Pharmacological Potential and Therapeutic Insights. Phytomedicine 2021, 90, 153647. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, X.; Zhu, Y.; Chen, S.; Zhou, D.; Wang, Y. Honokiol Inhibits the Inflammatory Reaction during Cerebral Ischemia Reperfusion by Suppressing NF-κB Activation and Cytokine Production of Glial Cells. Neurosci. Lett. 2013, 534, 123–127. [Google Scholar] [CrossRef]
- Chen, C.M.; Liu, S.H.; Lin-Shiau, S.Y. Honokiol, a Neuroprotectant against Mouse Cerebral Ischaemia, Mediated by Preserving Na+, K+-ATPase Activity and Mitochondrial Functions. Basic Clin. Pharmacol. Toxicol. 2007, 101, 108–116. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, D.Y.; Yun, Y.P.; Han, S.B.; Kim, H.M.; Lee, K.; Choi, S.H.; Yang, M.P.; Jeon, H.S.; Jeong, J.H.; et al. Ethanol Extract of Magnolia Officinalis Prevents Lipopolysaccharide-Induced Memory Deficiency via Its Antineuroinflammatory and Antiamyloidogenic Effects. Phytother. Res. 2013, 27, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Scanlan, A.; Koneru, R.; Morrell, C.R.; Reece, M.D.; Edwards, E.; Roa, S.; Gavegnano, C.; Bimonte-Nelson, H.; Arbiser, J.; et al. Honokiol Hexafluoro Confers Reversal of Neuropathological Markers of HIV Infection in a Murine SCID Model. Neurotherapeutics 2024, 21, e00329. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Microglia and the Aging Brain: Are Senescent Microglia the Key to Neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Borgonetti, V.; Galeotti, N. Rosmarinic Acid Reduces Microglia Senescence: A Novel Therapeutic Approach for the Management of Neuropathic Pain Symptoms. Biomedicines 2022, 10, 1468. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.; Kasinski, A. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio Protoc. 2016, 6. [Google Scholar] [CrossRef]
- Pardella, E.; Pranzini, E.; Nesi, I.; Parri, M.; Spatafora, P.; Torre, E.; Muccilli, A.; Castiglione, F.; Fambrini, M.; Sorbi, F.; et al. Therapy-Induced Stromal Senescence Promoting Aggressiveness of Prostate and Ovarian Cancer. Cells 2022, 11, 4026. [Google Scholar] [CrossRef]
- Galeotti, N.; Ghelardini, C.; Zoppi, M.; Del Bene, E.; Raimondi, L.; Beneforti, E.; Bartolini, A. Hypofunctionality of Gi Proteins as Aetiopathogenic Mechanism for Migraine and Cluster Headache. Cephalalgia 2001, 21, 38–45. [Google Scholar] [CrossRef]
- Borgonetti, V.; Galeotti, N. Microglia Senescence Is Related to Neuropathic Pain-Associated Comorbidities in the Spared Nerve Injury Model. Pain 2023, 164, 1106–1117. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Anerillas, C.; Abdelmohsen, K.; Gorospe, M. Regulation of Senescence Traits by MAPKs. Geroscience 2020, 42, 397–408. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The Senescence-Associated Secretory Phenotype and Its Regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Finkel, T. Free Radicals and Senescence. Exp. Cell Res. 2008, 314, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.Y.; Chan, M.H.; Zong, Y.; Sheng, W.; He, Y.; Jiang, J.H.; Simonyi, A.; Gu, Z.; Fritsche, K.L.; Cui, J.; et al. Magnolia Polyphenols Attenuate Oxidative and Inflammatory Responses in Neurons and Microglial Cells. J. Neuroinflamm. 2013, 10, 786. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Chigurupati, S.; Arumugam, T.V.; Jo, D.G.; Li, H.; Chan, S.L. Notch Activation Enhances the Microglia-Mediated Inflammatory Response Associated with Focal Cerebral Ischemia. Stroke 2011, 42, 2589–2594. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Narita, M. Notch and Senescence. Adv. Exp. Med. Biol. 2018, 1066, 299–318. [Google Scholar] [CrossRef]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997–1007.e5. [Google Scholar] [CrossRef]
- Greenwood, E.K.; Brown, D.R. Senescent Microglia: The Key to the Ageing Brain? Int. J. Mol. Sci. 2021, 22, 4402. [Google Scholar] [CrossRef]
- Ng, P.Y.; McNeely, T.L.; Baker, D.J. Untangling Senescent and Damage-Associated Microglia in the Aging and Diseased Brain. FEBS J. 2023, 290, 1326–1339. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, Y.; Xiao, F. Comparison of Several Commonly Used Detection Indicators of Cell Senescence. Drug Chem. Toxicol. 2020, 43, 213–218. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-Associated (Beta)-Galactosidase Reflects an Increase in Lysosomal Mass during Replicative Ageing of Human Endothelial Cells. J. Cell Sci. 2000, 113 Pt 20, 3613–3622. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling Pathway of MAPK/ERK in Cell Proliferation, Differentiation, Migration, Senescence and Apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Ben-Neriah, Y. Senescence-Associated Inflammatory Responses: Aging and Cancer Perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-κB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell Surface-Bound IL-1alpha Is an Upstream Regulator of the Senescence-Associated IL-6/IL-8 Cytokine Network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Ríos, J.L.; Francini, F.; Schinella, G.R. Natural Products for the Treatment of Type 2 Diabetes Mellitus. Planta Med. 2015, 81, 975–994. [Google Scholar] [CrossRef]
- Woodbury, A.; Yu, S.P.; Wei, L.; García, P. Neuro-Modulating Effects of Honokiol: A Review. Front. Neurol. 2013, 4, 130. [Google Scholar] [CrossRef]
- Tan, Y.; Yu, H.; Sun, S.; Gan, S.; Gong, R.; Mou, K.J.; Xue, J.; Xu, S.; Wu, J.; Ma, L. Honokiol Exerts Protective Effects on Neural Myelin Sheaths after Compressed Spinal Cord Injury by Inhibiting Oligodendrocyte Apoptosis through Regulation of ER-Mitochondrial Interactions. J. Spinal Cord. Med. 2022, 45, 595–604. [Google Scholar] [CrossRef]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an Activator of Sirtuin-3 (SIRT3) Preserves Mitochondria and Protects the Heart from Doxorubicin-Induced Cardiomyopathy in Mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, A.; Li, G.; Ramanathan, A.; Mwangi, S.M.; Hart, C.M.; Arbiser, J.L.; Srinivasan, S. SIRT3 Activation Promotes Enteric Neurons Survival and Differentiation. Sci. Rep. 2022, 12, 22076. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular Senescence in Brain Aging and Neurodegenerative Diseases: Evidence and Perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch Signaling Pathway: Architecture, Disease, and Therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 Mediates a Switch between Two Distinct Secretomes during Senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Kagawa, S.; Natsuizaka, M.; Whelan, K.A.; Facompre, N.; Naganuma, S.; Ohashi, S.; Kinugasa, H.; Egloff, A.M.; Basu, D.; Gimotty, P.A.; et al. Cellular Senescence Checkpoint Function Determines Differential Notch1-Dependent Oncogenic and Tumor-Suppressor Activities. Oncogene 2015, 34, 2347–2359. [Google Scholar] [CrossRef]
- Parry, A.J.; Hoare, M.; Bihary, D.; Hänsel-Hertsch, R.; Smith, S.; Tomimatsu, K.; Mannion, E.; Smith, A.; D’Santos, P.; Russell, I.A.; et al. NOTCH-Mediated Non-Cell Autonomous Regulation of Chromatin Structure during Senescence. Nat. Commun. 2018, 9, 1840. [Google Scholar] [CrossRef]
- Kaushik, G.; Venugopal, A.; Ramamoorthy, P.; Standing, D.; Subramaniam, D.; Umar, S.; Jensen, R.A.; Anant, S.; Mammen, J.M.V. Honokiol Inhibits Melanoma Stem Cells by Targeting Notch Signaling. Mol. Carcinog. 2015, 54, 1710–1721. [Google Scholar] [CrossRef]
- Ponnurangam, S.; Mammen, J.M.V.; Ramalingam, S.; He, Z.; Zhang, Y.; Umar, S.; Subramaniam, D.; Anant, S. Honokiol in Combination with Radiation Targets Notch Signaling to Inhibit Colon Cancer Stem Cells. Mol. Cancer Ther. 2012, 11, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Borgonetti, V.; Galeotti, N. Honokiol-Rich Magnolia Officinalis Bark Extract Attenuates Trauma-Induced Neuropathic Pain. Antioxidants 2023, 12, 1518. [Google Scholar] [CrossRef] [PubMed]
- Dovey, H.F.; John, V.; Anderson, J.P.; Chen, L.Z.; De Saint Andrieu, P.; Fang, L.Y.; Freedman, S.B.; Folmer, B.; Goldbach, E.; Holsztynska, E.J.; et al. Functional Gamma-Secretase Inhibitors Reduce Beta-Amyloid Peptide Levels in Brain. J. Neurochem. 2001, 76, 173–181. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IL-6 (reverse) | 5′-TGCATCATCGTTGTTCATAC-3′ |
| IL-6 (forward) | 5′-CTTCCATCCAGTTGCCTTCT-3′ |
| IL-1β (reverse) | 5′-CCCATCAGAGGCAAGGAGGAA-3′ |
| IL-1β (forward) | 5′-CCTGCAGCTGGAGAGTGTGGA-3′ |
| β2M (reverse) | 5′-GTCATGCTTAACTCTGCAGG-3′ |
| β2M (forward) | 5′-TGCTATCCAGAAAACCCCTC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasia, C.; Borgonetti, V.; Mancini, C.; Lori, G.; Arbiser, J.L.; Taddei, M.L.; Galeotti, N. The Neolignan Honokiol and Its Synthetic Derivative Honokiol Hexafluoro Reduce Neuroinflammation and Cellular Senescence in Microglia Cells. Cells 2024, 13, 1652. https://doi.org/10.3390/cells13191652
Sasia C, Borgonetti V, Mancini C, Lori G, Arbiser JL, Taddei ML, Galeotti N. The Neolignan Honokiol and Its Synthetic Derivative Honokiol Hexafluoro Reduce Neuroinflammation and Cellular Senescence in Microglia Cells. Cells. 2024; 13(19):1652. https://doi.org/10.3390/cells13191652
Chicago/Turabian StyleSasia, Chiara, Vittoria Borgonetti, Caterina Mancini, Giulia Lori, Jack L. Arbiser, Maria Letizia Taddei, and Nicoletta Galeotti. 2024. "The Neolignan Honokiol and Its Synthetic Derivative Honokiol Hexafluoro Reduce Neuroinflammation and Cellular Senescence in Microglia Cells" Cells 13, no. 19: 1652. https://doi.org/10.3390/cells13191652
APA StyleSasia, C., Borgonetti, V., Mancini, C., Lori, G., Arbiser, J. L., Taddei, M. L., & Galeotti, N. (2024). The Neolignan Honokiol and Its Synthetic Derivative Honokiol Hexafluoro Reduce Neuroinflammation and Cellular Senescence in Microglia Cells. Cells, 13(19), 1652. https://doi.org/10.3390/cells13191652