
Identification of hsa_circ_0018905 as a New Potential Biomarker for Multiple Sclerosis

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. RNA Extraction
2.3. RNA Digestion, Amplification, Labeling, and Hybridization
2.4. Reverse Transcription (RT)-Quantitative (q)PCR Analysis
2.5. Prediction of circRNA-miRNA and circRNA-RBP Interactions
2.6. CircRNA-microRNA-mRNA Network
2.7. Statistical Analysis
2.8. Statistics and Data Representation
3. Results
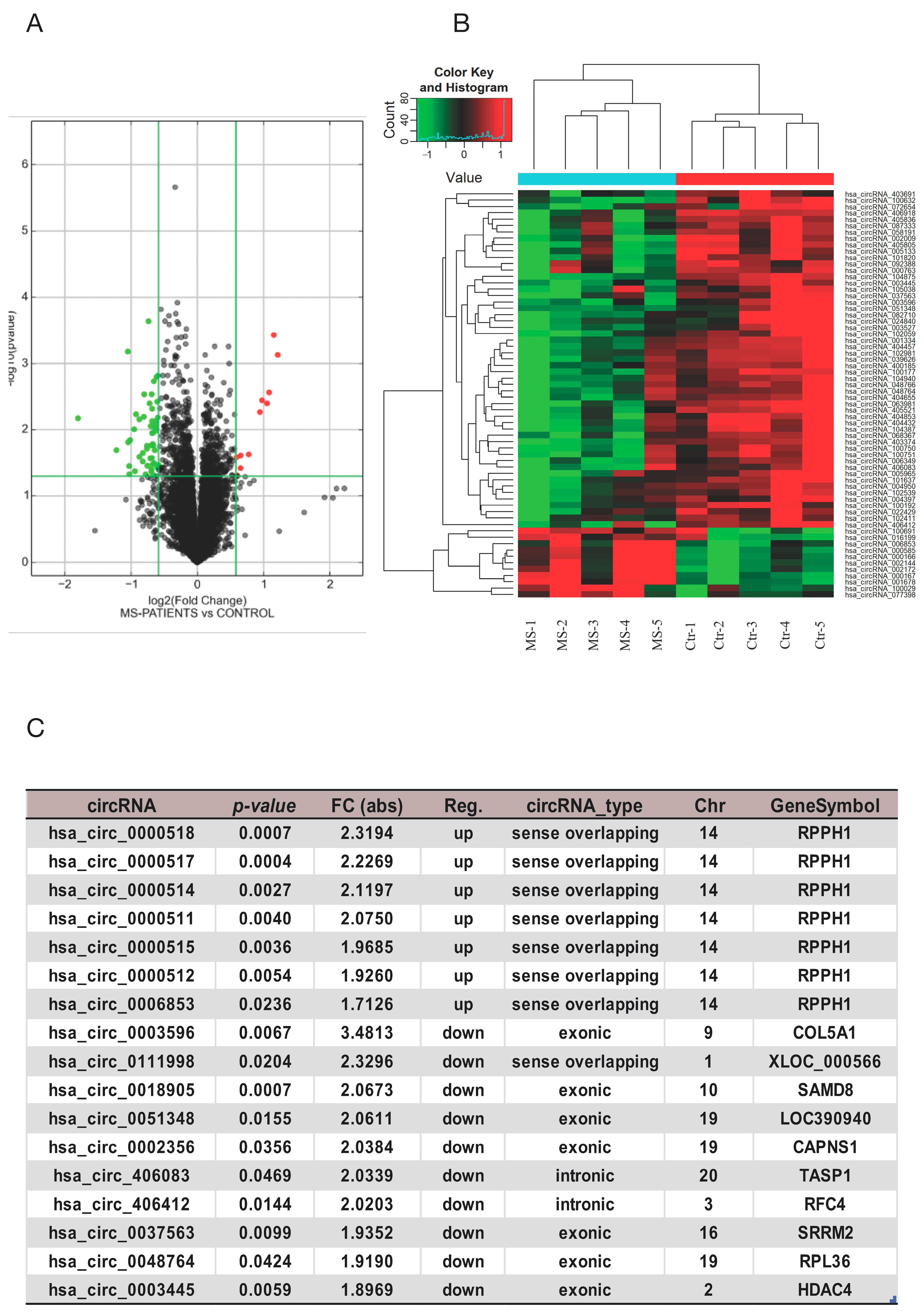
3.1. Analysis of circRNAs Expressed in MS Patients Using circRNA Arrays
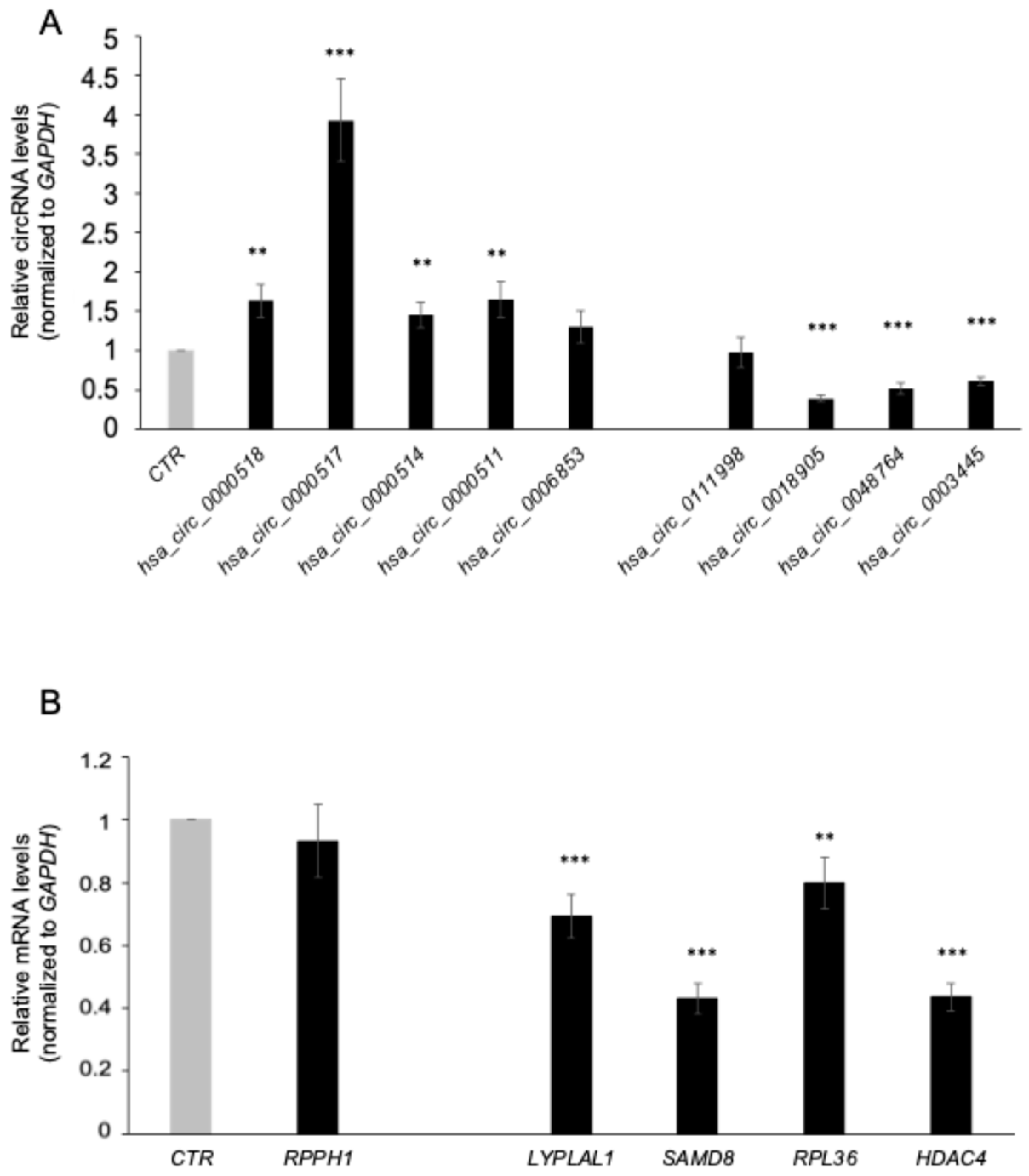
3.2. Validation of circRNA Expression Profiles by RT-qPCR
3.3. PBMC circRNA Expression Correlation with Serum
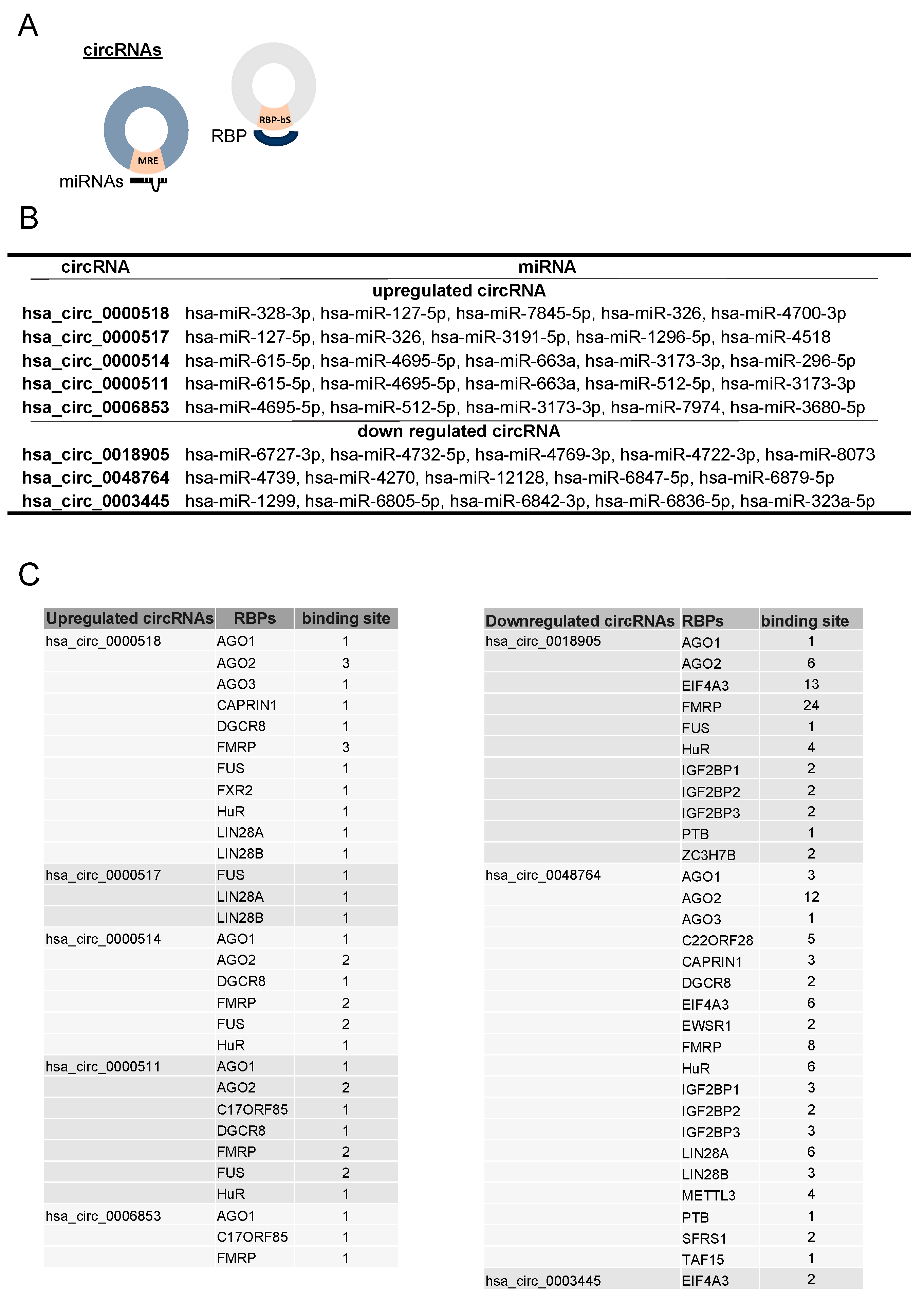
3.4. Identification of the miRNA and RBP Targets and circRNA-miRNA-mRNA Network
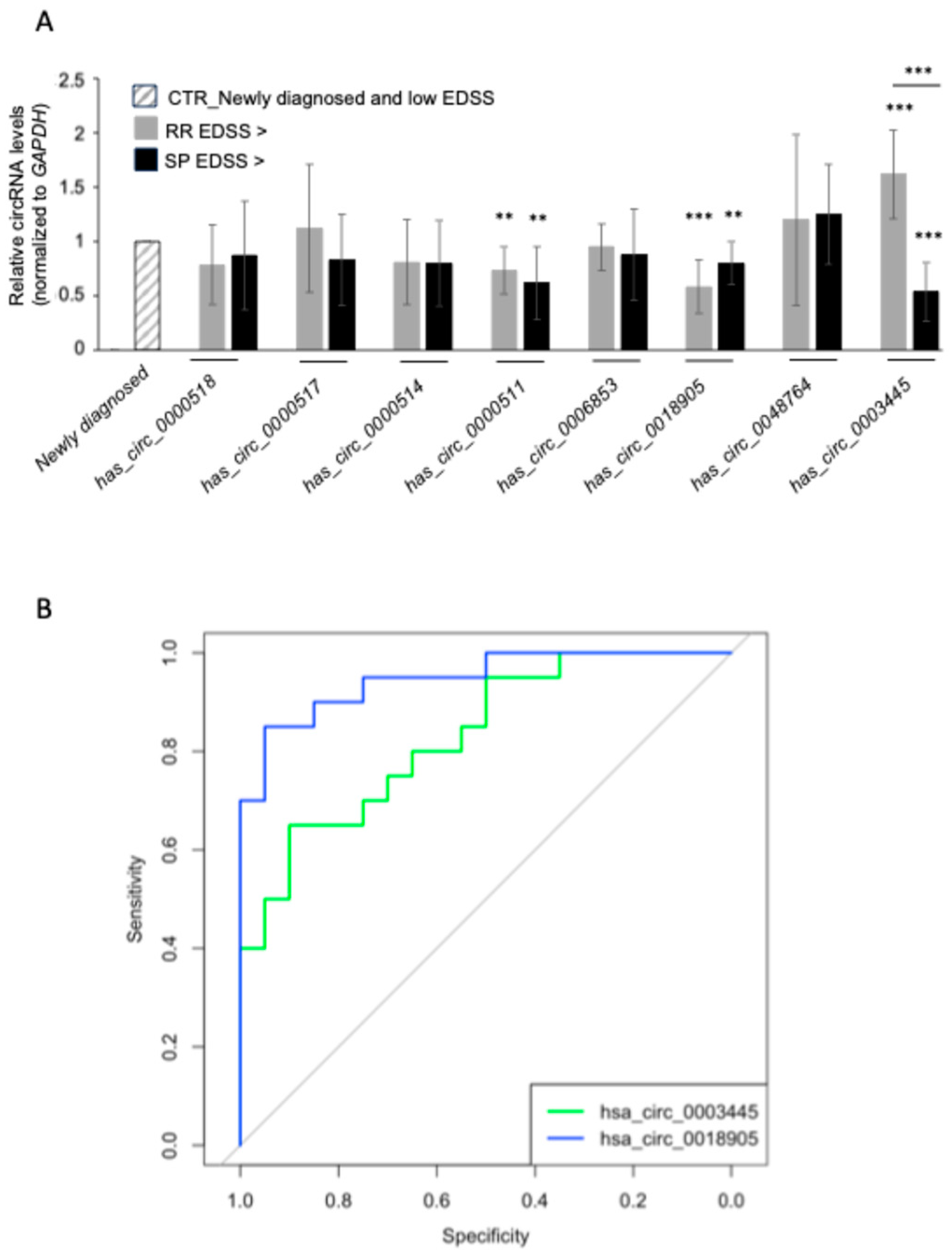
3.5. PBMC circRNA Expression and Correlation with Disease Severity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shah, A.; Panchal, V.; Patel, K.; Alimohamed, Z.; Kaka, N.; Sethi, Y.; Patel, N. Pathogenesis and management of multiple sclerosis revisited. Dis.-A-Mon. 2023, 69, 101497. [Google Scholar] [CrossRef]
- Sand, I.K. Classification, diagnosis, and differential diagnosis of multiple sclerosis. Curr. Opin. Neurol. 2015, 28, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Coghe, G.; Lorefice, L.; Fenu, G.; Cocco, E. Infections and Multiple Sclerosis: From the World to Sardinia, From Sardinia to the World. Front. Immunol. 2021, 12, 728677. [Google Scholar] [CrossRef] [PubMed]
- Urru, S.A.M.; Antonelli, A.; Sechi, G.M.; MS Working Group. Prevalence of multiple sclerosis in Sardinia: A systematic cross-sectional multi-source survey. Mult. Scler. J. 2019, 26, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2016, 13, 25–36. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Steri, M.; Orrù, V.; Idda, M.L.; Pitzalis, M.; Pala, M.; Zara, I.; Sidore, C.; Faà, V.; Floris, M.; Deiana, M.; et al. Overexpression of the cytokine BAFF and autoimmunity risk. N. Engl. J. Med. 2017, 376, 1615–1626. [Google Scholar] [CrossRef]
- Mohammed, E.M. Environmental Influencers, MicroRNA, and Multiple Sclerosis. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573519894955. [Google Scholar] [CrossRef] [PubMed]
- Paraboschi, E.M.; Cardamone, G.; Soldà, G.; Duga, S.; Asselta, R. Interpreting Non-coding Genetic Variation in Multiple Sclerosis Genome-Wide Associated Regions. Front. Genet. 2018, 9, 647. [Google Scholar] [CrossRef]
- Mohammed, E.M. Circular RNA in Multiple Sclerosis: Pathogenicity and Potential Biomarker Development: A Systematic Review. Epigenetics Insights 2023, 16, 25168657231213195. [Google Scholar] [CrossRef]
- Li, H.; Ma, X.; Li, H. Intriguing circles: Conflicts and controversies in circular RNA research. Wiley Interdiscip. Rev. RNA 2019, 10, e1538. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Guo, X.Y.; Shang, Q.J.; Gao, P. The biogenesis, biological functions and modification of Circular RNAs. Exp. Mol. Pathol. 2023, 131, 104861. [Google Scholar] [CrossRef] [PubMed]
- Hanan, M.; Soreq, H.; Kadener, S. CircRNAs in the brain. RNA Biol. 2016, 14, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.B.; Tsitsipatis, D.; Gorospe, M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol. Cell 2022, 82, 2252–2266. [Google Scholar] [CrossRef] [PubMed]
- Cardamone, G.; Paraboschi, E.M.; Rimoldi, V.; Duga, S.; Soldà, G.; Asselta, R. The characterization of GSDMB splicing and backsplicing profiles identifies novel isoforms and a circular rna that are dysregulated in multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 576. [Google Scholar] [CrossRef] [PubMed]
- Iparraguirre, L.; Muñoz-Culla, M.; Prada-Luengo, I.; Castillo-Triviño, T.; Olascoaga, J.; Otaegui, D. Circular RNA profiling reveals that circular RNAs from ANXA2 can be used as new biomarkers for multiple sclerosis. Hum. Mol. Genet. 2017, 26, 3564–3572. [Google Scholar] [CrossRef]
- Holdt, L.M.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.A.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.H.; Nicolaou, A.; et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat. Commun. 2016, 7, 12429. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.J.; Esteller, M. Non-coding RNAs, epigenetics, and cancer: Tying it all together. Cancer Metastasis Rev. 2018, 37, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. Enright-2003-Genomebiol. Pdf. Genome Biol. 2003, 5, R1. [Google Scholar]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Lin, Y.-C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA–target interactions. Nucleic Acids Res. 2021, 50, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models. Genome Res. 1971, 13, 426. [Google Scholar] [CrossRef]
- Hecker, M.; Fitzner, B.; Boxberger, N.; Putscher, E.; Engelmann, R.; Bergmann, W.; Müller, M.; Ludwig-Portugall, I.; Schwartz, M.; Meister, S.; et al. Transcriptome alterations in peripheral blood B cells of patients with multiple sclerosis receiving immune reconstitution therapy. J. Neuroinflamm. 2023, 20, 181. [Google Scholar] [CrossRef]
- Ikeda, Y.; Morikawa, S.; Nakashima, M.; Yoshikawa, S.; Taniguchi, K.; Sawamura, H.; Suga, N.; Tsuji, A.; Matsuda, S. CircRNAs and RNA-Binding Proteins Involved in the Pathogenesis of Cancers or Central Nervous System Disorders. Non-Coding RNA 2023, 9, 23. [Google Scholar] [CrossRef]
- Griffin, E.N.; Jucius, T.; Sim, S.-E.; Harris, B.S.; Heinz, S.; Ackerman, S.L. RREB1 regulates neuronal proteostasis and the microtubule network. Sci. Adv. 2024, 10, eadh3929. [Google Scholar] [CrossRef]
- Perga, S.; Montarolo, F.; Martire, S.; Berchialla, P.; Malucchi, S.; Bertolotto, A. Anti-inflammatory genes associated with multiple sclerosis: A gene expression study. J. Neuroimmunol. 2015, 279, 75–78. [Google Scholar] [CrossRef]
- Grant, S.F.; Qu, H.-Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Taback, S.P.; Frackelton, E.C.; Eckert, A.W.; et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 Diabetes. Diabetes 2009, 58, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Perdigones, N.; Martín, E.; Robledo, G.; Lamas, J.R.; Taxonera, C.; Díaz-Rubio, M.; de la Concha, E.G.; López-Nevot, M.A.; García, A.; Gómez-García, M.; et al. Study of chromosomal region 5p13.1 in Crohn’s disease, ulcerative colitis, and rheumatoid arthritis. Hum. Immunol. 2010, 71, 826–828. [Google Scholar] [CrossRef]
- Medici, M.; Porcu, E.; Pistis, G.; Teumer, A.; Brown, S.J.; Jensen, R.A.; Rawal, R.; Roef, G.L.; Plantinga, T.S.; Vermeulen, S.H.; et al. Identification of Novel Genetic Loci Associated with Thyroid Peroxidase Antibodies and Clinical Thyroid Disease. PLoS Genet. 2014, 10, e1004123. [Google Scholar] [CrossRef]
- Zurawska, A.E.; Mycko, M.P.; Selmaj, I.; Raine, C.S.; Selmaj, K.W. Multiple Sclerosis. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e1041. [Google Scholar] [CrossRef]
- Potokar, M.; Jorgačevski, J. Plectin in the central nervous system and a putative role in brain astrocytes. Cells 2021, 10, 2353. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.; Marquez, S.M.; Pace, N.R. RNase P: Interface of the RNA and protein worlds. Trends Biochem. Sci. 2006, 31, 333–341. [Google Scholar] [CrossRef]
- Wang, H.; Gao, X.; Yu, S.; Wang, W.; Liu, G.; Jiang, X.; Sun, D. Circular RNAs regulate parental gene expression: A new direction for molecular oncology research. Front. Oncol. 2022, 12, 947775. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, X.; Cui, X.; Hu, J.; Wang, L.; Xue, F.; Guo, S.; Wang, X. Circ_0000518 Promotes Macrophage/Microglia M1 Polarization via the FUS/CaMKKβ/AMPK Pathway to Aggravate Multiple Sclerosis. Neuroscience 2022, 490, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Peng, R.; Chen, W.; Fu, X.; Zhang, L.; Peng, H.; Zhang, Z. Long non-coding RNA Rpph1 promotes inflammation and proliferation of mesangial cells in diabetic nephropathy via an interaction with Gal-3. Cell Death Dis. 2019, 10, 526. [Google Scholar] [CrossRef]
- Liang, Z.X.; Liu, H.S.; Wang, F.W.; Xiong, L.; Zhou, C.; Hu, T.; He, X.W.; Wu, X.J.; Xie, D.; Wu, X.R.; et al. LncRNA RPPH1 promotes colorectal cancer metastasis by interacting with TUBB3 and by promoting exosomes-mediated macrophage M2 polarization. Cell Death Dis. 2019, 10, 829. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Sun, Z.; Jia, H.; Luo, H.; Ye, X.; Wu, Q.; Xiong, Y.; Zhang, W.; Wan, J. Rpph1 upregulates CDC42 expression and promotes hippocampal neuron dendritic spine formation by competing with miR-330-5p. Front. Mol. Neurosci. 2017, 10, 27. [Google Scholar] [CrossRef]
- Gu, R.; Wang, L.; Tang, M.; Li, S.-R.; Liu, R.; Hu, X. LncRNA Rpph1 protects amyloid-β induced neuronal injury in SK-N-SH cells via miR-122/Wnt1 axis. Int. J. Neurosci. 2019, 130, 443–453. [Google Scholar] [CrossRef]
- Tafesse, F.G.; Vacaru, A.M.; Bosma, E.F.; Hermansson, M.; Jain, A.; Hilderink, A.; Somerharju, P.; Holthuis, J.C.M. Sphingomyelin synthase-related protein SMSr is a suppressor of ceramide-induced mitochondrial apoptosis. J. Cell Sci. 2013, 127, 445–454. [Google Scholar] [CrossRef]
- Cabukusta, B.; Nettebrock, N.T.; Kol, M.; Hilderink, A.; Tafesse, F.G.; Holthuis, J.C. Ceramide phosphoethanolamine synthase SMSr is a target of caspase-6 during apoptotic cell death. Biosci. Rep. 2017, 37, BSR20170867. [Google Scholar] [CrossRef]
- Filippatou, A.G.; Moniruzzaman, M.; Sotirchos, E.S.; Fitzgerald, K.C.; Kalaitzidis, G.; Lambe, J.; Vasileiou, E.; Saidha, S.; Prince, J.L.; Haughey, N.; et al. Serum ceramide levels are altered in multiple sclerosis. Mult. Scler. J. 2020, 27, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; Hogan, E. Molecular and immunogenic features of myelin lipids: Incitants or modulators of multiple sclerosis? Mult. Scler. J. 2009, 15, 1011–1029. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mangano, K.; Di Marco, R.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Upregulated expression of macrophage migration inhibitory factor, its analogue D-dopachrome tautomerase, and the CD44 receptor in peripheral CD4 T cells from clinically isolated syndrome patients with rapid conversion to clinical defined multiple sclerosis. Medicina 2019, 55, 667. [Google Scholar] [CrossRef]
- Alaya, F.; Baraket, G.; Adediran, D.A.; Cuttler, K.; Ajiboye, I.; Kivumbi, M.T.; Sitharam, N.; Awe, O.I. Multiple Sclerosis Stages and their Differentially Expressed Genes: A Bioinformatics Analysis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef]
- Camelo, S.; Iglesias, A.H.; Hwang, D.; Due, B.; Ryu, H.; Smith, K.; Gray, S.G.; Imitola, J.; Duran, G.; Assaf, B.; et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 164, 10–21. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Hu, Z.; Xiao, H.; Zhou, F.; Yang, B. The tale of histone modifications and its role in multiple sclerosis. Hum. Genom. 2018, 12, 31. [Google Scholar] [CrossRef]
- Maltby, V.E.; Lea, R.A.; Burnard, S.; Xavier, A.; Van Cao, T.; White, N.; Kennedy, D.; Groen, K.; Sanders, K.A.; Seeto, R.; et al. Epigenetic differences at the HTR2A locus in progressive multiple sclerosis patients. Sci. Rep. 2020, 10, 22217. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, B.; Huang, S.; Zhao, L. Roles of circular RNAs in immune regulation and autoimmune diseases. Cell Death Dis. 2019, 10, 503. [Google Scholar] [CrossRef]
- Carlos-Reyes, Á.; Romero-Garcia, S.; Contreras-Sanzón, E.; Ruiz, V.; Prado-Garcia, H. Role of Circular RNAs in the Regulation of Immune Cells in Response to Cancer Therapies. Front. Genet. 2022, 13, 823238. [Google Scholar] [CrossRef]
- Chen, X.; Yang, T.; Wang, W.; Xi, W.; Zhang, T.; Li, Q.; Yang, A.; Wang, T. Circular RNAs in immune responses and immune diseases. Theranostics 2019, 9, 588–607. [Google Scholar] [CrossRef]
- Gaffo, E.; Boldrin, E.; Molin, A.D.; Bresolin, S.; Bonizzato, A.; Trentin, L.; Frasson, C.; Debatin, K.-M.; Meyer, L.H.; Kronnie, G.T.; et al. Circular RNA differential expression in blood cell populations and exploration of circRNA deregulation in pediatric acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 14670. [Google Scholar] [CrossRef]
- Maass, P.G.; Glažar, P.; Memczak, S.; Dittmar, G.; Hollfinger, I.; Schreyer, L.; Sauer, A.V.; Toka, O.; Aiuti, A.; Luft, F.C.; et al. A map of human circular RNAs in clinically relevant tissues. J. Mol. Med. 2017, 95, 1179–1189. [Google Scholar] [CrossRef]
- Junker, A.; Hohlfeld, R.; Meinl, E. The emerging role of microRNAs in multiple sclerosis. Nat. Rev. Neurol. 2010, 7, 56–59. [Google Scholar] [CrossRef]
- Zang, J.; Lu, D.; Xu, A. The interaction of circRNAs and RNA binding proteins: An important part of circRNA maintenance and function. J. Neurosci. Res. 2018, 98, 87–97. [Google Scholar] [CrossRef]
- Lodde, V.; Floris, M.; Zoroddu, E.; Zarbo, I.R.; Idda, M.L. RNA-binding proteins in autoimmunity: From genetics to molecular biology. Wiley Interdiscip. Rev. RNA 2023, 14, e1772. [Google Scholar] [CrossRef]
- Li, J.; Durose, W.W.; Ito, J.; Kakita, A.; Iguchi, Y.; Katsuno, M.; Kunisawa, K.; Shimizu, T.; Ikenaka, K. Exploring the factors underlying remyelination arrest by studying the post-transcriptional regulatory mechanisms of cystatin F gene. J. Neurochem. 2020, 157, 2070–2090. [Google Scholar] [CrossRef]
- Kiritsi, D.; Tsakiris, L.; Schauer, F. Plectin in Skin Fragility Disorders. Cells 2021, 10, 2738. [Google Scholar] [CrossRef]
- Orton, S.M.; Sangha, A.; Gupta, M.; Martens, K.; Metz, L.M.; de Koning, A.P.J.; Pfeffer, G. Expression of risk genes linked to vitamin D receptor super-enhancer regions and their association with phenotype severity in multiple sclerosis. Front. Neurol. 2022, 13, 1064008. [Google Scholar] [CrossRef]
- Najafi, S.; Zarch, S.M.A.; Majidpoor, J.; Pordel, S.; Aghamiri, S.; Rasul, M.F.; Asemani, Y.; Vakili, O.; Mohammadi, V.; Movahedpour, A.; et al. Recent insights into the roles of circular RNAs in human brain development and neurologic diseases. Int. J. Biol. Macromol. 2023, 225, 1038–1048. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, J.; Yang, W.; Ye, W.-C. CircRNAs in colorectal cancer: Potential biomarkers and therapeutic targets. Cell Death Dis. 2023, 14, 353. [Google Scholar] [CrossRef]
- Lodde, V.; Murgia, G.; Simula, E.R.; Steri, M.; Floris, M.; Idda, M.L. Long noncoding RNAs and circular RNAs in autoimmune diseases. Biomolecules 2020, 10, 1044. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT-(q)PCR Primers | Primer Sequences (5′-3′) |
|---|---|
| hsa_circ_0000518 | FW: CATGCCTACATTGCCCCAGA |
| RV: CAGACCTTCCCAAGGGACAT | |
| hsa_circ_0000517 | FW: GGGAGGTGAGTTCCCAGAG |
| RV: CAGGGAGAGCCCTGTTAGG | |
| hsa_circ_0000514 | FW: GAGCTTGGAACAGACTCACG |
| RV: CATCTCCTGCCCAGTCTGA | |
| hsa_circ_0000511 | FW: CCTCCTTTGCCGGAGCTT |
| RV: GGTCCACGGCATCTCCTG | |
| hsa_circ_0006853 | FW: TGAGTTCAATGGCTGAGGTG |
| RV: GTTCCAAGCTCCGGCAAA | |
| hsa_circ_0111998 | FW: CAGCACTCCACAGCATCCACTA |
| RV: TCCATTTCAATGGTAGCCTGCA | |
| hsa_circ_0018905 | FW: TGTTTGTGACCTCCCTCTCC |
| RV: CTTCATCCTTCAGCCACACA | |
| hsa_circ_0048764 | FW: AAGCTGCTGCCAAGAAAGAC |
| RV: ATCCTCCAACCTGCACAGAG | |
| hsa_circ_0003445 | FW: AGGAGCAGGAGCTGGAGAA |
| RV: GTGCTGGGCATGTGGTTC | |
| RPPH1 | FW: CTGTCACTCCACTCCCATGT |
| RV: TTCTCTGGGAACTCACCTCC | |
| LYLPLAL1 | FW: CTGCCCAGAACACCTTGAATC |
| RV: TCCTGTTCTTCTTGATGCCAC | |
| SAMD8 | FW: TATGATCTCCGGTCTCCTCCT |
| RV: TGTCACTGTTGTAGCCCATCT | |
| RPL36 | FW: GACCAAACACACCAAGTTCGT |
| RV: TAAATTTGAGGGCCCGTTTGT | |
| HDAC4 | FW: AAAACGCAGCACAGTTCCC |
| RV: GTCATCTTTGGCGTCGTACA | |
| GAPDH | FW: ATTTGGTCGTATTGGGCGCC |
| RV: TTGAGGTCAATGAAGGGGTC |
| Discovery Set | HC | MS |
| No. tot | 5 | 5 |
| Female | 3 | 3 |
| Male | 2 | 2 |
| Age, yrs, mean ± SD | 34.8 ± 12.49 | 34.8 ± 12.49 |
| Disease duration, mean ± SD | na | at diagnosis |
| EDSS, mean ± SD | na | 2.3 ± 1.01 |
| Validation Set | HC | MS |
| No. tot | 20 | 20 |
| Female | 10 | 12 |
| Male | 10 | 8 |
| Age, yrs, mean ± SD | 48.75 ± 15.71 | 43.3 ± 14.23 |
| Disease duration, mean ± SD | na | 3.21 ± 3.89 |
| EDSS, mean ± SD | na | 2.5 ± 0.81 |
| MS Type | RRMS |
| Disease Type | RRMS Edss < 4.5 | RRMS Edss > 4.5 | SPMS Edss > 4.5 |
|---|---|---|---|
| No. tot | 8 | 6 | 8 |
| Female | 6 | 5 | 4 |
| Male | 2 | 1 | 4 |
| Age, yrs, mean ± SD | 34.5 ± 11.95 | 64.3 ± 3.8 | 67.4 ± 7.2 |
| Disease duration, mean ± SD | at diagnosis | 25.8 ± 16.6 | 27.4 ± 12.1 |
| EDSS, mean ± SD | 2.3 ± 0.51 | 6 ± 0.70 | 6.3 ± 0.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodde, V.; Zarbo, I.R.; Farina, G.; Masia, A.; Solla, P.; Campesi, I.; Delogu, G.; Muroni, M.R.; Tsitsipatis, D.; Gorospe, M.; et al. Identification of hsa_circ_0018905 as a New Potential Biomarker for Multiple Sclerosis. Cells 2024, 13, 1668. https://doi.org/10.3390/cells13191668
Lodde V, Zarbo IR, Farina G, Masia A, Solla P, Campesi I, Delogu G, Muroni MR, Tsitsipatis D, Gorospe M, et al. Identification of hsa_circ_0018905 as a New Potential Biomarker for Multiple Sclerosis. Cells. 2024; 13(19):1668. https://doi.org/10.3390/cells13191668
Chicago/Turabian StyleLodde, Valeria, Ignazio Roberto Zarbo, Gabriele Farina, Aurora Masia, Paolo Solla, Ilaria Campesi, Giuseppe Delogu, Maria Rosaria Muroni, Dimitrios Tsitsipatis, Myriam Gorospe, and et al. 2024. "Identification of hsa_circ_0018905 as a New Potential Biomarker for Multiple Sclerosis" Cells 13, no. 19: 1668. https://doi.org/10.3390/cells13191668
APA StyleLodde, V., Zarbo, I. R., Farina, G., Masia, A., Solla, P., Campesi, I., Delogu, G., Muroni, M. R., Tsitsipatis, D., Gorospe, M., Floris, M., & Idda, M. L. (2024). Identification of hsa_circ_0018905 as a New Potential Biomarker for Multiple Sclerosis. Cells, 13(19), 1668. https://doi.org/10.3390/cells13191668