
Investigating the p21 Ubiquitin-Independent Degron Reveals a Dual Degron Module Regulating p21 Degradation and Function



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Cloning and Construct Design
2.3. Transfection and Cell Harvest
2.3.1. Immunoblot Analysis
2.3.2. Luciferase Complementation Assay
2.3.3. p21-CRISPR and Construct Design
2.3.4. Cell Proliferation Assay (XTT Based)
2.3.5. Cell-Cycle Analysis
2.3.6. Trapper Binding to Peptide Array
2.4. Statistical Analysis
3. Results
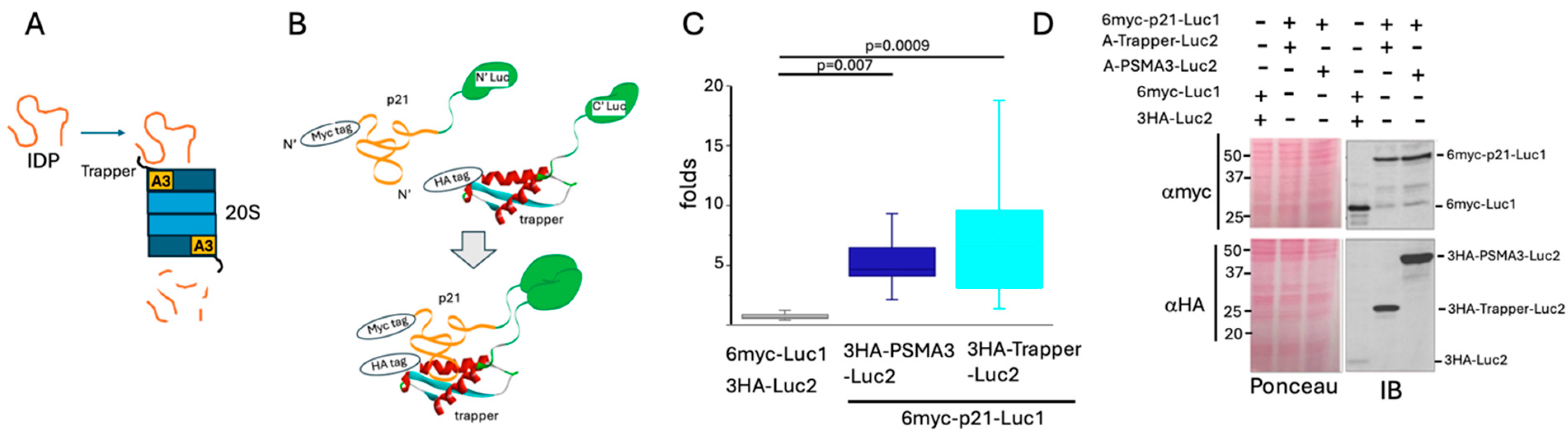
3.1. p21 Interacts with the PSMA3 Trapper in the Cells
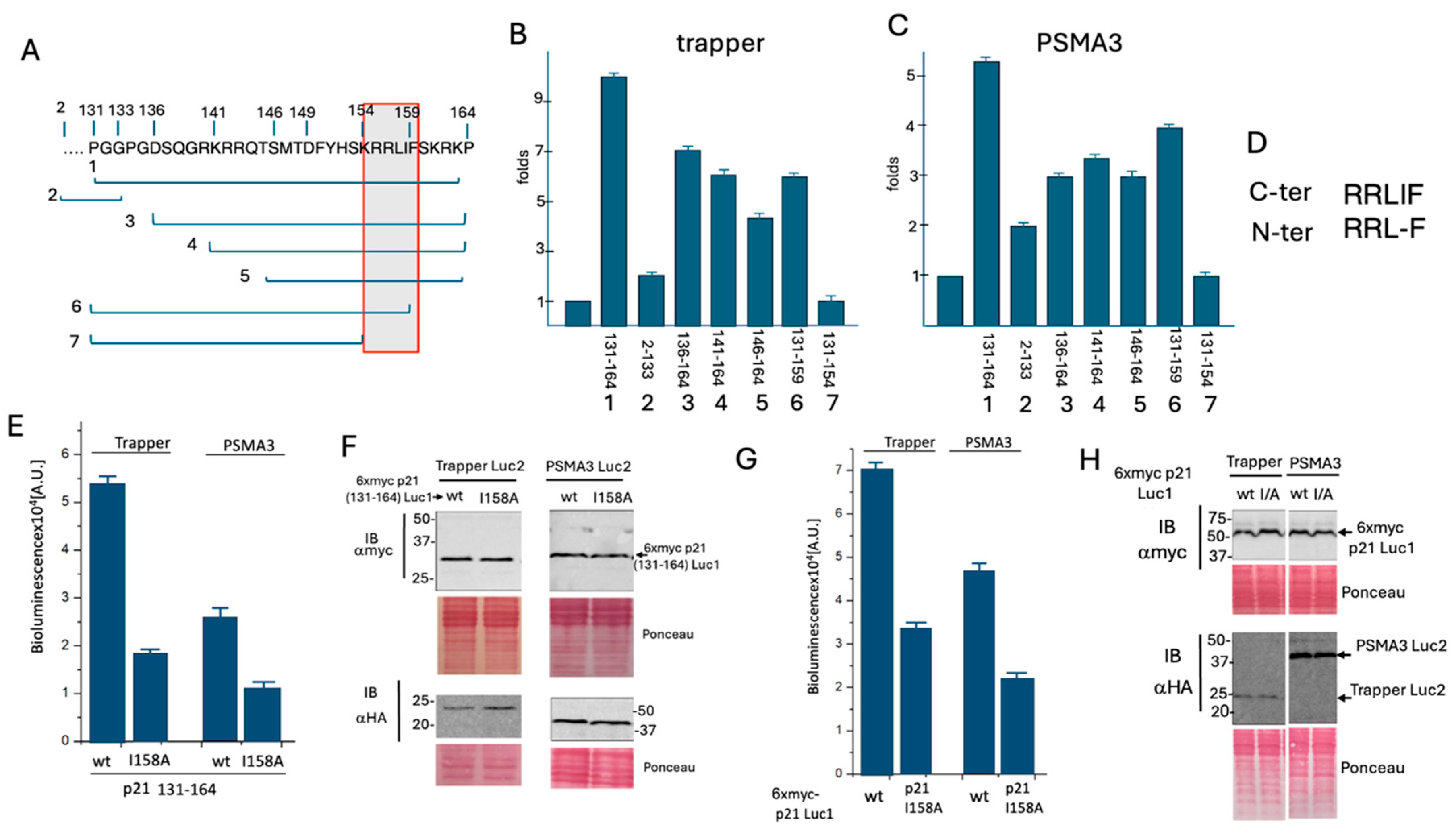
3.2. Delineation of the Critical p21 Motif in Binding the PSMA3 Trapper
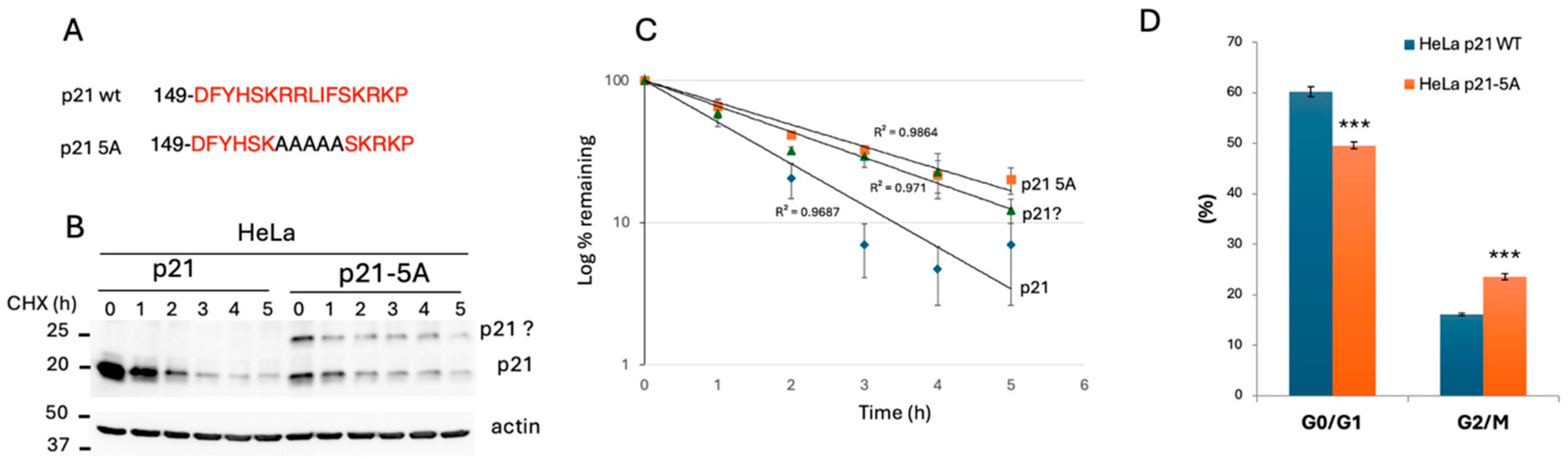
3.3. Analysis of Edited HEK293 Cells Highlighted the Important Role of the p21 C-Terminus in Regulating p21 Decay and Cell Growth
3.4. Under DNA Damage Conditions, the Stable p21 Mutants Efficiently Induce the Expression of Senescence Hallmark Genes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Dutto, I.; Tillhon, M.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Biology of the cell cycle inhibitor p21(CDKN1A): Molecular mechanisms and relevance in chemical toxicology. Arch. Toxicol. 2015, 89, 155–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fisher, J.C.; Mathew, R.; Ou, L.; Otieno, S.; Sublet, J.; Xiao, L.; Chen, J.; Roussel, M.F.; Kriwacki, R.W. Intrinsic disorder mediates the diverse regulatory functions of the Cdk inhibitor p21. Nat. Chem. Biol. 2011, 7, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Adams, P.D.; Sellers, W.R.; Sharma, S.K.; Wu, A.D.; Nalin, C.M.; Kaelin, W.G. Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol. Cell. Biol. 1996, 16, 6623–6633. [Google Scholar] [CrossRef]
- Chen, J.; Saha, P.; Kornbluth, S.; Dynlacht, B.D.; Dutta, A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol. Cell. Biol. 1996, 16, 4673–4682. [Google Scholar] [CrossRef]
- Mansilla, S.F.; de la Vega, M.B.; Calzetta, N.L.; Siri, S.O.; Gottifredi, V. CDK-Independent and PCNA-Dependent Functions of p21 in DNA Replication. Genes 2020, 11, 593. [Google Scholar] [CrossRef]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef]
- Zhu, H.; Nie, L.; Maki, C.G. Cdk2-dependent Inhibition of p21 stability via a C-terminal cyclin-binding motif. J. Biol. Chem. 2005, 280, 29282–29288. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Touitou, R.; Richardson, J.; Bose, S.; Nakanishi, M.; Rivett, J.; Allday, M.J. A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J. 2001, 20, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGγ proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hunter, T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 2010, 9, 2342–2352. [Google Scholar] [CrossRef]
- Sheaff, R.J.; Singer, J.D.; Swanger, J.; Smitherman, M.; Roberts, J.M.; Clurman, B.E. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol. Cell 2000, 5, 403–410. [Google Scholar] [CrossRef]
- Bloom, J.; Amador, V.; Bartolini, F.; DeMartino, G.; Pagano, M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell 2003, 115, 71–82. [Google Scholar] [CrossRef]
- Chen, X.; Chi, Y.; Bloecher, A.; Aebersold, R.; Clurman, B.E.; Roberts, J.M. N-acetylation and ubiquitin-independent proteasomal degradation of p21(Cip1). Mol. Cell 2004, 16, 839–847. [Google Scholar] [CrossRef]
- Bendjennat, M.; Boulaire, J.; Jascur, T.; Brickner, H.; Barbier, V.; Sarasin, A.; Fotedar, A.; Fotedar, R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003, 114, 599–610. [Google Scholar] [CrossRef]
- Lee, H.; Zeng, S.X.; Lu, H. UV Induces p21 rapid turnover independently of ubiquitin and Skp2. J. Biol. Chem. 2006, 281, 26876–26883. [Google Scholar] [CrossRef]
- Liu, C.-W.; Corboy, M.J.; DeMartino, G.N.; Thomas, P.J. Endoproteolytic activity of the proteasome. Science 2003, 299, 408–411. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Myers, N.; Moscovitz, O.; Sharon, M.; Prilusky, J.; Shaul, Y. Thermo-resistant intrinsically disordered proteins are efficient 20S proteasome substrates. Mol. Biosyst. 2012, 8, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Sdek, P.; Ying, H.; Chang, D.L.F.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Xiao, Z.-X.J. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 2005, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Myers, N.; Steinberger, S.; Adler, J.; Riutin, M.; Broennimann, K.; Reuven, N.; Shaul, Y. The C-Terminus of the PSMA3 Proteasome Subunit Preferentially Traps Intrinsically Disordered Proteins for Degradation. Cells 2022, 11, 3231. [Google Scholar] [CrossRef] [PubMed]
- Bond, S.R.; Naus, C.C. RF-Cloning.org: An online tool for the design of restriction-free cloning projects. Nucleic Acids Res. 2012, 40, W209–W213. [Google Scholar] [CrossRef] [PubMed]
- Michnick, S.W.; Ear, P.H.; Manderson, E.N.; Remy, I.; Stefan, E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 2007, 6, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.; Adler, J.; Broennimann, K.; Myers, N.; Shaul, Y. Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing. Biomolecules 2019, 9, 584. [Google Scholar] [CrossRef]
- Reuven, N.; Adler, J.; Myers, N.; Shaul, Y. CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing. Int. J. Mol. Sci. 2021, 22, 3741. [Google Scholar] [CrossRef]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novák, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 14728. [Google Scholar] [CrossRef]
- Ando, T.; Kawabe, T.; Ohara, H.; Ducommun, B.; Itoh, M.; Okamoto, T. Involvement of the interaction between p21 and proliferating cell nuclear antigen for the maintenance of G2/M arrest after DNA damage. J. Biol. Chem. 2001, 276, 42971–42977. [Google Scholar] [CrossRef]
- Chang, B.-D.; Watanabe, K.; Broude, E.V.; Fang, J.; Poole, J.C.; Kalinichenko, T.V.; Roninson, I.B. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 4291–4296. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Asher, G.; Paz, A.; Reuven, N.; Sussman, J.L.; Silman, I.; Shaul, Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins 2008, 70, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef]
- Kroker, A.J.; Bruning, J.B. p21 Exploits Residue Tyr151 as a Tether for High-Affinity PCNA Binding. Biochemistry 2015, 54, 3483–3493. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Walter, J.C. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 2009, 35, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. The nanny model for IDPs. Nat. Chem. Biol. 2009, 5, 778–781. [Google Scholar] [CrossRef]
- Nakakido, M.; Deng, Z.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. PRMT6 increases cytoplasmic localization of p21CDKN1A in cancer cells through arginine methylation and makes more resistant to cytotoxic agents. Oncotarget 2015, 6, 30957–30967. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Altschuler, S.J.; Wu, L.F. Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 2019, 178, 361–373.e12. [Google Scholar] [CrossRef] [PubMed]
- May, E.; Jenkins, J.R.; May, P. Endogenous HeLa p53 proteins are easily detected in HeLa cells transfected with mouse deletion mutant p53 gene. Oncogene 1991, 6, 1363–1365. [Google Scholar]
- Campisi, J.; di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Stewart-Ornstein, J.; Lahav, G. Dynamics of CDKN1A in single cells defined by an endogenous fluorescent tagging toolkit. Cell Rep. 2016, 14, 1800–1811. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riutin, M.; Erez, P.; Adler, J.; Biran, A.; Myers, N.; Shaul, Y. Investigating the p21 Ubiquitin-Independent Degron Reveals a Dual Degron Module Regulating p21 Degradation and Function. Cells 2024, 13, 1670. https://doi.org/10.3390/cells13191670
Riutin M, Erez P, Adler J, Biran A, Myers N, Shaul Y. Investigating the p21 Ubiquitin-Independent Degron Reveals a Dual Degron Module Regulating p21 Degradation and Function. Cells. 2024; 13(19):1670. https://doi.org/10.3390/cells13191670
Chicago/Turabian StyleRiutin, Marianna, Pnina Erez, Julia Adler, Assaf Biran, Nadav Myers, and Yosef Shaul. 2024. "Investigating the p21 Ubiquitin-Independent Degron Reveals a Dual Degron Module Regulating p21 Degradation and Function" Cells 13, no. 19: 1670. https://doi.org/10.3390/cells13191670