
Comparative Analysis of Plasma Protein Dynamics in Women with ST-Elevation Myocardial Infarction and Takotsubo Syndrome

 , , , , ,
, , , , ,  , , , and
, , , and
Abstract
1. Introduction
2. Methods
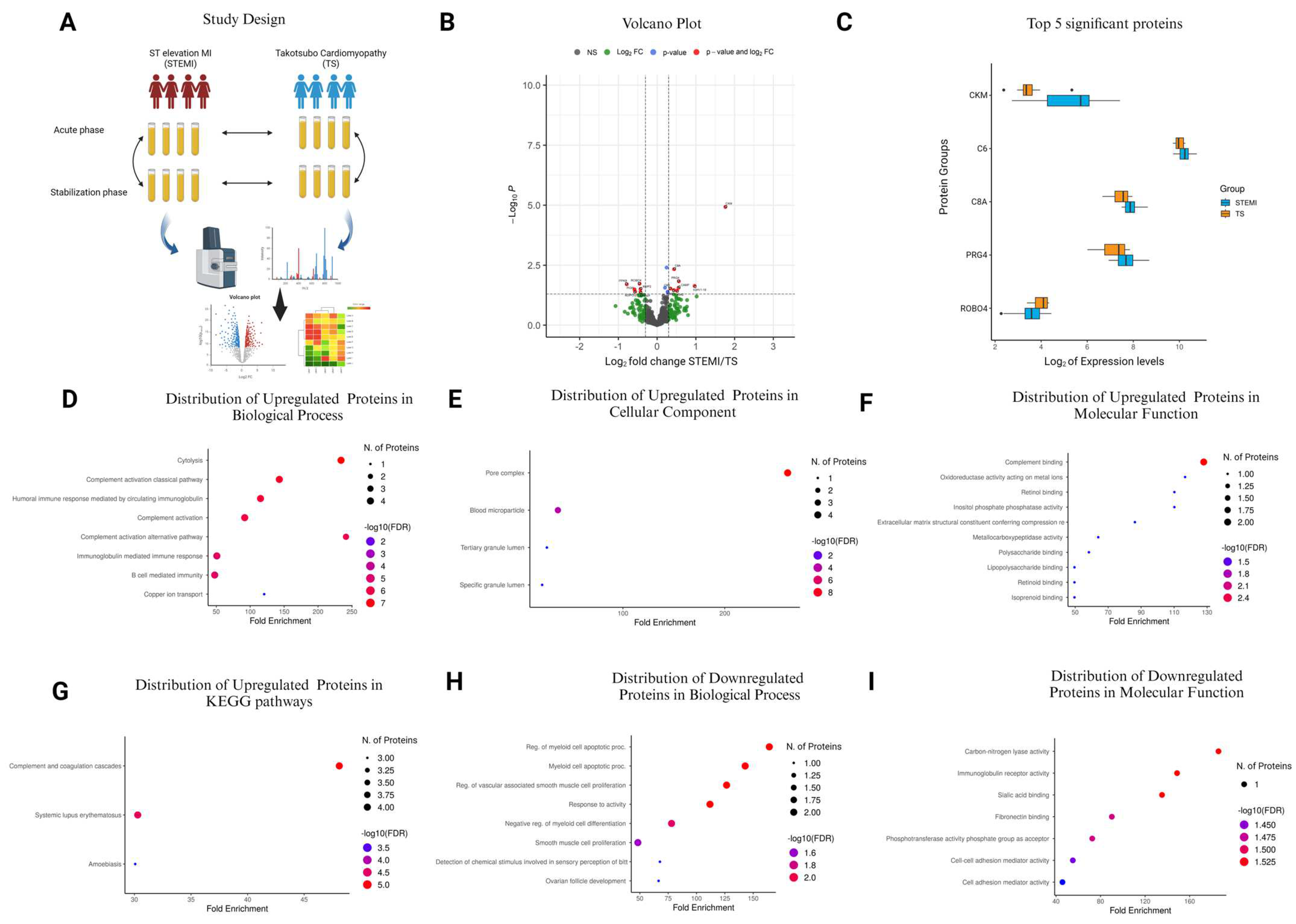
2.1. Study Design
2.2. Participant Recruitment and Selection
2.3. Baseline Characteristics
2.4. Blood Sampling and Plasma Collection
2.5. Proteomic Analysis
2.5.1. Sample Preparation
2.5.2. Identification and Quantification
2.5.3. Statistical Analysis
2.6. Bioinformatic Analysis
3. Results
3.1. Patient Characteristics
3.2. Clinical Outcomes
3.3. Laboratory Markers in the Acute Phase
3.4. Plasma Proteome of Women with STEMI and TS
3.5. Comparison of STEMI vs. TS in the Acute Phase
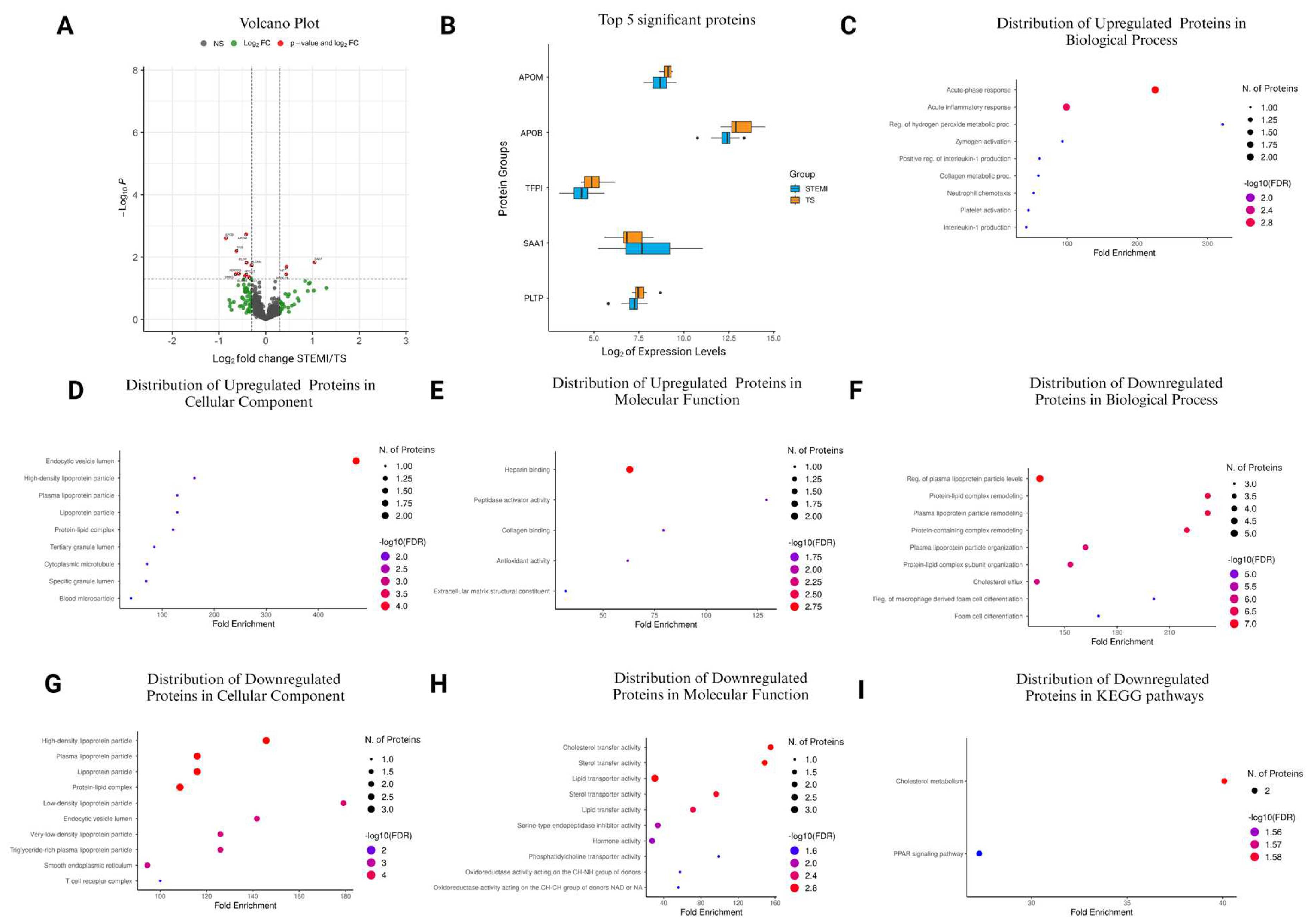
3.6. Comparison of STEMI vs. TS in the Stabilization Phase
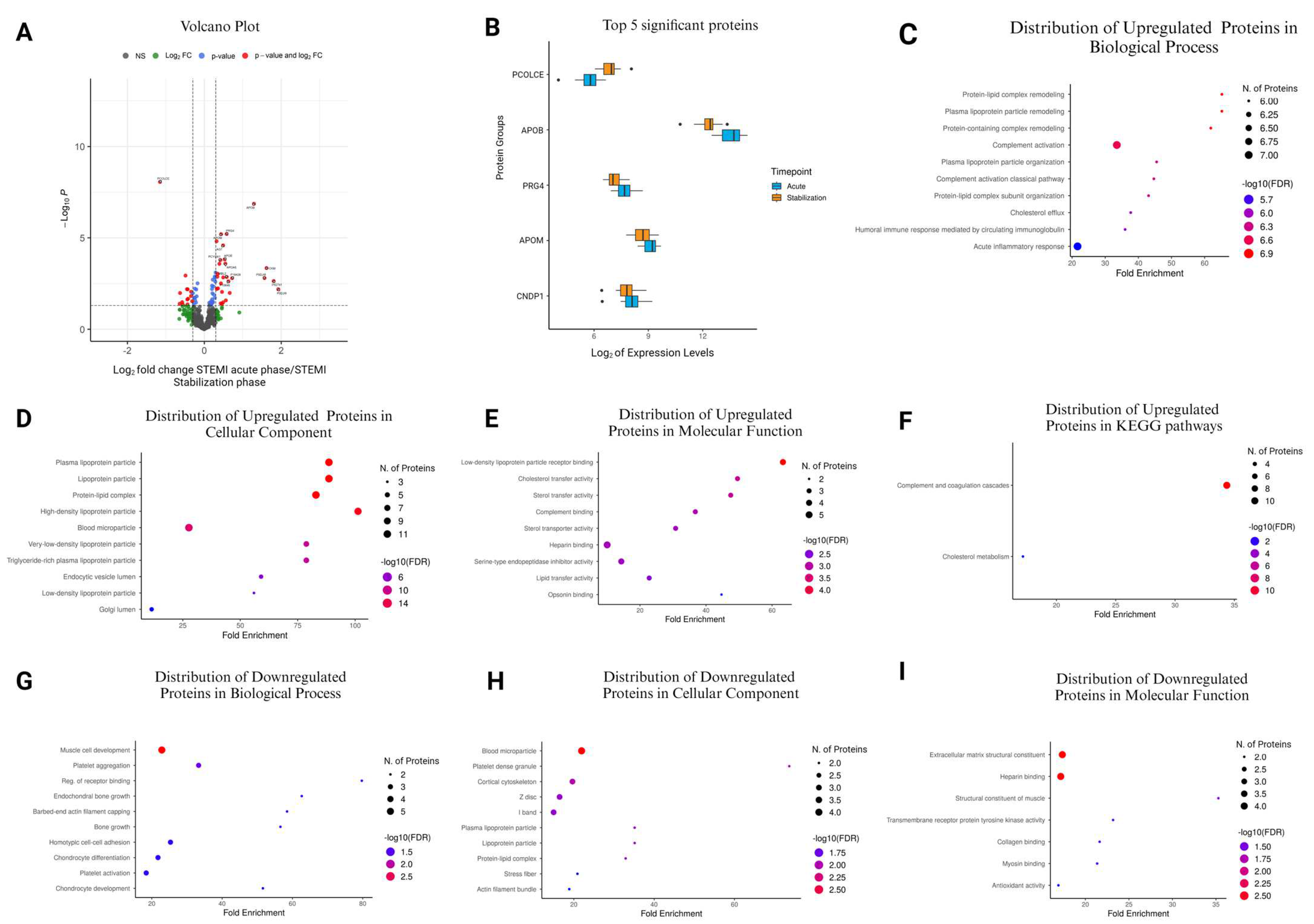
3.7. Comparison of STEMI Acute Phase vs. STEMI Stabilization Phase
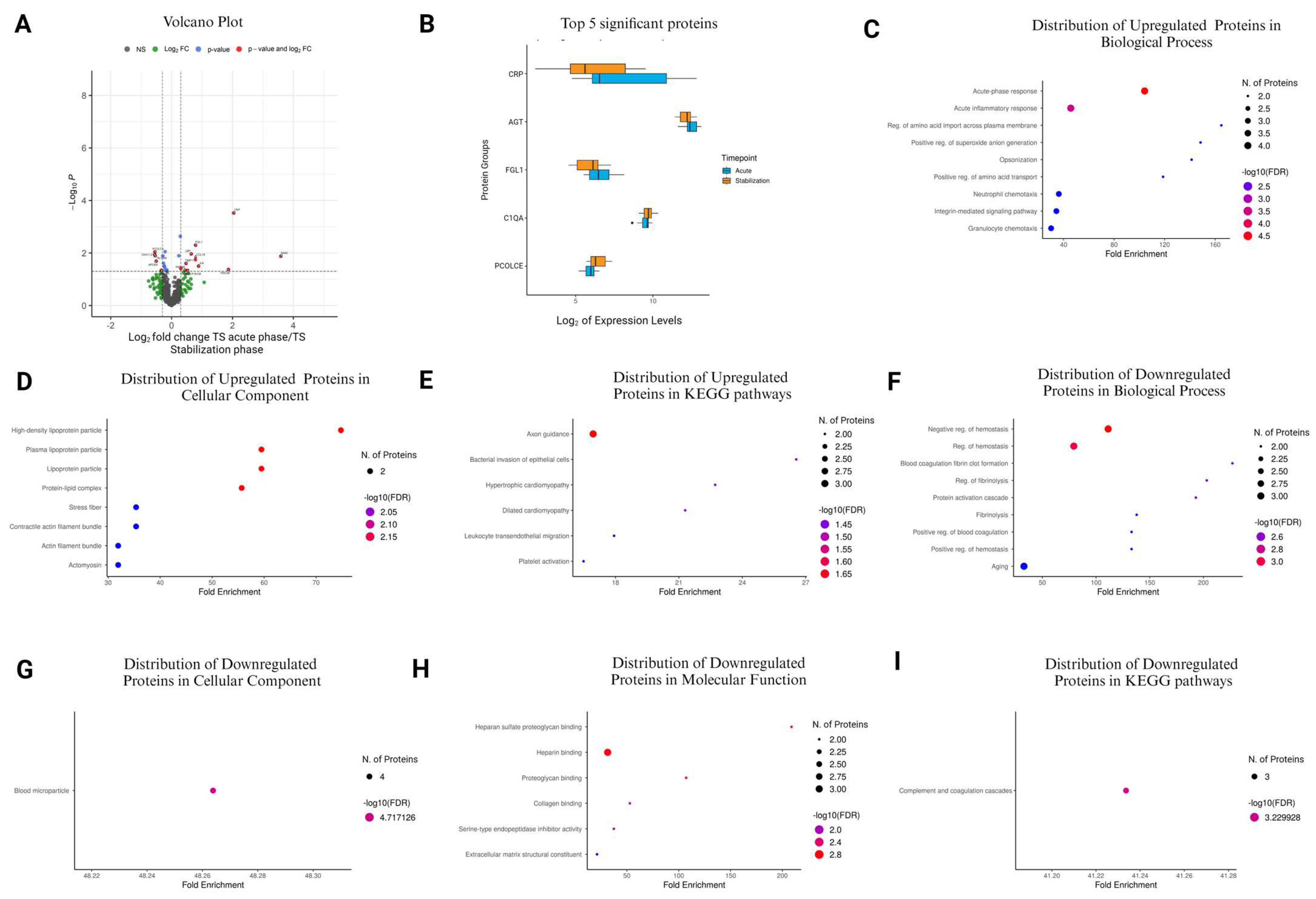
3.8. Comparison of TS Acute Phase vs. TS Stabilization Phase
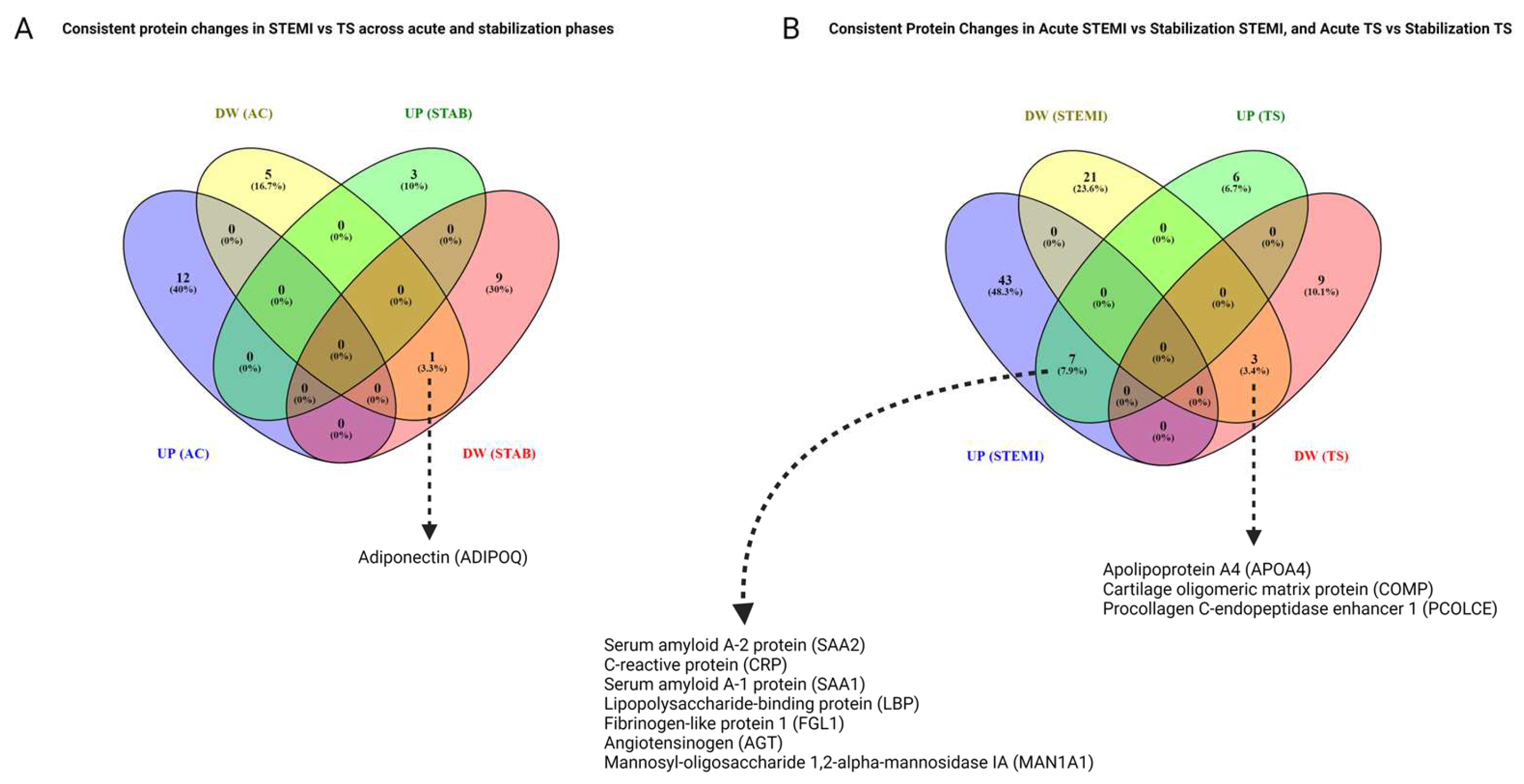
3.9. Consistent Proteomic Changes in the Acute Phase and Stabilization Phase, and Comparisons of STEMI and TS
3.10. Common Protein Changes in STEMI Acute Phase vs. Stabilization Phase and TS Acute Phase vs. Stabilization Phase
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Committee Approval
Abbreviations
| MI | Myocardial infarction |
| TS | Takotsubo syndrome |
| PCI | Percutaneous coronary intervention |
| STEMI | ST-elevation myocardial infarction |
| RI | Reperfusion injury |
| EDTA | Ethylenediaminetetraacetic acid |
| DTT | DL-dithiothreitol |
| IAA | Iodoacetamide |
| LC-MS/MS | Tandem mass spectrometry |
| GO | Gene Ontology |
| FDR | False discovery rate |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| HDL | High-density lipoprotein |
| COPD | Chronic obstructive pulmonary disease |
| hs-cTnI | High-sensitivity cardiac troponin I |
| hs-cTnT | High-sensitivity cardiac troponin T |
| NT-proBNP | N-terminal pro B-type natriuretic peptide |
| IQR | Interquartile range |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| HF | Heart failure |
| SMD | Standardized mean difference |
| BMI | Body mass index |
| ACEi | Angiotensin-converting enzyme inhibitor |
| ARB | Angiotensin receptor blocker |
| ASA | Acetylsalicylic acid |
| OAC | Oral anticoagulant |
| P2Y12i | P2Y12 receptor inhibitor |
| TIA | Transient ischemic attack |
References
- Lyon, A.R.; Citro, R.; Schneider, B.; Morel, O.; Ghadri, J.R.; Templin, C.; Omerovic, E. Pathophysiology of Takotsubo Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 902–921. [Google Scholar] [CrossRef] [PubMed]
- Pelliccia, F.; Kaski, J.C.; Crea, F.; Camici, P.G. Pathophysiology of Takotsubo Syndrome. Circulation 2017, 135, 2426–2441. [Google Scholar] [CrossRef] [PubMed]
- Budnik, M.; Kochanowski, J.; Piatkowski, R.; Peller, M.; Wojtera, K.; Gaska-Dzwonkowska, M.; Glowacka, P.; Karolczak, P.; Ochijewicz, D.; Opolski, G. Comparison of Complications and In-Hospital Mortality in Female Patients with Takotsubo Syndrome and ST-Segment Elevation Myocardial Infarction. J. Womens Health 2018, 27, 1513–1518. [Google Scholar] [CrossRef]
- Rawish, E.; Stiermaier, T.; Santoro, F.; Brunetti, N.D.; Eitel, I. Current Knowledge and Future Challenges in Takotsubo Syndrome: Part 1-Pathophysiology and Diagnosis. J. Clin. Med. 2021, 10, 479. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Mayr, M.; Gomes, A.V.; Delles, C.; Arrell, D.K.; Murphy, A.M.; Lange, R.A.; Costello, C.E.; Jin, Y.F.; Laskowitz, D.T.; et al. Transformative Impact of Proteomics on Cardiovascular Health and Disease: A Scientific Statement from the American Heart Association. Circulation 2015, 132, 852–872. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, D.; Li, Z.; Zhou, L.; Cen, T.; Wei, B.; Wei, L.; Wu, H.; Su, L.; Sooranna, S.R.; et al. A plasma proteomic approach in patients with heart failure after acute myocardial infarction: Insights into the pathogenesis and progression of the disease. Front. Cardiovasc. Med. 2023, 10, 1153625. [Google Scholar] [CrossRef]
- Chen, S.; Redfors, B.; Serruys, P.W.; Pieter Kappetein, A.; Crowley, A.; Ben-Yehuda, O.; Srdanovic, I.; Lembo, N.J.; Brown, W.M., 3rd; Sabik, J.F., 3rd; et al. Impact of renin-angiotensin system inhibitors after revascularization of patients with left main coronary artery disease. Coron. Artery Dis. 2022, 31, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Bossone, E.; Schneider, B.; Sechtem, U.; Citro, R.; Underwood, S.R.; Sheppard, M.N.; Figtree, G.A.; Parodi, G.; Akashi, Y.J.; et al. Current state of knowledge on Takotsubo syndrome: A Position Statement from the Taskforce on Takotsubo Syndrome of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2016, 18, 8–27. [Google Scholar] [CrossRef]
- Skowronek, P.; Thielert, M.; Voytik, E.; Tanzer, M.C.; Hansen, F.M.; Willems, S.; Karayel, O.; Brunner, A.D.; Meier, F.; Mann, M. Rapid and In-Depth Coverage of the (Phospho-)Proteome with Deep Libraries and Optimal Window Design for dia-PASEF. Mol. Cell Proteom. 2022, 21, 100279. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Watanabe, M.; Okamura, T.; Kokubo, Y.; Higashiyama, A.; Okayama, A. Elevated serum creatine kinase predicts first-ever myocardial infarction: A 12-year population-based cohort study in Japan, the Suita study. Int. J. Epidemiol. 2009, 38, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, K.S.; Park, J.L.; Tanhehco, E.J.; Booth, E.A.; Marks, R.M.; Lucchesi, B.R. Attenuation of interleukin-8 expression in C6-deficient rabbits after myocardial ischemia/reperfusion. J. Mol. Cell Cardiol. 1998, 30, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, H.; Huang, T. Quantitative proteomics reveal three potential biomarkers for risk assessment of acute myocardial infarction. Bioengineered 2022, 13, 4939–4950. [Google Scholar] [CrossRef] [PubMed]
- Squire, I.B.; Evans, J.; Ng, L.L.; Loftus, I.M.; Thompson, M.M. Plasma MMP-9 and MMP-2 following acute myocardial infarction in man: Correlation with echocardiographic and neurohumoral parameters of left ventricular dysfunction. J. Card. Fail. 2004, 10, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Li, H.; Shen, X.; Liu, R. Increased serum adiponectin predicts improved coronary flow and clinical outcomes in patients with ST-segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J. Clin. Lab. Anal. 2019, 33, e22864. [Google Scholar] [CrossRef]
- Song, C.; Hsu, K.; Yamen, E.; Yan, W.; Fock, J.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar] [CrossRef]
- Chiang, K.H.; Kao, Y.T.; Leu, H.B.; Huang, P.H.; Huang, S.S.; Cheng, T.M.; Pan, J.P. Higher post-acute myocardial infarction plasma haptoglobin level is associated with poor long-term overall survival. Int. J. Cardiol. 2017, 229, 102–107. [Google Scholar] [CrossRef]
- Ramunddal, T.; Lindbom, M.; Scharin-Tang, M.; Stillemark-Billton, P.; Boren, J.; Omerovic, E. Overexpression of apolipoprotein-B improves cardiac function and increases survival in mice with myocardial infarction. Biochem. Biophys. Res. Commun. 2009, 385, 336–340. [Google Scholar] [CrossRef]
- Cavusoglu, E.; Marmur, J.D.; Chhabra, S.; Hojjati, M.R.; Yanamadala, S.; Chopra, V.; Eng, C.; Jiang, X.C. Elevated baseline plasma phospholipid protein (PLTP) levels are an independent predictor of long-term all-cause mortality in patients with diabetes mellitus and known or suspected coronary artery disease. Atherosclerosis 2015, 239, 503–508. [Google Scholar] [CrossRef]
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left ventricular remodelling post-myocardial infarction: Pathophysiology, imaging, and novel therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef]
- Zhang, M.; Hou, Y.J.; Cavusoglu, E.; Lee, D.C.; Steffensen, R.; Yang, L.; Bashari, D.; Villamil, J.; Moussa, M.; Fernaine, G.; et al. MASP-2 activation is involved in ischemia-related necrotic myocardial injury in humans. Int. J. Cardiol. 2013, 166, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Orn, S.; Manhenke, C.; Ueland, T.; Damas, J.K.; Mollnes, T.E.; Edvardsen, T.; Aukrust, P.; Dickstein, K. C-reactive protein, infarct size, microvascular obstruction, and left-ventricular remodelling following acute myocardial infarction. Eur. Heart J. 2009, 30, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Orrem, H.L.; Nilsson, P.H.; Pischke, S.E.; Grindheim, G.; Garred, P.; Seljeflot, I.; Husebye, T.; Aukrust, P.; Yndestad, A.; Andersen, G.O.; et al. Acute heart failure following myocardial infarction: Complement activation correlates with the severity of heart failure in patients developing cardiogenic shock. ESC Heart Fail. 2018, 5, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Pagowska-Klimek, I.; Cedzynski, M. Mannan-binding lectin in cardiovascular disease. BioMed Res. Int. 2014, 2014, 616817. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Renee Ruhaak, L.; van der Laarse, A.; Cobbaert, C.M. Apolipoprotein profiling as a personalized approach to the diagnosis and treatment of dyslipidaemia. Ann. Clin. Biochem. 2019, 56, 338–356. [Google Scholar] [CrossRef]
- Hein, S.; Block, T.; Zimmermann, R.; Kostin, S.; Scheffold, T.; Kubin, T.; Klovekorn, W.P.; Schaper, J. Deposition of nonsarcomeric alpha-actinin in cardiomyocytes from patients with dilated cardiomyopathy or chronic pressure overload. Exp. Clin. Cardiol. 2009, 14, e68–e75. [Google Scholar]
- Bloomekatz, J.; Diaz, J.T.; Yelon, D.; Chi, N.C. Cardiac Morphogenesis: Crowding and Tension Resolved through Social Distancing. Dev. Cell. 2021, 56, 159–160. [Google Scholar] [CrossRef]
- Miyasaka, K.Y.; Kida, Y.S.; Sato, T.; Minami, M.; Ogura, T. Csrp1 regulates dynamic cell movements of the mesendoderm and cardiac mesoderm through interactions with Dishevelled and Diversin. Proc. Natl. Acad. Sci. USA 2007, 104, 11274–11279. [Google Scholar] [CrossRef]
- Posey, K.L.; Coustry, F.; Hecht, J.T. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 2018, 71–72, 161–173. [Google Scholar] [CrossRef]
- Wang, L.; Du, A.; Lu, Y.; Zhao, Y.; Qiu, M.; Su, Z.; Shu, H.; Shen, H.; Sun, W.; Kong, X. Peptidase Inhibitor 16 Attenuates Left Ventricular Injury and Remodeling After Myocardial Infarction by Inhibiting the HDAC1-Wnt3a-beta-Catenin Signaling Axis. J. Am. Heart Assoc. 2023, 12, e028866. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.H.; Mouton, A.J.; DeLeon-Pennell, K.Y.; Genovese, F.; Karsdal, M.; Lindsey, M.L. Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biol. 2019, 75–76, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Kessler-Icekson, G.; Schlesinger, H.; Freimann, S.; Kessler, E. Expression of procollagen C-proteinase enhancer-1 in the remodeling rat heart is stimulated by aldosterone. Int. J. Biochem. Cell Biol. 2006, 38, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.A.; Mabotuwana, N.S.; Murtha, L.A.; Coulter, B.; Sanchez-Bezanilla, S.; Al-Omary, M.S.; Senanayake, T.; Loering, S.; Starkey, M.; Lee, R.J.; et al. Novel role of extracellular matrix protein 1 (ECM1) in cardiac aging and myocardial infarction. PLoS ONE 2019, 14, e0212230. [Google Scholar] [CrossRef]
- Murtha, L.A.; Hardy, S.A.; Mabotuwana, N.S.; Bigland, M.J.; Bailey, T.; Raguram, K.; Liu, S.; Ngo, D.T.; Sverdlov, A.L.; Tomin, T.; et al. Fibulin-3 is necessary to prevent cardiac rupture following myocardial infarction. Sci. Rep. 2023, 13, 14995. [Google Scholar] [CrossRef]
- Scally, C.; Abbas, H.; Ahearn, T.; Srinivasan, J.; Mezincescu, A.; Rudd, A.; Spath, N.; Yucel-Finn, A.; Yuecel, R.; Oldroyd, K.; et al. Myocardial and Systemic Inflammation in Acute Stress-Induced (Takotsubo) Cardiomyopathy. Circulation 2019, 139, 1581–1592. [Google Scholar] [CrossRef]
- Jumeau, C.; Awad, F.; Assrawi, E.; Cobret, L.; Duquesnoy, P.; Giurgea, I.; Valeyre, D.; Grateau, G.; Amselem, S.; Bernaudin, J.F.; et al. Expression of SAA1, SAA2 and SAA4 genes in human primary monocytes and monocyte-derived macrophages. PLoS ONE 2019, 14, e0217005. [Google Scholar] [CrossRef]
- Moady, G.; Yelin, B.; Sweid, R.; Atar, S. C-Reactive Protein Can Predict Outcomes in Patients with Takotsubo Syndrome. Int. J. Heart Fail. 2024, 6, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cooley, B.C.; Akinc, A.; Butler, J.; Borodovsky, A. Knockdown of liver-derived factor XII by GalNAc-siRNA ALN-F12 prevents thrombosis in mice without impacting hemostatic function. Thromb. Res. 2020, 196, 200–205. [Google Scholar] [CrossRef]
- Wolberg, A.S.; Sang, Y. Fibrinogen and Factor XIII in Venous Thrombosis and Thrombus Stability. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 931–941. [Google Scholar] [CrossRef]
- Graham, L.C.; Kocalis, H.E.; Soto, I.; Howell, G.R. Deficiency of Complement Component C1Q Prevents Cerebrovascular Damage and White Matter Loss in a Mouse Model of Chronic Obesity. eNeuro 2020, 7, ENEURO.0057-20.2020. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhang, Q.; Chang, S.; Zhao, X.; Wang, M.; Li, W. Adiponectin protects against myocardial ischemia-reperfusion injury: A systematic review and meta-analysis of preclinical animal studies. Lipids Health Dis. 2024, 23, 51. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Espliguero, R.; Viana-Llamas, M.C.; Silva-Obregon, A.; Avanzas, P. The Role of C-reactive Protein in Patient Risk Stratification and Treatment. Eur. Cardiol. 2021, 16, e28. [Google Scholar] [CrossRef]
- Sakura, T.; Morioka, T.; Shioi, A.; Kakutani, Y.; Miki, Y.; Yamazaki, Y.; Motoyama, K.; Mori, K.; Fukumoto, S.; Shoji, T.; et al. Lipopolysaccharide-binding protein is associated with arterial stiffness in patients with type 2 diabetes: A cross-sectional study. Cardiovasc. Diabetol. 2017, 16, 62. [Google Scholar] [CrossRef]
- Qian, W.; Zhao, M.; Wang, R.; Li, H. Fibrinogen-like protein 1 (FGL1): The next immune checkpoint target. J. Hematol. Oncol. 2021, 14, 147. [Google Scholar] [CrossRef]
- Legler, K.; Rosprim, R.; Karius, T.; Eylmann, K.; Rossberg, M.; Wirtz, R.M.; Muller, V.; Witzel, I.; Schmalfeldt, B.; Milde-Langosch, K.; et al. Reduced mannosidase MAN1A1 expression leads to aberrant N-glycosylation and impaired survival in breast cancer. Br. J. Cancer 2018, 118, 847–856. [Google Scholar] [CrossRef]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. N. Am. J. Med. Sci. 2011, 4, 183–190. [Google Scholar] [CrossRef]
- Qu, J.; Ko, C.W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhang, J. Cartilage Oligomeric Matrix Protein, Diseases, and Therapeutic Opportunities. Int. J. Mol. Sci. 2022, 23, 9253. [Google Scholar] [CrossRef]
- Lagoutte, P.; Bettler, E.; Vadon-Le Goff, S.; Moali, C. Procollagen C-proteinase enhancer-1 (PCPE-1), a potential biomarker and therapeutic target for fibrosis. Matrix Biol. Plus 2021, 11, 100062. [Google Scholar] [CrossRef]
- Núñez-Gil, I.J.; Santoro, F.; Vazirani, R.; Novo, G.; Blanco-Ponce, E.; Arcari, L.; Uribarri, A.; Cacciotti, L.; Guerra, F.; Salamanca, J.; et al. Smoking influence in Takotsubo syndrome: Insights from an international cohort. Front. Cardiovasc. Med. 2023, 10, 1282018. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, L.; Landi, A.; Maurizi, N.; Pizzi, C.; Leo, L.A.; Arangalage, D.; Iglesias, J.F.; Eeckhout, E.; Schwitter, J.; Valgimigli, M.; et al. Acute response of the noninfarcted myocardium and surrounding tissue assessed by T2 mapping after STEMI. JACC: Cardiovasc. Imaging 2024, 17, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | STEMI (n = 24) | Takotsubo (n = 12) |
|---|---|---|
| Age (years) | 68.5 ± 11.3 | 67.5 ± 9.7 |
| BMI (kg/m2) | 26.9 ± 5.3 | 26.4 ± 6.4 |
| Atrial Fibrillation | 0/24 (0.0%) | 1/12 (8.3%) |
| COPD | 3/24 (12.5%) | 0/12 (0.0%) |
| Diabetes | 1/24 (4.2%) | 0/12 (0.0%) |
| Hyperlipidemia | 1/24 (4.2%) | 1/12 (8.3%) |
| Hypertension | 9/24 (37.5%) | 5/12 (41.7%) |
| Peripheral Vascular Disease | 1/24 (4.2%) | 0/12 (0.0%) |
| Former Smoker | 5/24 (20.8%) | 1/12 (8.3%) |
| Current Smoker | 5/24 (20.8%) | 1/12 (8.3%) |
| Trigger | ||
| Somatic | N/A | 4/12 (33.3%) |
| Emotional | N/A | 4/12 (33.3%) |
| None | N/A | 4/12 (33.3%) |
| Heart Rate (BPM) | 78.1 ± 19.7 | 86.4 ± 16.7 |
| Systolic Blood Pressure (mmHg) | 130 ± 22.4 | 142 ± 34.9 |
| Diastolic Blood Pressure (mmHg) | 78.5 ± 13.3 | 86.6 ± 16.6 |
| Oxygen at Admission | 5/24 (20.8%) | 6/12 (50.0%) |
| Baseline Drugs | ||
| ARB | 4/24 (16.7%) | 6/12 (50.0%) |
| Beta Blocker | 4/24 (16.7%) | 4/12 (33.3%) |
| Calcium Channel Blocker | 2/24 (8.3%) | 1/12 (8.3%) |
| Corticosteroids | 2/24 (8.3%) | 0/12 (0.0%) |
| Paracetamol | 2/24 (8.3%) | 1/12 (8.3%) |
| Loop Diuretic | 0/24 (0.0%) | 1/12 (8.3%) |
| Metformin | 1/24 (4.2%) | 0/12 (0.0%) |
| ASA | 0/24 (0.0%) | 3/12 (25.0%) |
| P2Y12 | 0/24 (0.0%) | 0/12 (0.0%) |
| OAC | 1/24 (4.2%) | 2/12 (16.7%) |
| Statins | 2/24 (8.3%) | 4/12 (33.3%) |
| Ezetrol | 0/24 (0.0%) | 1/12 (8.3%) |
| Discharge Drugs Taken Over 30 Days | ||
| ASA | 18/24 (75.0%) | 5/12 (41.7%) |
| P2Y12 | 24/24 (100.0%) | 0/12 (0.0%) |
| OAC | 4/24 (16.7%) | 4/12 (33.3%) |
| Statins | 24/24 (100.0%) | 6/12 (50.0%) |
| Clinical Outcomes | STEMI | Takotsubo |
|---|---|---|
| (n = 24) | (n = 12) | |
| Outcomes (3 days) | ||
| Pre- or intra-procedure sustained ventricular tachycardia | 0/24 (0.0%) | 0/12 (0.0%) |
| Pre- or intra-procedure ventricular fibrillation | 1/24 (4.2%) | 0/12 (0.0%) |
| Pre- or intra-procedure AV-block grade III | 1/24 (4.2%) | 0/12 (0.0%) |
| Pre- or intra-procedure sinus bradycardia | 3/24 (12.5%) | 1/12 (8.3%) |
| Pre- or intra-procedure cardiogenic shock | 1/24 (4.2%) | 3/12 (25.0%) |
| Post-procedure sustained ventricular tachycardia within 3 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Post-procedure ventricular fibrillation within 3 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Post-procedure III degree AV-block within 3 days | 1/24 (4.2%) | 0/12 (0.0%) |
| Post-procedure II degree AV-block within 3 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Post-procedure cardiogenic shock within 3 days | 1/24 (4.2%) | 2/12 (16.7%) |
| Post-procedure death within 3 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Outcomes (30 days) | ||
| Death within 30 days | 0/24 (0.0%) | 0/12 (0.0%) |
| HF rehospitalization within 30 days | 1/24 (4.2%) | 0/12 (0.0%) |
| Additional myocardial infarction within 30 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Stroke or TIA within 30 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Thromboembolization within 30 days | 0/24 (0.0%) | 0/12 (0.0%) |
| Laboratory Markers | STEMI | Takotsubo |
|---|---|---|
| n = 24 | n = 12 | |
| Peak hs-cTnI (ng/L) | 32,000 (25,000, 46,000) | 1700 (550, 2300) |
| Peak hs-cTnT (ng/L) | 2630 (1985, 4460) | 280 (196, 433.5) |
| Peak NT-proBNP (ng/L) | 2540 (2020, 4180) | 6140 (4990, 7620) |
| Peak creatinine (μmol/L) | 73 (60, 84) | 68.5 (55.8, 84.2) |
| Baseline cholesterol (mmol/L) | 5.2 (4.4, 6) | 5.7 (4.9, 6) |
| Baseline LDL (mmol/L) | 3.8 (3.2, 4.6) | 3.6 (3, 4.5) |
| Baseline HDL (mmol/L) | 1.2 (1, 1.4) | 1.4 (1.3, 1.8) |
| Baseline triglycerides (mmol/L) | 0.9 (0.8, 1.1) | 0.8 (0.8, 1.2) |
| Accession No. | Protein Name | Protein Symbol | Log2FC | p-Value |
|---|---|---|---|---|
| P06732 | Creatine kinase M-type | CKM | 1.765922 | 1.18 × 10−5 |
| A0A0C4DH31 | Immunoglobulin heavy variable 1–18 | IGHV1-18 | 0.969459 | 0.0234 |
| Q92954 | Proteoglycan 4 | PRG4 | 0.563158 | 0.0148 |
| P49913 | Cathelicidin antimicrobial peptide | CAMP | 0.554811 | 0.0277 |
| P0DOX2 | Immunoglobulin alpha-2 heavy chain | IGA2 | 0.513252 | 0.0367 |
| P07357 | Complement component C8 alpha chain | C8A | 0.441414 | 0.00467 |
| P07360 | Complement component C8 gamma chain | C8G | 0.432728 | 0.0345 |
| P07358 | Complement component C8 beta chain | C8B | 0.346922 | 0.0301 |
| P15169 | Carboxypeptidase N catalytic chain | CPN1 | 0.272563 | 0.0406 |
| Q9UNW1 | Multiple inositol polyphosphate phosphatase 1 | MINPP1 | 0.272294 | 0.0423 |
| P13671 | Complement component C6 | C6 | 0.248847 | 0.004 |
| P00450 | Ceruloplasmin | CP | 0.204697 | 0.0277 |
| Q9HDC9 | Adipocyte plasma membrane-associated protein | APMAP | −0.42631 | 0.0418 |
| P08253 | Matrix metalloproteinase-2 | MMP2 | −0.42791 | 0.031 |
| Q8WZ75 | Roundabout homolog 4 | ROBO4 | −0.45055 | 0.0189 |
| Q15848 | Adiponectin | ADIPOQ | −0.56441 | 0.0399 |
| P01833 | Polymeric immunoglobulin receptor | PIGR | −0.58109 | 0.0322 |
| P27987 | Inositol-trisphosphate 3-kinase B | ITPKB | −0.77973 | 0.0197 |
| Accession No. | Protein Name | Protein Symbol | Log2FC | p-Value |
|---|---|---|---|---|
| P0DJI8 | Serum amyloid A-1 protein | SAA1 | 1.045935 | 0.0147 |
| P00738 | Haptoglobin | HP | 0.446092 | 0.0206 |
| Q15113 | Procollagen C-endopeptidase enhancer 1 | PCOLCE | 0.438112 | 0.0355 |
| Q13740 | Activated leukocyte cell adhesion molecule | ALCAM | −0.30117 | 0.0182 |
| P01019 | Angiotensinogen | AGT | −0.35355 | 0.0436 |
| P55058 | Phospholipid transfer protein | PLTP | −0.40843 | 0.0152 |
| Q9Y4L1 | Hypoxia up-regulated protein 1 | HYOU1 | −0.41781 | 0.0371 |
| O95445 | Apolipoprotein M | APOM | −0.41958 | 0.00186 |
| P30043 | Flavin reductase | BLVRB | −0.45002 | 0.0421 |
| Q15848 | Adiponectin | ADIPOQ | −0.57104 | 0.0343 |
| P10646 | Tissue factor pathway inhibitor | TFPI | −0.6277 | 0.00644 |
| P04278 | Sex hormone-binding globulin | SHBG | −0.63928 | 0.0348 |
| P04114 | Apolipoprotein B-100 | APOB | −0.85254 | 0.0025 |
| Accession No. | Protein Name | Protein Symbol | Log2FC | p-Value |
|---|---|---|---|---|
| P0DJI9 | Serum amyloid A-2 protein | SAA2 | 1.925515 | 0.00653 |
| P02741 | C-reactive protein | CRP | 1.804223 | 0.00229 |
| P06732 | Creatine kinase M-type | CKM | 1.619238 | 0.000446 |
| P0DJI8 | Serum amyloid A-1 protein | SAA1 | 1.567947 | 0.00157 |
| P04114 | Apolipoprotein B-100 | APOB | 1.293007 | 1.37 × 10⁻7 |
| P18428 | Lipopolysaccharide-binding protein | LBP | 0.7243 | 0.0016 |
| Q08830 | Fibrinogen-like protein 1 | FGL1 | 0.661358 | 0.0102 |
| P10646 | Tissue factor pathway inhibitor | TFPI | 0.631156 | 0.00238 |
| P11226 | Mannose-binding protein C | MBL2 | 0.584484 | 0.00137 |
| Q92954 | Proteoglycan 4 | PRG4 | 0.579881 | 6.16 × 10⁻6 |
| Q5XPI4 | E3 ubiquitin-protein ligase RNF123 | RNF123 | 0.561965 | 0.0263 |
| Q6Q788 | Apolipoprotein A-V | APOA5 | 0.550032 | 0.000257 |
| P02649 | Apolipoprotein E | APOE | 0.528982 | 0.000149 |
| P11597 | Cholesteryl ester transfer protein | CETP | 0.493641 | 0.00147 |
| P07195 | L-lactate dehydrogenase B chain | LDHB | 0.489102 | 0.0337 |
| P01019 | Angiotensinogen | AGT | 0.489055 | 2.59 × 10⁻5 |
| Q9Y4L1 | Hypoxia up-regulated protein 1 | HYOU1 | 0.459645 | 0.00906 |
| P00915 | Carbonic anhydrase 1 | CA1 | 0.458761 | 0.0408 |
| O95445 | Apolipoprotein M | APOM | 0.439817 | 6.28 × 10⁻6 |
| O95497 | Pantetheinase | VNN1 | 0.433483 | 0.00309 |
| A0A0B4J1U3 | Immunoglobulin lambda variable 1–36 | IGLV1-36 | 0.428394 | 0.0382 |
| Q9UHG3 | Prenylcysteine oxidase 1 | PCYOX1 | 0.417686 | 0.000164 |
| Q9UK55 | Protein Z-dependent protease inhibitor | SERPINA10 | 0.392472 | 0.000264 |
| Q15166 | Serum paraoxonase/lactonase 3 | PON3 | 0.357544 | 0.0058 |
| P33908 | Mannosyl-oligosaccharide 1,2-alpha-mannosidase IA | MAN1A1 | 0.350637 | 0.000921 |
| O14791 | Apolipoprotein L1 | APOL1 | 0.340909 | 0.00133 |
| P22352 | Glutathione peroxidase 3 | GPX3 | 0.324418 | 0.0062 |
| Q96KN2 | Beta-Ala-His dipeptidase | CNDP1 | 0.320214 | 1.52 × 10⁻5 |
| Q15582 | Transforming growth factor-beta-induced protein ig-h3 | TGFBI | 0.296124 | 0.000792 |
| P22792 | Carboxypeptidase N subunit 2 | CPN2 | 0.290145 | 0.00251 |
| O75636 | Ficolin-3 | FCN3 | 0.280598 | 0.0015 |
| Q8WWA0 | Intelectin-1 | ITLN1 | 0.276047 | 0.0189 |
| P27169 | Serum paraoxonase/arylesterase 1 | PON1 | 0.272015 | 0.0013 |
| P04070 | Vitamin K-dependent protein C | PROC | 0.244853 | 0.0125 |
| P35858 | Insulin-like growth factor-binding protein complex acid labile subunit | IGFALS | 0.241407 | 0.00452 |
| P15169 | Carboxypeptidase N catalytic chain | CPN1 | 0.228361 | 0.0339 |
| P08709 | Coagulation factor VII | F7 | 0.226092 | 0.0313 |
| O75882 | Attractin | ATRN | 0.219264 | 0.00193 |
| P22891 | Vitamin K-dependent protein Z | PROZ | 0.210335 | 0.0108 |
| O00187 | Mannan-binding lectin serine protease 2 | MASP2 | 0.205538 | 0.0228 |
| P00736 | Complement C1r subcomponent | C1R | 0.201275 | 0.0041 |
| P00740 | Coagulation factor IX | F9 | 0.199953 | 0.00631 |
| P12259 | Coagulation factor V | F5 | 0.189057 | 0.0417 |
| P07360 | Complement component C8 gamma chain | C8G | 0.181396 | 0.035 |
| P07358 | Complement component C8 beta chain | C8B | 0.180535 | 0.029 |
| P06276 | Cholinesterase | BCHE | 0.17415 | 0.0479 |
| P00739 | Haptoglobin-related protein | HPR | 0.17372 | 0.0403 |
| Q9NPH3 | Interleukin-1 receptor accessory protein | IL1RAP | 0.15913 | 0.0362 |
| P19827 | Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH1 | 0.136853 | 0.0452 |
| P09871 | Complement C1s subcomponent | C1S | 0.128955 | 0.0357 |
| P02774 | Vitamin D-binding protein | GC | −0.17533 | 0.00304 |
| Q16610 | Extracellular matrix protein 1 | ECM1 | −0.19871 | 0.0156 |
| P49908 | Selenoprotein P | SELENOP | −0.20905 | 0.0066 |
| P13591 | Neural cell adhesion molecule 1 | NCAM1 | −0.21476 | 0.0342 |
| P61769 | Beta-2-microglobulin | B2M | −0.23769 | 0.0255 |
| P01871 | Immunoglobulin heavy constant mu | IGHM | −0.24231 | 0.00603 |
| P61626 | Lysozyme C | LYZ | −0.24325 | 0.0183 |
| P06396 | Gelsolin | GSN | −0.26045 | 0.0189 |
| Q03591 | Complement factor H-related protein 1 | CFHR1 | −0.26836 | 0.0426 |
| P06727 | Apolipoprotein A-IV | APOA4 | −0.29485 | 0.0106 |
| Q96QR1 | Secretoglobin family 3A member 1 | SCGB3A1 | −0.31331 | 0.0292 |
| P01619 | Immunoglobulin kappa variable 3–20 | IGKV3-20 | −0.33987 | 0.00893 |
| O14786 | Neuropilin-1 | NRP1 | −0.3454 | 0.0153 |
| Q6UXB8 | Peptidase inhibitor 16 | PI16 | −0.42234 | 0.0214 |
| Q9NPY3 | Complement component C1q receptor | CD93 | −0.43177 | 0.0065 |
| P09211 | Glutathione S-transferase P | GSTP1 | −0.43399 | 0.0435 |
| P08519 | Apolipoprotein(a) | LPA | −0.44072 | 0.0234 |
| Q12805 | EGF-containing fibulin-like extracellular matrix protein 1 | EFEMP1 | −0.44838 | 0.00641 |
| P49747 | Cartilage oligomeric matrix protein | COMP | −0.48649 | 0.00115 |
| P47756 | F-actin-capping protein subunit beta | CAPZB | −0.55793 | 0.0442 |
| P12814 | Alpha-actinin-1 | ACTN1 | −0.57426 | 0.0327 |
| P21291 | Cysteine and glycine-rich protein 1 | CSRP1 | −0.63131 | 0.0104 |
| P24844 | Myosin regulatory light polypeptide 9 | MYL9 | −0.63826 | 0.04 |
| Q15113 | Procollagen C-endopeptidase enhancer 1 | PCOLCE | −1.15362 | 8.69 × 10⁻9 |
| Accession No. | Protein Name | Protein Symbol | Log2FC | p-Value |
|---|---|---|---|---|
| P0DJI9 | Serum amyloid A-2 protein | SAA2 | 3.586989 | 0.0133 |
| P02741 | C-reactive protein | CRP | 2.040108 | 0.000303 |
| P0DJI8 | Serum amyloid A-1 protein | SAA1 | 1.869815 | 0.0423 |
| Q13418 | Integrin-linked protein kinase | ILK | 0.885547 | 0.0319 |
| Q08830 | Fibrinogen-like protein 1 | FGL1 | 0.786728 | 0.00509 |
| P55774 | C-C motif chemokine 18 | CCL18 | 0.782166 | 0.018 |
| P18428 | Lipopolysaccharide-binding protein | LBP | 0.649046 | 0.011 |
| O14950;P19105 | Myosin regulatory light chain 12B | MYL12B | 0.527264 | 0.0464 |
| P01033 | Metalloproteinase inhibitor 1 | TIMP1 | 0.473518 | 0.0251 |
| P05556 | Integrin beta-1 | ITGB1 | 0.447755 | 0.0455 |
| O95810 | Caveolae-associated protein 2 | CAVIN2 | 0.305359 | 0.0396 |
| P01019 | Angiotensinogen | AGT | 0.286575 | 0.00233 |
| P33908 | Mannosyl-oligosaccharide 1,2-alpha-mannosidase IA | MAN1A1 | 0.236024 | 0.0128 |
| P05160 | Coagulation factor XIII B chain | F13B | −0.15496 | 0.0435 |
| P02745 | Complement C1q subcomponent subunit A | C1QA | −0.21297 | 0.00893 |
| P00748 | Coagulation factor XII | F12 | −0.22025 | 0.0456 |
| P01700 | Immunoglobulin lambda variable 1–47 | IGLV1-47 | −0.22973 | 0.0347 |
| P19823 | Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | −0.23373 | 0.0315 |
| P02786 | Transferrin receptor protein 1 | TFRC | −0.26734 | 0.0246 |
| P04196 | Histidine-rich glycoprotein | HRG | −0.28371 | 0.0129 |
| P49747 | Cartilage oligomeric matrix protein | COMP | −0.33966 | 0.0454 |
| P06727 | Apolipoprotein A-IV | APOA4 | −0.5037 | 0.0205 |
| Q13740 | CD166 antigen | ALCAM | −0.53791 | 0.0127 |
| Q15113 | Procollagen C-endopeptidase enhancer 1 | PCOLCE | −0.551 | 0.00924 |
| P23083 | Immunoglobulin heavy variable 1–2 | IGHV1-2 | −0.55475 | 0.0115 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, S.; Jha, S.; Berger, E.; Molander, L.; Sevastianova, V.; Sheybani, Z.; Espinosa, A.S.; Elmahdy, A.; Al-Awar, A.; Kakaei, Y.; et al. Comparative Analysis of Plasma Protein Dynamics in Women with ST-Elevation Myocardial Infarction and Takotsubo Syndrome. Cells 2024, 13, 1764. https://doi.org/10.3390/cells13211764
Hussain S, Jha S, Berger E, Molander L, Sevastianova V, Sheybani Z, Espinosa AS, Elmahdy A, Al-Awar A, Kakaei Y, et al. Comparative Analysis of Plasma Protein Dynamics in Women with ST-Elevation Myocardial Infarction and Takotsubo Syndrome. Cells. 2024; 13(21):1764. https://doi.org/10.3390/cells13211764
Chicago/Turabian StyleHussain, Shafaat, Sandeep Jha, Evelin Berger, Linnea Molander, Valentyna Sevastianova, Zahra Sheybani, Aaron Shekka Espinosa, Ahmed Elmahdy, Amin Al-Awar, Yalda Kakaei, and et al. 2024. "Comparative Analysis of Plasma Protein Dynamics in Women with ST-Elevation Myocardial Infarction and Takotsubo Syndrome" Cells 13, no. 21: 1764. https://doi.org/10.3390/cells13211764
APA StyleHussain, S., Jha, S., Berger, E., Molander, L., Sevastianova, V., Sheybani, Z., Espinosa, A. S., Elmahdy, A., Al-Awar, A., Kakaei, Y., Kalani, M., Zulfaj, E., Nejat, A., Jha, A., Pylova, T., Krasnikova, M., Andersson, E. A., Omerovic, E., & Redfors, B. (2024). Comparative Analysis of Plasma Protein Dynamics in Women with ST-Elevation Myocardial Infarction and Takotsubo Syndrome. Cells, 13(21), 1764. https://doi.org/10.3390/cells13211764