
Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review

 , ,
, ,
Abstract
:1. Introduction
2. Background
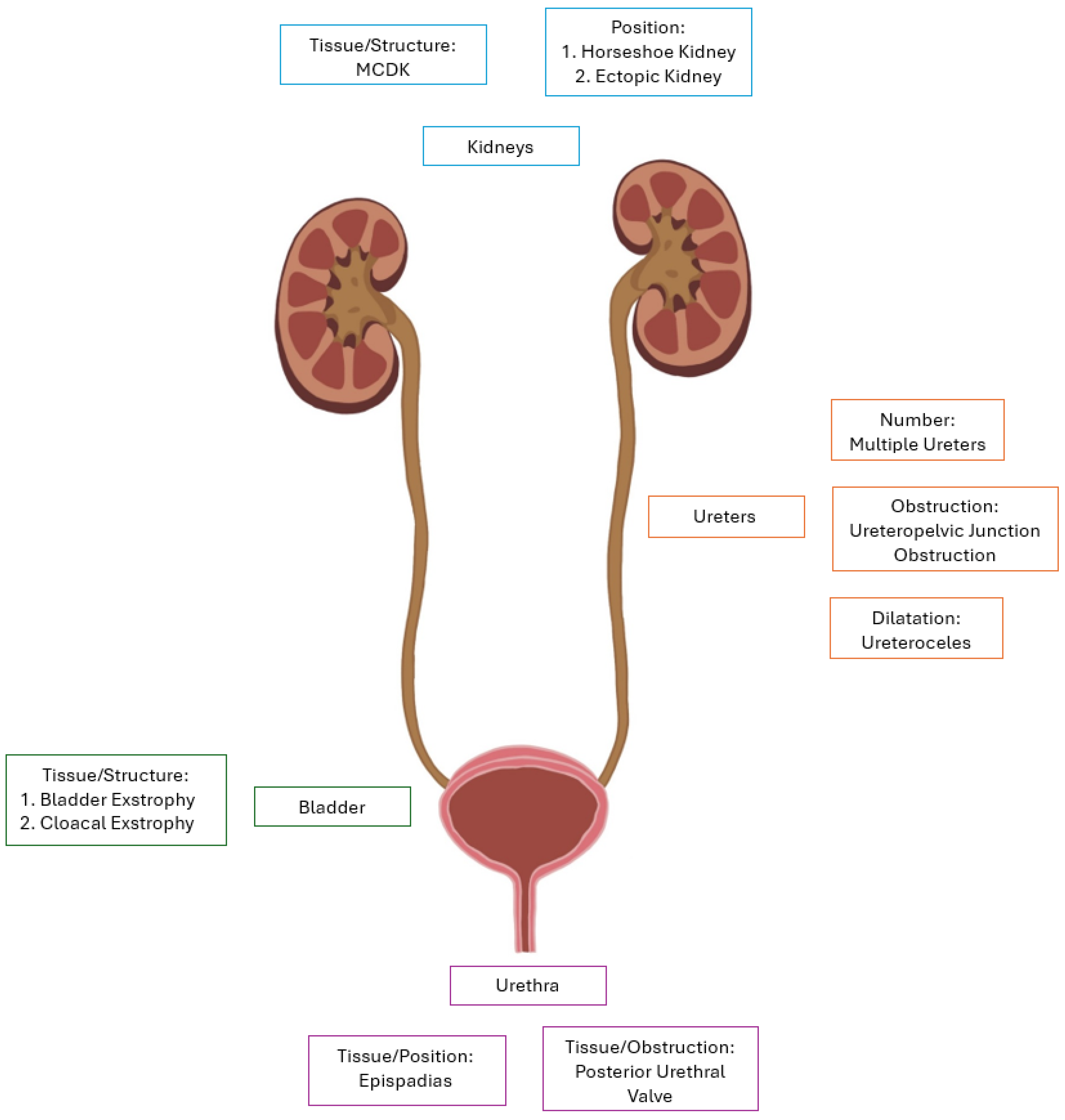
3. CAKUT Diagnoses
3.1. Nephropathies
3.1.1. Aplasia and Dysplasia
Epidemiology
Clinical Presentation
Pathophysiology
3.1.2. Hypoplasia and Oligomeganephronia
Epidemiology
Clinical Presentation
Pathophysiology
3.1.3. Positional Anomalies of the Kidneys (Horseshoe Kidney, Ectopic Kidney, Pancake Kidney, Malrotation)
Epidemiology
Clinical Presentation
Pathophysiology
3.2. Uropathies
3.2.1. Multiple Ureters and Vesicoureteral Reflux
Epidemiology
Clinical Presentation
Pathophysiology
3.2.2. Bladder-Exstrophy-Epispadias-Complex
Epidemiology
Clinical Presentation
Pathophysiology
3.2.3. Obstructive Uropathy (Posterior Urethral Valves, Ureteropelvic Junction Obstruction)
Epidemiology
Clinical Presentation
Pathophysiology
3.2.4. Ureteroceles
Epidemiology
Clinical Presentation
Pathophysiology
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murugapoopathy, V.; Gupta, I.R. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin. J. Am. Soc. Nephrol. CJASN 2020, 15, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Kolvenbach, C.M.; Shril, S.; Hildebrandt, F. The genetics and pathogenesis of CAKUT. Nat. Rev. Nephrol. 2023, 19, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Dressler, G.R. Advances in early kidney specification, development and patterning. Dev. Camb. Engl. 2009, 136, 3863–3874. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chen, F. Developmental pathology of congenital kidney and urinary tract anomalies. Clin. Kidney J. 2019, 12, 382–399. [Google Scholar] [CrossRef]
- Talati, A.N.; Webster, C.M.; Vora, N.L. Prenatal genetic considerations of congenital anomalies of the kidney and urinary tract (CAKUT). Prenat. Diagn. 2019, 39, 679–692. [Google Scholar] [CrossRef]
- Rehman, S.; Ahmed, D. Embryology, Kidney, Bladder, and Ureter. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK547747/ (accessed on 29 September 2024).
- Lin, C.-C.; Sheu, J.-C.; Tsai, P.-S.; Lee, M.-D.; Lin, T.-H.; Tsai, J.-D. Zinner syndrome in children: Clinical presentation, imaging findings, diagnosis, and outcome. Pediatr. Nephrol. 2022, 37, 3075–3084. [Google Scholar] [CrossRef]
- Acién, P.; Armiñana, E.; Garcia-Ontiveros, E. Unilateral renal agenesis associated with ipsilateral blind vagina. Arch. Gynecol. 1987, 240, 1–8. [Google Scholar] [CrossRef]
- Acién, P.; Acién, M. Renal agenesis, associated genital malformations, and responsible genes. Fertil. Steril. 2021, 116, 1370–1371. [Google Scholar] [CrossRef]
- Herlin, M.K.; Petersen, M.B.; Brännström, M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: A comprehensive update. Orphanet J. Rare Dis. 2020, 15, 214. [Google Scholar] [CrossRef]
- Raina, R.; Chakraborty, R.; Sethi, S.K.; Kumar, D.; Gibson, K.; Bergmann, C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2021, 78, 125–141. [Google Scholar] [CrossRef]
- Lu, Y.; Xie, Y.; Li, M.; Zuo, N.; Ning, S.; Luo, B.; Ning, M.; Song, J.; Liang, Y.; Qin, Y. A novel ADGRG2 truncating variant associated with X-linked obstructive azoospermia in a large Chinese pedigree. J. Assist. Reprod. Genet. 2023, 40, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Weiske, W.-H.; Sälzler, N.; Schroeder-Printzen, I.; Weidner, W. Clinical findings in congenital absence of the vasa deferentia. Andrologia 2000, 32, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chu, C.; Li, S.; Lu, D.; Zheng, P.; Sheng, J.; Luo, L.-J.; Wu, X.; Zhang, Y.-D.; Yin, C.; et al. Renal agenesis-related genes are associated with Herlyn-Werner-Wunderlich syndrome. Fertil. Steril. 2021, 116, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhao, S.; Jolly, A.; Wang, L.; Pan, H.; Yuan, J.; Chen, S.; Koch, A.; Ma, C.; Tian, W.; et al. Perturbations of genes essential for Müllerian duct and Wölffian duct development in Mayer-Rokitansky-Küster-Hauser syndrome. Am. J. Hum. Genet. 2021, 108, 337–345. [Google Scholar] [CrossRef]
- Yoshino, M.; Shimabukuro, W.; Takeichi, M.; Omura, J.; Yokota, C.; Yamamoto, J.; Nakanishi, K.; Morisada, N.; Nozu, K.; Iijima, K.; et al. A case of Potter sequence with WT1 mutation. CEN Case Rep. 2022, 12, 184–188. [Google Scholar] [CrossRef]
- Sugiyama, H. WT1 (Wilms’ Tumor Gene 1): Biology and Cancer Immunotherapy. Jpn. J. Clin. Oncol. 2010, 40, 377–387. [Google Scholar] [CrossRef]
- Mrowka, C.; Schedl, A. Wilms’ tumor suppressor gene WT1: From structure to renal pathophysiologic features. J. Am. Soc. Nephrol. JASN 2000, 11 (Suppl. 16), S106–S115. [Google Scholar] [CrossRef]
- Hu, Y.; Bouloux, P.-M. X-linked GnRH deficiency: Role of KAL-1 mutations in GnRH deficiency. Mol. Cell. Endocrinol. 2011, 346, 13–20. [Google Scholar] [CrossRef]
- Smith, R.J. Branchiootorenal Spectrum Disorder. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1380/ (accessed on 29 September 2024).
- Skinner, M.A.; Safford, S.D.; Reeves, J.G.; Jackson, M.E.; Freemerman, A.J. Renal aplasia in humans is associated with RET mutations. Am. J. Hum. Genet. 2008, 82, 344–351. [Google Scholar] [CrossRef]
- Cain, J.E.; Di Giovanni, V.; Smeeton, J.; Rosenblum, N.D. Genetics of renal hypoplasia: Insights into the mechanisms controlling nephron endowment. Pediatr. Res. 2010, 68, 91–98. [Google Scholar] [CrossRef]
- Majumdar, A.; Vainio, S.; Kispert, A.; McMahon, J.; McMahon, A.P. Wnt11 and Ret/Gdnf pathways cooperate in regulating ureteric branching during metanephric kidney development. Dev. Camb. Engl. 2003, 130, 3175–3185. [Google Scholar] [CrossRef]
- Belk, R.A.; Thomas, D.F.M.; Mueller, R.F.; Godbole, P.; Markham, A.F.; Weston, M.J. A family study and the natural history of prenatally detected unilateral multicystic dysplastic kidney. J. Urol. 2002, 167, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Tripathi, P.; Manson, S.R.; Austin, P.F.; Chen, F. Transcriptional Dysregulation in the Ureteric Bud Causes Multicystic Dysplastic Kidney by Branching Morphogenesis Defect. J. Urol. 2015, 193, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Bonsib, S.M. Renal Hypoplasia, From Grossly Insufficient to Not Quite Enough: Consideration for Expanded Concepts Based Upon the Author’s Perspective With Historical Review. Adv. Anat. Pathol. 2020, 27, 311. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Tellier, A.L.; Attie-Bitach, T.; Amiel, J.; Vekemans, M.; Lyonnet, S.; Dureau, P.; Niaudet, P.; Gubler, M.C.; Broyer, M. PAX2 mutations in oligomeganephronia. Kidney Int. 2001, 59, 457–462. [Google Scholar] [CrossRef]
- Broyer, M.; Soto, B.; Gagnadoux, M.F.; Adi, M.; Rica, C.; Gubler, M.C. Oligomeganephronic renal hypoplasia. Adv. Nephrol. Necker Hosp. 1997, 26, 47–63. [Google Scholar]
- Detre, Z.; Miltényi, M. Oligomeganephronic renal hypoplasia. Pathol. Res. Pract. 1984, 178, 416–419. [Google Scholar] [CrossRef]
- Shindo, S.; Bernstein, J.; Arant, B.S. Evolution of renal segmental atrophy (Ask-Upmark kidney) in children with vesicoureteric reflux: Radiographic and morphologic studies. J. Pediatr. 1983, 102, 847–854. [Google Scholar] [CrossRef]
- Di Giovanni, V.; Alday, A.; Chi, L.; Mishina, Y.; Rosenblum, N.D. Alk3 controls nephron number and androgen production via lineage-specific effects in intermediate mesoderm. Development 2011, 138, 2717–2727. [Google Scholar] [CrossRef]
- Kopan, R.; Chen, S.; Little, M. Nephron progenitor cells: Shifting the balance of self-renewal and differentiation. Curr. Top. Dev. Biol. 2014, 107, 293–331. [Google Scholar] [CrossRef]
- Kazama, I.; Mahoney, Z.; Miner, J.H.; Graf, D.; Economides, A.N.; Kreidberg, J.A. Podocyte-Derived BMP7 Is Critical for Nephron Development. J. Am. Soc. Nephrol. 2008, 19, 2181. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, M.; Fukushima, K.; Kawada, M.; Onishi, S.; Furuta, Y.; Yonemura, S.; Kitamura, T.; Nosaka, T.; Sasai, Y. Cv2, functioning as a pro-BMP factor via twisted gastrulation, is required for early development of nephron precursors. Dev. Biol. 2010, 337, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.A. Renal coloboma syndrome. Eur. J. Hum. Genet. 2011, 19, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, S.M.; Ohlemiller, K.K.; Yang, J.; McDill, B.W.; Kohlhase, J.; Rauchman, M. Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum. Mol. Genet. 2003, 12, 2221–2227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Quinlan, J.; Hoy, W.; Hughson, M.D.; Lemire, M.; Hudson, T.; Hueber, P.-A.; Benjamin, A.; Roy, A.; Pascuet, E.; et al. A Common RET Variant Is Associated with Reduced Newborn Kidney Size and Function. J. Am. Soc. Nephrol. JASN 2008, 19, 2027–2034. [Google Scholar] [CrossRef]
- Cuccurullo, C.; Miele, G.; Piccolo, G.; Bilo, L.; Accogli, A.; D’Amico, A.; Fratta, M.; Guerrisi, S.; Iacomino, M.; Salpietro, V.; et al. Hydranencephaly in CENPJ-related Seckel syndrome. Eur. J. Med. Genet. 2022, 65, 104659. [Google Scholar] [CrossRef]
- Anderson, C.E.; Wallerstein, R.; Zamerowski, S.T.; Witzleben, C.; Hoyer, J.R.; Gibas, L.; Jackson, L.G. Ring chromosome 4 mosaicism coincidence of oligomeganephronia and signs of Seckel syndrome. Am. J. Med. Genet. 1997, 72, 281–285. [Google Scholar] [CrossRef]
- Jung, M.; Rai, A.; Wang, L.; Puttmann, K.; Kukreja, K.; Koh, C.J. Nephrolithiasis in a 17-Year-Old Male With Seckel Syndrome and Horseshoe Kidneys: Case Report and Review of the Literature. Urology 2018, 120, 241–243. [Google Scholar] [CrossRef]
- Arant, B.S.; Sotelo-Avila, C.; Bernstein, J. Segmental “hypoplasia” of the kidney (Ask-Upmark). J. Pediatr. 1979, 95, 931–939. [Google Scholar] [CrossRef]
- Sugimoto, T.; Tanaka, Y.; Nitta, N.; Uzu, T.; Nishio, Y.; Kashiwagi, A. Renal Segmental Hypoplasia, Ask-Upmark Kidney, in a Patient with Adult-onset Hypertension. Intern. Med. 2006, 45, 1101–1102. [Google Scholar] [CrossRef]
- Kirkpatrick, J.J.; Leslie, S.W. Horseshoe Kidney. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK431105/ (accessed on 29 September 2024).
- Bingham, G.; Leslie, S.W. Pelvic Kidney. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK563239/ (accessed on 29 September 2024).
- Neville, H.; Ritchey, M.L.; Shamberger, R.C.; Haase, G.; Perlman, S.; Yoshioka, T. The occurrence of Wilms tumor in horseshoe kidneys: A report from the National Wilms Tumor Study Group (NWTSG). J. Pediatr. Surg. 2002, 37, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Arslan, H.; Aydogan, C.; Orcen, C.; GonIllu, E. A rare case: Congenital thoracic ectopic kidney with diaphragmatic eventration. JPMA J. Pak. Med. Assoc. 2016, 66, 339–341. [Google Scholar] [PubMed]
- Divjak, N.; Birraux, J.; Chehade, H.; Sanchez, O. Hydronephrosis caused by kidney malrotation. Urol. Case Rep. 2021, 36, 101564. [Google Scholar] [CrossRef] [PubMed]
- Benz-Bohm, G. Anomalies of Kidney Rotation, Position and Fusion. In Pediatric Uroradiology; Fotter, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 55–60. [Google Scholar] [CrossRef]
- Bakshi, S. Incidentally detected pancake kidney: A case report. J. Med. Case Rep. 2020, 14, 129. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Choudhary, A.K.; Khowal, H.; Chaudhary, P.; Arora, M.P. Pancake kidney: A rare developmental anomaly. Can. Urol. Assoc. J. J. Assoc. Urol. Can. 2014, 8, E451–E452. [Google Scholar] [CrossRef]
- Subramani, A.; Kulkarni, V. Tandem Kidney: A Case of Left Crossed Fused Renal Ectopia. J. Med. Sci. Health 2022, 8, 189–192. [Google Scholar] [CrossRef]
- Humphries, A.; Speroni, S.; Eden, K.; Nolan, M.; Gilbert, C.; McNamara, J. Horseshoe kidney: Morphologic features, embryologic and genetic etiologies, and surgical implications. Clin. Anat. 2023, 36, 1081–1088. [Google Scholar] [CrossRef]
- Taghavi, K.; Kirkpatrick, J.; Mirjalili, S.A. The horseshoe kidney: Surgical anatomy and embryology. J. Pediatr. Urol. 2016, 12, 275–280. [Google Scholar] [CrossRef]
- Shah, H.U.; Ojili, V. Multimodality imaging spectrum of complications of horseshoe kidney. Indian J. Radiol. Imaging 2017, 27, 133–140. [Google Scholar] [CrossRef]
- Dretler, S.P.; Olsson, C.; Pfister, R.C. The anatomic, radiologic and clinical characteristics of the pelvic kidney: An analysis of 86 cases. J. Urol. 1971, 105, 623–627. [Google Scholar] [CrossRef]
- Eid, S.; Iwanaga, J.; Loukas, M.; Oskouian, R.J.; Tubbs, R.S. Pelvic Kidney: A Review of the Literature. Cureus 2018, 10, e2775. [Google Scholar] [CrossRef] [PubMed]
- Yener, S.; Pehlivanoğlu, C.; Akis Yıldız, Z.; Ilce, H.T.; Ilce, Z. Duplex Kidney Anomalies and Associated Pathologies in Children: A Single-Center Retrospective Review. Cureus 2022, 14, e25777. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, F.; Niglio, F.; Pastore, V.; Campanella, V.; Leggio, S.; Aceto, G.; Germano, M.; D’Addato, O.; Penza, R. Polydimethylsiloxane (macroplastique®) injection for vesicoureteral reflux in duplex ureters: A comparison with single renal systems. J. Pediatr. Urol. 2011, 7, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Doery, A.J.; Ang, E.; Ditchfield, M.R. Duplex kidney: Not just a drooping lily. J. Med. Imaging Radiat. Oncol. 2015, 59, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, V.M.; Schedl, A. Duplex kidney formation: Developmental mechanisms and genetic predisposition. F1000Research 2020, 9, F1000 Faculty Rev-2. [Google Scholar] [CrossRef]
- Ninoa, F.; Ilaria, M.; Noviello, C.; Santoro, L.; Rätsch, I.M.; Martino, A.; Cobellis, G. Genetics of Vesicoureteral Reflux. Curr. Genomics 2016, 17, 70. [Google Scholar] [CrossRef]
- Lu, W.; van Eerde, A.M.; Fan, X.; Quintero-Rivera, F.; Kulkarni, S.; Ferguson, H.; Kim, H.-G.; Fan, Y.; Xi, Q.; Li, Q.; et al. Disruption of ROBO2 Is Associated with Urinary Tract Anomalies and Confers Risk of Vesicoureteral Reflux. Am. J. Hum. Genet. 2007, 80, 616–632. [Google Scholar] [CrossRef]
- Bridgewater, D.; Cox, B.; Cain, J.; Lau, A.; Athaide, V.; Gill, P.S.; Kuure, S.; Sainio, K.; Rosenblum, N.D. Canonical WNT/beta-catenin signaling is required for ureteric branching. Dev. Biol. 2008, 317, 83–94. [Google Scholar] [CrossRef]
- Schultza, K.; Todab, L.Y. Genetic Basis of Ureterocele. Curr. Genomics 2016, 17, 62–69. [Google Scholar] [CrossRef]
- Gbadegesin, R.A.; Brophy, P.D.; Adeyemo, A.; Hall, G.; Gupta, I.R.; Hains, D.; Bartkowiak, B.; Rabinovich, C.E.; Chandrasekharappa, S.; Homstad, A.; et al. TNXB Mutations Can Cause Vesicoureteral Reflux. J. Am. Soc. Nephrol. JASN 2013, 24, 1313. [Google Scholar] [CrossRef]
- Anand, S.; Lotfollahzadeh, S. Bladder Exstrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK563156/ (accessed on 29 September 2024).
- Beaman, G.M.; Cervellione, R.M.; Keene, D.; Reutter, H.; Newman, W.G. The Genomic Architecture of Bladder Exstrophy Epispadias Complex. Genes 2021, 12, 1149. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Borer, J. Exstrophy-Epispadias Complex. Urol. Clin. N. Am. 2023, 50, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Sinatti, C.; Spinoit, A.-F.; Raes, A.; Laecke, E.V.; Hoebeke, P. Long-Term fate of the upper urinary tract and ITS association with continence in exstrophy patients. J. Pediatr. Urol. 2021, 17, 655.e1–655.e7. [Google Scholar] [CrossRef] [PubMed]
- Woo, L.L.; Thomas, J.C.; Brock, J.W. Cloacal exstrophy: A comprehensive review of an uncommon problem. J. Pediatr. Urol. 2010, 6, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, S.; Simon, L.; Bargy, F. Anatomical basis of a common embryological origin for epispadias and bladder or cloacal exstrophies. Surg. Radiol. Anat. SRA 1997, 19, 11–16. [Google Scholar] [CrossRef]
- Draaken, M.; Reutter, H.; Schramm, C.; Bartels, E.; Boemers, T.M.; Ebert, A.-K.; Rösch, W.; Schröder, A.; Stein, R.; Moebus, S.; et al. Microduplications at 22q11.21 are associated with non-syndromic classic bladder exstrophy. Eur. J. Med. Genet. 2010, 53, 55–60. [Google Scholar] [CrossRef]
- Wilkins, S.; Zhang, K.W.; Mahfuz, I.; Quantin, R.; D’Cruz, N.; Hutson, J.; Ee, M.; Bagli, D.; Aitken, K.; Fong, F.N.-Y.; et al. Insertion/deletion polymorphisms in the ΔNp63 promoter are a risk factor for bladder exstrophy epispadias complex. PLoS Genet. 2012, 8, e1003070. [Google Scholar] [CrossRef]
- Carroll, T.J.; Park, J.-S.; Hayashi, S.; Majumdar, A.; McMahon, A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 2005, 9, 283–292. [Google Scholar] [CrossRef]
- Baranowska Körberg, I.; Hofmeister, W.; Markljung, E.; Cao, J.; Nilsson, D.; Ludwig, M.; Draaken, M.; Holmdahl, G.; Barker, G.; Reutter, H.; et al. WNT3 involvement in human bladder exstrophy and cloaca development in zebrafish. Hum. Mol. Genet. 2015, 24, 5069–5078. [Google Scholar] [CrossRef]
- Draaken, M.; Knapp, M.; Pennimpede, T.; Schmidt, J.M.; Ebert, A.-K.; Rösch, W.; Stein, R.; Utsch, B.; Hirsch, K.; Boemers, T.M.; et al. Genome-wide association study and meta-analysis identify ISL1 as genome-wide significant susceptibility gene for bladder exstrophy. PLoS Genet. 2015, 11, e1005024. [Google Scholar] [CrossRef]
- Zhang, R.; Knapp, M.; Suzuki, K.; Kajioka, D.; Schmidt, J.M.; Winkler, J.; Yilmaz, Ö.; Pleschka, M.; Cao, J.; Kockum, C.C.; et al. ISL1 is a major susceptibility gene for classic bladder exstrophy and a regulator of urinary tract development. Sci. Rep. 2017, 7, 42170. [Google Scholar] [CrossRef] [PubMed]
- Ching, S.T.; Infante, C.R.; Du, W.; Sharir, A.; Park, S.; Menke, D.B.; Klein, O.D. Isl1 mediates mesenchymal expansion in the developing external genitalia via regulation of Bmp4, Fgf10 and Wnt5a. Hum. Mol. Genet. 2018, 27, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Capone, V.; Persico, N.; Berrettini, A.; Decramer, S.; De Marco, E.A.; De Palma, D.; Familiari, A.; Feitz, W.; Herthelius, M.; Kazlauskas, V.; et al. Definition, diagnosis and management of fetal lower urinary tract obstruction: Consensus of the ERKNet CAKUT-Obstructive Uropathy Work Group. Nat. Rev. Urol. 2022, 19, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Malin, G.; Tonks, A.; Morris, R.; Gardosi, J.; Kilby, M. Congenital lower urinary tract obstruction: A population-based epidemiological study. BJOG Int. J. Obstet. Gynaecol. 2012, 119, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Al Aaraj, M.S.; Badreldin, A.M. Ureteropelvic Junction Obstruction. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK560740/ (accessed on 29 September 2024).
- Krajewski, W.; Wojciechowska, J.; Dembowski, J.; Zdrojowy, R.; Szydełko, T. Hydronephrosis in the course of ureteropelvic junction obstruction—An underestimated problem?Current opinion on pathogenesis, diagnosis and treatment. Adv. Clin. Exp. Med. 2017, 26, 857–864. [Google Scholar] [CrossRef]
- Ebadi, M.; Kajbafzadeh, A.-M.; Tourchi, A.; Mousavian, A.-A. Endoureterotomy as the Initial Management of Concurrent Ureteropelvic and Ureterovesical Junction Obstruction After Failed Conservative Therapy. Urology 2013, 82, 214–219. [Google Scholar] [CrossRef]
- Merlini, E.; Spina, P. Primary non-refluxing megaureters. J. Pediatr. Urol. 2005, 1, 409–417. [Google Scholar] [CrossRef]
- Adams, M.C.; Hendren, W.H. Chapter 119—Megaureter and Prune-Belly Syndrome. In Pediatric Surgery, 7th ed.; Coran, A.G., Ed.; Mosby: Philadelphia, PA, USA, 2012; pp. 1497–1514. Available online: https://www.sciencedirect.com/science/article/pii/B9780323072557001197 (accessed on 6 November 2024).
- Bingham, G.; Leslie, S.W.; Rentea, R.M. Posterior Urethral Valves. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK560881/ (accessed on 29 September 2024).
- Nasir, A.A.; Ameh, E.A.; Abdur-Rahman, L.O.; Adeniran, J.O.; Abraham, M.K. Posterior urethral valve. World J. Pediatr. WJP 2011, 7, 205–216. [Google Scholar] [CrossRef]
- van der Zanden, L.F.M.; Maj, C.; Borisov, O.; van Rooij, I.A.L.M.; Quaedackers, J.S.L.T.; Steffens, M.; Schierbaum, L.; Schneider, S.; Waffenschmidt, L.; Kiemeney, L.A.L.M.; et al. Genome-wide association study in patients with posterior urethral valves. Front. Pediatr. 2022, 10, 988374. [Google Scholar] [CrossRef]
- Woolf, A.S.; Lopes, F.M.; Ranjzad, P.; Roberts, N.A. Congenital Disorders of the Human Urinary Tract: Recent Insights From Genetic and Molecular Studies. Front. Pediatr. 2019, 7, 136. Available online: https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2019.00136/full (accessed on 25 October 2024). [CrossRef]
- Chan, M.M.; Sadeghi-Alavijeh, O.; Lopes, F.M.; Hilger, A.C.; Stanescu, H.C.; Voinescu, C.D.; Beaman, G.M.; Newman, W.G.; Zaniew, M.; Weber, S.; et al. Diverse ancestry whole-genome sequencing association study identifies TBX5 and PTK7 as susceptibility genes for posterior urethral valves. eLife 2022, 11, e74777. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, C.; Bommarito, D.; Zambaiti, E.; Antona, V.; Li Voti, G. Genetic Basis of Posterior Urethral Valves Inheritance. Urology 2016, 95, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Grasso, M.; Caruso, R.P.; Phillips, C.K. UPJ Obstruction in the Adult Population: Are Crossing Vessels Significant? Rev. Urol. 2001, 3, 42–51. [Google Scholar] [PubMed]
- Kohno, M.; Ogawa, T.; Kojima, Y.; Sakoda, A.; Johnin, K.; Sugita, Y.; Nakane, A.; Noguchi, M.; Moriya, K.; Hattori, M.; et al. Pediatric congenital hydronephrosis (ureteropelvic junction obstruction): Medical management guide. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2020, 27, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L.; Thornhill, B.A.; Forbes, M.S.; Kiley, S.C. Mechanisms of renal injury and progression of renal disease in congenital obstructive nephropathy. Pediatr. Nephrol. Berl. Ger. 2010, 25, 687–697. [Google Scholar] [CrossRef]
- Pohl, H.G. Embryology, Treatment, and Outcomes of Ureteroceles in Children. Urol. Clin. N. Am. 2023, 50, 371–389. [Google Scholar] [CrossRef]
- Ghanem, K.; Leslie, S.W.; Badreldin, A.M. Ureterocele. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK597362/ (accessed on 29 September 2024).
- Merlini, E.; Lelli Chiesa, P. Obstructive ureterocele—An ongoing challenge. World J. Urol. 2004, 22, 107–114. [Google Scholar] [CrossRef]
- Ghaffari, N. Ectopic ureterocele. Am. J. Obstet. Gynecol. 2021, 225, B14–B15. [Google Scholar] [CrossRef]
- Tuncer, K.; Kilinc, G.; Sert, I.; Akpinar, G.; Tugmen, C. Ectopic ureter associated with Zinner’s syndrome in a kidney recipient: Case report and literature review. Rev. Assoc. Médica Bras. 2020, 66, 692–695. [Google Scholar] [CrossRef]
- Sözübir, S.; Ewalt, D.; Strand, W.; Baker, L.A. Familial ureteroceles: An evidence for genetic background? Turk. J. Pediatr. 2005, 47, 255–260. [Google Scholar] [CrossRef]

{kind=link}
| Nephropathies | Uropathies |
|---|---|
| Unilateral renal agenesis | Multiple ureters |
| Bilateral renal agenesis | Epispadias |
| Multicystic dysplastic kidneys (MCDK) | Bladder exstrophy |
| Simple renal hypoplasia | Cloacal exstrophy |
| Oligomeganephronia | Posterior urethral valves (PUV) |
| Horseshoe kidney | Ureteropelvic junction obstruction (UPJO) |
| Ectopic kidney | Ureterocele |
| Malrotated kidney |
| Disease | Genetic Mutations | Mechanism | Symptoms/ Outcome | Associated Syndromes/Notes |
|---|---|---|---|---|
| Unilateral renal agenesis | CFTR *, ADGRG2 *, WT1 *, RET ^, CHD1L *, TRIM32 *, WNT, WNT4 ^, PAX8 * | Failure of the ureteric bud to induce metanephric mesenchyme | Frequently asymptomatic. Less often hypertension, compensatory hypertrophy of solitary kidney, CKD, UTI | Zinner syndrome, OHVIRA, VACTERL |
| Bilateral renal agenesis | RET^, GDNF, WNT11, DSTYK, ITGA8, FGF20, GREB1L, WT1, ANOS1 *, EYA1 *, SIX1 *, SIX5 * | Developmental failure of both ureteric buds | Rapidly fatal, oligohydramnios, pulmonary hypoplasia, Potter phenotype | |
| Multicystic dysplastic kidneys | PAX2, TCF2, calcineurin-NFAT, BMPER | Abnormal interaction between the ureteric bud and the metanephric mesenchyme | VUR, UTI, Potter phenotype, hypertension, progression to renal failure | VACTERL, renal coloboma, branchio-oto-renal syndrome, Mayer-Rokitansky |
| Simple renal hypoplasia | BMP2, BMP4, ALK3 | An underdevelopment of renal tissue due to insufficient nephron formation in utero | Frequently asymptomatic. VUR, hypertension | |
| Oligomeganephronia | PAX2 *, EAY1 *, SALL1 *, RET1, chromosome 4 * | Unknown, but hypothesized as reduced nephron number during renal development | Short stature, polyuria, polydipsia, proteinuria, progressive CKD, hypertension, glomerular hypertrophy | Renal-coloboma syndrome, branchio-oto-renal syndrome, Townes-Brocks syndrome, acrorenal syndrome, and Seckel syndrome, |
| Horseshoe kidney | No known genetic etiology | Fusion of inferior poles of kidney | Frequently asymptomatic. VUR, nephrolithiasis, UTI, hydronephrosis | Turner Syndrome, Trisomy 18 |
| Ectopic kidney | No known genetic etiology | Abnormal migration of the kidney during development | Frequently asymptomatic. VUR, nephrolithiasis, urinary incontinence, UTI, hydronephrosis. | Omphalocele–Exstrophy-Imperforate Anus–Spinal Defects Syndrome (OEIS), MCDK, Ureterocele |
| Multiple ureters | RET, GDNF, GATA3, SLIT2/ROBO2, FOXC1, FOXC2, SOX11, GREM1, BMP4, beta-catenin | Premature bifurcation of the ureteric bud or two distinct ureteral buds. | VUR, UTI, hydronephrosis, ureteroceles, nephrolithiasis | Associated with ureteroceles |
| Epispadias | WNT3, WNT9B, TP63, ISL1 | Failure of midline fusion of the genetic tubercle | Urinary incontinence, cosmetic concerns, sexual dysfunction | Frequently presents with bladder exstrophy |
| Bladder exstrophy | WNT3, WNT9B, TP63, ISL1 | Improper closure of the mesoderm development between bladder and abdominal wall | Urinary incontinence, hydronephrosis, UTI, sexual dysfunction | Epispadias, pelvic floor defects |
| Cloacal exstrophy | WNT3, WNT9B, TP63, ISL1 | Severe disruption in the closure of the ventral abdominal wall and cloacal membrane | Urinary + bowel incontinence, obstructive uropathy, renal failure, fistula formation | Spinal anomalies, genital malformation |
| Posterior urethral valve | PCDH9, SALL1, BNC2, TBX5, PTK7 | Formed by remnants of the Wolffian duct or failure of the urogenital membrane to dissolve | Oligohydramnios, hydronephrosis, Potter phenotype, UTI, CKD | Exclusive to males |
| Ureteropelvic junction obstruction | No known genetic etiology | Aperistalsis of ureteral segments due to hypertrophy or absence of ureteral smooth muscles | Oligohydramnios, hydronephrosis, Potter phenotype, renal failure | |
| Ureteroceles | RET/GDNF, EYA1, PAX2, SALL1, FOXC1, FOXC2, SLIT2/ROBO2, AGTR1 | Failure of membrane at the distal ureter to completely dissolve and its subsequent dilation | Asymptomatic, bladder distension, urosepsis | Dual renal collecting systems |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brockwell, M.; Hergenrother, S.; Satariano, M.; Shah, R.; Raina, R. Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review. Cells 2024, 13, 1866. https://doi.org/10.3390/cells13221866
Brockwell M, Hergenrother S, Satariano M, Shah R, Raina R. Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review. Cells. 2024; 13(22):1866. https://doi.org/10.3390/cells13221866
Chicago/Turabian StyleBrockwell, Maximilian, Sean Hergenrother, Matthew Satariano, Raghav Shah, and Rupesh Raina. 2024. "Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review" Cells 13, no. 22: 1866. https://doi.org/10.3390/cells13221866
APA StyleBrockwell, M., Hergenrother, S., Satariano, M., Shah, R., & Raina, R. (2024). Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review. Cells, 13(22), 1866. https://doi.org/10.3390/cells13221866