
Impact of Intrauterine Insults on Fetal and Postnatal Cerebellar Development in Humans and Rodents

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Scope
3. Cerebellar Anatomy
4. Embryology of the Cerebellum
4.1. Neurogenesis
4.1.1. Glutamatergic Neuron Development
4.1.2. GABAergic Neuron Development
4.2. Embryology of the Precerebellar System
5. Extrinsic Deterrents Influencing Cerebellar Development
5.1. Maternal Substance (Ab)use and Cerebellar Maturation
5.1.1. Maternal Alcohol Consumption—Impact on Cerebellar Maturation
5.1.2. Maternal Smoking—Impact on Cerebellar Maturation
5.1.3. Maternal Cannabinoid Usage—Impact on Cerebellar Maturation
5.1.4. Maternal Opioid Usage—Impact on Cerebellar Maturation
5.2. Stress and Sleep and Cerebellar Development
5.2.1. Perinatal Stress—Impact on Cerebellar Maturation
5.2.2. Impact of Sleep Deprivation on Cerebellar Maturation
5.3. Intrauterine Growth Restriction—Impacts on Cerebellar Maturation
5.4. Intrauterine Infections and the Impact on Cerebellar Maturation
6. Vulnerable and Critical Periods in Cerebellar Development Affected by Intrauterine Insults
6.1. First Trimester (Until gw13 in Humans, Until E12/13 in Rodents)
6.2. Second Trimester (gw13–26 in Humans, E13–Birth in Rodents)
6.3. Third Trimester (gw27–gw40 in Humans)
6.4. Two Weeks Postnatal Rodents
6.5. Differential Spatiotemporal Vulnerability of Cerebellar Development
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dietrich, K.N.; Eskenazi, B.; Schantz, S.; Yolton, K.; Rauh, V.A.; Johnson, C.B.; Alkon, A.; Canfield, R.L.; Pessah, I.N.; Berman, R.F. Principles and Practices of Neurodevelopmental Assessment in Children: Lessons Learned from the Centers for Children’s Environmental Health and Disease Prevention Research. Environ. Health Perspect. 2005, 113, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Koning, I.V.; Dudink, J.; Groenenberg, I.A.L.; Willemsen, S.P.; Reiss, I.K.M.; Steegers-Theunissen, R.P.M. Prenatal Cerebellar Growth Trajectories and the Impact of Periconceptional Maternal and Fetal Factors. Hum. Reprod. 2017, 32, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Mwaniki, M.K.; Atieno, M.; Lawn, J.E.; Newton, C.R. Long-Term Neurodevelopmental Outcomes after Intrauterine and Neonatal Insults: A Systematic Review. Lancet 2012, 379, 445–452. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.J.; Meaney, M.J. Fetal Origins of Mental Health: The Developmental Origins of Health and Disease Hypothesis. Am. J. Psychiatry 2017, 174, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Carletti, B.; Rossi, F. Neurogenesis in the Cerebellum. Neuroscientist 2008, 14, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, B.; Mangold, U.; Sievers, J.; Berry, M. Derivation of Cerebellar Golgi Neurons from the External Granular Layer: Evidence from Explantation of External Granule Cells In Vivo. J. Comp. Neurol. 1985, 232, 511–522. [Google Scholar] [CrossRef]
- Haldipur, P.; Bharti, U.; Alberti, C.; Sarkar, C.; Gulati, G.; Iyengar, S.; Gressens, P.; Mani, S. Preterm Delivery Disrupts the Developmental Program of the Cerebellum. PLoS ONE 2011, 6, e23449. [Google Scholar] [CrossRef]
- van Essen, M.J.; Nayler, S.; Becker, E.B.E.; Jacob, J. Deconstructing Cerebellar Development Cell by Cell. PLoS Genet. 2020, 16, e1008630. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Osório, C.; White, J.J.; van Zoomeren, S.; van der Stok, H.; Xiong, B.; Nettersheim, I.H.; Mak, W.A.; Runge, M.; Fiocchi, F.R.; et al. Differential Spatiotemporal Development of Purkinje Cell Populations and Cerebellum-Dependent Sensorimotor Behaviors. Elife 2021, 10, e63668. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Gornati, S.V.; Canto, C.B.; Libster, A.M.; Schonewille, M.; De Zeeuw, C.I.; Hoebeek, F.E. Activity of Cerebellar Nuclei Neurons Correlates with ZebrinII Identity of Their Purkinje Cell Afferents. Cells 2021, 10, 2686. [Google Scholar] [CrossRef]
- Badura, A.; Verpeut, J.L.; Metzger, J.W.; Pereira, T.D.; Pisano, T.J.; Deverett, B.; Bakshinskaya, D.E.; Wang, S.S.-H. Normal Cognitive and Social Development Require Posterior Cerebellar Activity. Elife 2018, 7, e36401. [Google Scholar] [CrossRef] [PubMed]
- Bruchhage, M.M.K.; Bucci, M.-P.; Becker, E.B.E. Cerebellar Involvement in Autism and ADHD. Handb. Clin. Neurol. 2018, 155, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Sathyanesan, A.; Zhou, J.; Scafidi, J.; Heck, D.H.; Sillitoe, R.V.; Gallo, V. Emerging Connections between Cerebellar Development, Behaviour and Complex Brain Disorders. Nat. Rev. Neurosci. 2019, 20, 298–313. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.E.; Rey Hipolito, A.G.; Kim, L.H.; Kizek, D.J.; Perez, R.M.; Lin, T.; Sillitoe, R.V. Glutamatergic Cerebellar Neurons Differentially Contribute to the Acquisition of Motor and Social Behaviors. Nat. Commun. 2023, 14, 2771. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.; Bower, J.M.; Conforto, A.B.; Delgado-García, J.M.; da Guarda, S.N.F.; Gerwig, M.; Habas, C.; Hagura, N.; Ivry, R.B.; Mariën, P.; et al. Consensus Paper: Roles of the Cerebellum in Motor Control—The Diversity of Ideas on Cerebellar Involvement in Movement. Cerebellum 2012, 11, 457–487. [Google Scholar] [CrossRef]
- Schmahmann, J.D. Dysmetria of Thought: Clinical Consequences of Cerebellar Dysfunction on Cognition and Affect. Trends Cogn. Sci. 1998, 2, 362–371. [Google Scholar] [CrossRef]
- Andreasen, N.C.; Paradiso, S.; O’Leary, D.S. “Cognitive Dysmetria” as an Integrative Theory of Schizophrenia: A Dysfunction in Cortical-Subcortical-Cerebellar Circuitry? Schizophr. Bull. 1998, 24, 203–218. [Google Scholar] [CrossRef]
- Andreasen, N.C.; Pierson, R. The Role of the Cerebellum in Schizophrenia. Biol. Psychiatry 2008, 64, 81–88. [Google Scholar] [CrossRef]
- Wang, S.S.-H.; Kloth, A.D.; Badura, A. The Cerebellum, Sensitive Periods, and Autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef]
- Koning, I.V.; Tielemans, M.J.; Hoebeek, F.E.; Ecury-Goossen, G.M.; Reiss, I.K.M.; Steegers-Theunissen, R.P.M.; Dudink, J. Impacts on Prenatal Development of the Human Cerebellum: A Systematic Review. J. Matern. Fetal. Neonatal. Med. 2017, 30, 2461–2468. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y. Signaling Pathways in Cerebellar Granule Cells Development. Am. J. Stem Cells 2019, 8, 1–6. [Google Scholar] [PubMed]
- Haldipur, P.; Millen, K.J.; Aldinger, K.A. Human Cerebellar Development and Transcriptomics: Implications for Neurodevelopmental Disorders. Annu. Rev. Neurosci. 2022, 45, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Leto, K.; Arancillo, M.; Becker, E.B.E.; Buffo, A.; Chiang, C.; Ding, B.; Dobyns, W.B.; Dusart, I.; Haldipur, P.; Hatten, M.E.; et al. Consensus Paper: Cerebellar Development. Cerebellum 2015, 15, 789–828. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Hawkes, R. Cerebellar Cortical Organization: A One-Map Hypothesis. Nat. Rev. Neurosci. 2009, 10, 670–681. [Google Scholar] [CrossRef]
- Grodd, W.; Hülsmann, E.; Lotze, M.; Wildgruber, D.; Erb, M. Sensorimotor Mapping of the Human Cerebellum: FMRI Evidence of Somatotopic Organization. Hum. Brain Mapp. 2001, 13, 55–73. [Google Scholar] [CrossRef]
- Kelly, R.M.; Strick, P.L. Cerebellar Loops with Motor Cortex and Prefrontal Cortex of a Nonhuman Primate. J. Neurosci. 2003, 23, 8432–8444. [Google Scholar] [CrossRef]
- Schmahmann, J.D. The Cerebellum and Cognition. Neurosci. Lett. 2019, 688, 62–75. [Google Scholar] [CrossRef]
- Kansal, K.; Yang, Z.; Fishman, A.M.; Sair, H.I.; Ying, S.H.; Jedynak, B.M.; Prince, J.L.; Onyike, C.U. Structural Cerebellar Correlates of Cognitive and Motor Dysfunctions in Cerebellar Degeneration. Brain 2017, 140, 707–720. [Google Scholar] [CrossRef]
- Dudink, J.; Faneyte, S.J.; Hoebeek, F.E. Causes and Consequences of Structural Aberrations in Cerebellar Development. In Factors Affecting Neurodevelopment; Elsevier: Amsterdam, The Netherlands, 2021; pp. 371–382. [Google Scholar]
- De Zeeuw, C.I. Bidirectional Learning in Upbound and Downbound Microzones of the Cerebellum. Nat. Rev. Neurosci. 2021, 22, 92–110. [Google Scholar] [CrossRef]
- Brochu, G.; Maler, L.; Hawkes, R. Zebrin II: A Polypeptide Antigen Expressed Selectively by Purkinje Cells Reveals Compartments in Rat and Fish Cerebellum. J. Comp. Neurol. 1990, 291, 538–552. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Krueger-Naug, A.M.; Currie, R.W.; Hawkes, R. Constitutive Expression of the 25-KDa Heat Shock Protein Hsp25 Reveals Novel Parasagittal Bands of Purkinje Cells in the Adult Mouse Cerebellar Cortex. J. Comp. Neurol. 2000, 416, 383–397. [Google Scholar] [CrossRef]
- Hashizume, M.; Miyazaki, T.; Sakimura, K.; Watanabe, M.; Kitamura, K.; Kano, M. Disruption of Cerebellar Microzonal Organization in GluD2 (GluRδ2) Knockout Mouse. Front. Neural. Circuits 2013, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Marzban, H.; Watanabe, M.; Hawkes, R. Complementary Stripes of Phospholipase Cbeta3 and Cbeta4 Expression by Purkinje Cell Subsets in the Mouse Cerebellum. J. Comp. Neurol. 2006, 496, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.; Hawkes, R. Zones and Stripes. In Essentials of Cerebellum and Cerebellar Disorders A Primer for Graduate Students, 2nd ed.; Springer: Cham, Switzerland, 2023; pp. 99–106. [Google Scholar]
- Ament, S.A.; Cortes-Gutierrez, M.; Herb, B.R.; Mocci, E.; Colantuoni, C.; McCarthy, M.M. A Single-Cell Genomic Atlas for Maturation of the Human Cerebellum during Early Childhood. Sci. Transl. Med. 2023, 15, eade1283. [Google Scholar] [CrossRef] [PubMed]
- Coarelli, G.; Wirth, T.; Tranchant, C.; Koenig, M.; Durr, A.; Anheim, M. The Inherited Cerebellar Ataxias: An Update. J. Neurol. 2023, 270, 208–222. [Google Scholar] [CrossRef]
- Selimi, F.; Lohof, A.M.; Heitz, S.; Lalouette, A.; Jarvis, C.I.; Bailly, Y.; Mariani, J. Lurcher GRID2-Induced Death and Depolarization Can Be Dissociated in Cerebellar Purkinje Cells. Neuron 2003, 37, 813–819. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Yoshioka, Y.; Suzuki, K.; Miyazaki, T.; Koura, M.; Saigoh, K.; Kajimura, N.; Monobe, Y.; Kusunoki, S.; Matsuda, J.; et al. A New Mouse Allele of Glutamate Receptor Delta 2 with Cerebellar Atrophy and Progressive Ataxia. PLoS ONE 2014, 9, e107867. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Thomson, Z.; Phelps, I.G.; Haldipur, P.; Deng, M.; Timms, A.E.; Hirano, M.; Santpere, G.; Roco, C.; Rosenberg, A.B.; et al. Spatial and Cell Type Transcriptional Landscape of Human Cerebellar Development. Nat. Neurosci. 2021, 24, 1163–1175. [Google Scholar] [CrossRef]
- Zhong, S.; Wang, M.; Huang, L.; Chen, Y.; Ge, Y.; Zhang, J.; Shi, Y.; Dong, H.; Zhou, X.; Wang, B.; et al. Single-Cell Epigenomics and Spatiotemporal Transcriptomics Reveal Human Cerebellar Development. Nat. Commun. 2023, 14, 7613. [Google Scholar] [CrossRef]
- Hao, S.; Zhu, X.; Huang, Z.; Yang, Q.; Liu, H.; Wu, Y.; Zhan, Y.; Dong, Y.; Li, C.; Wang, H.; et al. Cross-Species Single-Cell Spatial Transcriptomic Atlases of the Cerebellar Cortex. Science 2024, 385, eado3927. [Google Scholar] [CrossRef]
- Voerman, S.; Urbanus, B.H.A.; Schonewille, M.; White, J.J.; De Zeeuw, C.I. Postsynaptic Plasticity of Purkinje Cells in Mice Is Determined by Molecular Identity. Commun. Biol. 2022, 5, 1328. [Google Scholar] [CrossRef] [PubMed]
- Voerman, S.; Broersen, R.; Swagemakers, S.M.A.; De Zeeuw, C.I.; van der Spek, P.J. Plasticity Mechanisms of Genetically Distinct Purkinje Cells. BioEssays 2024, 46, 2400008. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, Y.; Broussard, G.J.; Kislin, M.; Yuede, C.M.; Zhang, S.; Dietmann, S.; Gabel, H.; Zhao, G.; Wang, S.S.-H.; et al. Transcriptomic Mapping Uncovers Purkinje Neuron Plasticity Driving Learning. Nature 2022, 605, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Kozareva, V.; Martin, C.; Osorno, T.; Rudolph, S.; Guo, C.; Vanderburg, C.; Nadaf, N.; Regev, A.; Regehr, W.G.; Macosko, E. A Transcriptomic Atlas of Mouse Cerebellar Cortex Comprehensively Defines Cell Types. Nature 2021, 598, 214–219. [Google Scholar] [CrossRef]
- Zhou, H.; Lin, Z.; Voges, K.; Ju, C.; Gao, Z.; Bosman, L.W.; Ruigrok, T.J.; Hoebeek, F.E.; De Zeeuw, C.I.; Schonewille, M. Cerebellar Modules Operate at Different Frequencies. Elife 2014, 3, e02536. [Google Scholar] [CrossRef]
- Lackey, E.P.; Moreira, L.; Norton, A.; Hemelt, M.E.; Osorno, T.; Nguyen, T.M.; Macosko, E.Z.; Lee, W.-C.A.; Hull, C.A.; Regehr, W.G. Specialized Connectivity of Molecular Layer Interneuron Subtypes Leads to Disinhibition and Synchronous Inhibition of Cerebellar Purkinje Cells. Neuron 2024, 112, 2333–2348.e6. [Google Scholar] [CrossRef]
- Koops, R.N.; Canto, C.B.; Wu, B.; Schonewille, M.; Winkelman, B.H.J.; De Zeeuw, C.I. Role of Unipolar Brush Cells in the Vestibulocerebellum. In Essentials of Cerebellum and Cerebellar Disorders; Springer: Cham, Switzerland, 2023; pp. 243–258. [Google Scholar]
- Wang, X.; Novello, M.; Gao, Z.; Ruigrok, T.J.H.; De Zeeuw, C.I. Input and Output Organization of the Mesodiencephalic Junction for Cerebro-cerebellar Communication. J. Neurosci. Res. 2022, 100, 620–637. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Hoebeek, F.E.; Bosman, L.W.J.; Schonewille, M.; Witter, L.; Koekkoek, S.K. Spatiotemporal Firing Patterns in the Cerebellum. Nat. Rev. Neurosci. 2011, 12, 327–344. [Google Scholar] [CrossRef]
- Lange, W. Cell Number and Cell Density in the Cerebellar Cortex of Man and Some Other Mammals. Cell Tissue Res. 1975, 157, 115–124. [Google Scholar] [CrossRef]
- Jukic, A.M.; Baird, D.D.; Weinberg, C.R.; McConnaughey, D.R.; Wilcox, A.J. Length of Human Pregnancy and Contributors to Its Natural Variation. Hum. Reprod. 2013, 28, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Mukherjee, S.; Wilburn, A.N.; Cates, C.; Lewkowich, I.P.; Deshmukh, H.; Zacharias, W.J.; Chougnet, C.A. Pulmonary Consequences of Prenatal Inflammatory Exposures: Clinical Perspective and Review of Basic Immunological Mechanisms. Front. Immunol. 2020, 11, 1285. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, M.; Presicce, P.; Kallapur, S.G. Immunobiology of Acute Chorioamnionitis. Front. Immunol. 2020, 11, 649. [Google Scholar] [CrossRef] [PubMed]
- Haldipur, P.; Aldinger, K.A.; Bernardo, S.; Deng, M.; Timms, A.E.; Overman, L.M.; Winter, C.; Lisgo, S.N.; Razavi, F.; Silvestri, E.; et al. Spatiotemporal Expansion of Primary Progenitor Zones in the Developing Human Cerebellum. Science 2019, 366, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Joyner, A.L. Otx2 and Gbx2 Are Required for Refinement and Not Induction of Mid-Hindbrain Gene Expression. Development 2001, 128, 4979–4991. [Google Scholar] [CrossRef] [PubMed]
- Beckinghausen, J.; Sillitoe, R.V. Insights into Cerebellar Development and Connectivity. Neurosci. Lett. 2019, 688, 2–13. [Google Scholar] [CrossRef]
- Wingate, R.J.T. The Rhombic Lip and Early Cerebellar Development. Curr. Opin. Neurobiol. 2001, 11, 82–88. [Google Scholar] [CrossRef]
- Millet, S.; Campbell, K.; Epstein, D.J.; Losos, K.; Harris, E.; Joyner, A.L. A Role for Gbx2 in Repression of Otx2 and Positioning the Mid/Hindbrain Organizer. Nature 1999, 401, 161–164. [Google Scholar] [CrossRef]
- Broccoli, V.; Boncinelli, E.; Wurst, W. The Caudal Limit of Otx2 Expression Positions the Isthmic Organizer. Nature 1999, 401, 164–168. [Google Scholar] [CrossRef]
- Chizhikov, V.V.; Lindgren, A.G.; Mishima, Y.; Roberts, R.W.; Aldinger, K.A.; Miesegaes, G.R.; Currle, D.S.; Monuki, E.S.; Millen, K.J. Lmx1a Regulates Fates and Location of Cells Originating from the Cerebellar Rhombic Lip and Telencephalic Cortical Hem. Proc. Natl. Acad. Sci. USA 2010, 107, 10725–10730. [Google Scholar] [CrossRef]
- Corrales, J.D.; Rocco, G.L.; Blaess, S.; Guo, Q.; Joyner, A.L. Spatial Pattern of Sonic Hedgehog Signaling through Gli Genes during Cerebellum Development. Development 2004, 131, 5581–5590. [Google Scholar] [CrossRef]
- Belzunce, I.; Belmonte-Mateos, C.; Pujades, C. The Interplay of Atoh1 Genes in the Lower Rhombic Lip during Hindbrain Morphogenesis. PLoS ONE 2020, 15, e0228225. [Google Scholar] [CrossRef] [PubMed]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 Induces Cerebral Small-Vessel Disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef] [PubMed]
- Aldinger, K.A.; Lehmann, O.J.; Hudgins, L.; Chizhikov, V.V.; Bassuk, A.G.; Ades, L.C.; Krantz, I.D.; Dobyns, W.B.; Millen, K.J. FOXC1 Is Required for Normal Cerebellar Development and Is a Major Contributor to Chromosome 6p25.3 Dandy-Walker Malformation. Nat. Genet. 2009, 41, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Aldinger, K.A.; Timms, A.E.; Thomson, Z.; Mirzaa, G.M.; Bennett, J.T.; Rosenberg, A.B.; Roco, C.M.; Hirano, M.; Abidi, F.; Haldipur, P.; et al. Redefining the Etiologic Landscape of Cerebellar Malformations. Am. J. Hum. Genet. 2019, 105, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.R.; Musci, T.S.; Neumann, P.E.; Capecchi, M.R. Swaying Is a Mutant Allele of the Proto-Oncogene Wnt-1. Cell 1991, 67, 969–976. [Google Scholar] [CrossRef]
- Lowenstein, E.D.; Cui, K.; Hernandez-Miranda, L.R. Regulation of Early Cerebellar Development. FEBS J. 2023, 290, 2786–2804. [Google Scholar] [CrossRef]
- Martinez, S.; Andreu, A.; Mecklenburg, N.; Echevarria, D. Cellular and Molecular Basis of Cerebellar Development. Front. Neuroanat. 2013, 7, 18. [Google Scholar] [CrossRef]
- Akazawa, C.; Ishibashi, M.; Shimizu, C.; Nakanishi, S.; Kageyama, R. A Mammalian Helix-Loop-Helix Factor Structurally Related to the Product of Drosophila Proneural Gene Atonal Is a Positive Transcriptional Regulator Expressed in the Developing Nervous System. J. Biol. Chem. 1995, 270, 8730–8738. [Google Scholar] [CrossRef]
- Hoshino, M.; Nakamura, S.; Mori, K.; Kawauchi, T.; Terao, M.; Nishimura, Y.V.; Fukuda, A.; Fuse, T.; Matsuo, N.; Sone, M.; et al. Ptf1a, a BHLH Transcriptional Gene, Defines GABAergic Neuronal Fates in Cerebellum. Neuron 2005, 47, 201–213. [Google Scholar] [CrossRef]
- Machold, R.; Fishell, G. Math1 Is Expressed in Temporally Discrete Pools of Cerebellar Rhombic-Lip Neural Progenitors. Neuron 2005, 48, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sekerková, G.; Ilijic, E.; Mugnaini, E. Time of Origin of Unipolar Brush Cells in the Rat Cerebellum as Observed by Prenatal Bromodeoxyuridine Labeling. Neuroscience 2004, 127, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Chizhikov, V.; Millen, K.J. Development and Malformations of the Cerebellum in Mice. Mol. Genet. Metab. 2003, 80, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Consalez, G.G.; Goldowitz, D.; Casoni, F.; Hawkes, R. Origins, Development, and Compartmentation of the Granule Cells of the Cerebellum. Front. Neural. Circuits 2021, 14, 611841. [Google Scholar] [CrossRef] [PubMed]
- Komuro, H.; Yacubova, E.; Yacubova, E.; Rakic, P. Mode and Tempo of Tangential Cell Migration in the Cerebellar External Granular Layer. J. Neurosci. 2001, 21, 527–540. [Google Scholar] [CrossRef]
- Helms, A.W.; Gowan, K.; Abney, A.; Savage, T.; Johnson, J.E. Overexpression of MATH1 Disrupts the Coordination of Neural Differentiation in Cerebellum Development. Mol. Cell. Neurosci. 2001, 17, 671–682. [Google Scholar] [CrossRef]
- Wizeman, J.W.; Guo, Q.; Wilion, E.M.; Li, J.Y. Specification of Diverse Cell Types during Early Neurogenesis of the Mouse Cerebellum. Elife 2019, 8, e42388. [Google Scholar] [CrossRef]
- Zordan, P.; Croci, L.; Hawkes, R.; Consalez, G.G. Comparative Analysis of Proneural Gene Expression in the Embryonic Cerebellum. Dev. Dyn. 2008, 237, 1726–1735. [Google Scholar] [CrossRef]
- Miterko, L.N.; Sillitoe, R.V.; Hawkes, R. Zones and Stripes: Development of Cerebellar Topography. In Handbook of the Cerebellum and Cerebellar Disorders, 2nd ed.; Springer: Cham, Switzerland, 2021; Volume 3. [Google Scholar]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and Yotari Disrupt the Disabled Gene and Produce a Reeler-like Phenotype in Mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef]
- Howell, B.W.; Hawkes, R.; Soriano, P.; Cooper, J.A. Neuronal Position in the Developing Brain Is Regulated by Mouse Disabled-1. Nature 1997, 389, 733–737. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Scares, H.D.; Morgan, J.I.; Curran, T. A Protein Related to Extracellular Matrix Proteins Deleted in the Mouse Mutant Reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-H.; Marzban, H.; Croci, L.; Consalez, G.G.; Hawkes, R. Purkinje Cell Subtype Specification in the Cerebellar Cortex: Early B-Cell Factor 2 Acts to Repress the Zebrin II-Positive Purkinje Cell Phenotype. Neuroscience 2008, 153, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tran-Anh, K.; Hirata, T.; Sugihara, I. Striped Distribution Pattern of Purkinje Cells of Different Birthdates in the Mouse Cerebellar Cortex Studied with the Neurog2-CreER Transgenic Line. Neuroscience 2021, 462, 122–140. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.E.; Lackey, E.P.; Perez, R.; Ișleyen, F.S.; Brown, A.M.; Donofrio, S.G.; Lin, T.; Zoghbi, H.Y.; Sillitoe, R.V. Maturation of Purkinje Cell Firing Properties Relies on Neurogenesis of Excitatory Neurons. Elife 2021, 10, e68045. [Google Scholar] [CrossRef]
- McKay, B.E.; Turner, R.W. Physiological and Morphological Development of the Rat Cerebellar Purkinje Cell. J. Physiol. 2005, 567, 829–850. [Google Scholar] [CrossRef]
- Beekhof, G.C.; Schonewille, M. Lobule-Related Action Potential Shape- and History-Dependent Current Integration in Purkinje Cells of Adult and Developing Mice. Cells 2023, 12, 623. [Google Scholar] [CrossRef]
- Kapfhammer, J.P. Cellular and Molecular Control of Dendritic Growth and Development of Cerebellar Purkinje Cells. Prog. Histochem. Cytochem. 2004, 39, 131–182. [Google Scholar] [CrossRef]
- Busch, S.E.; Hansel, C. Climbing Fiber Multi-Innervation of Mouse Purkinje Dendrites with Arborization Common to Human. Science 2023, 381, 420–427. [Google Scholar] [CrossRef]
- Sultan, F.; Czubayko, U.; Thier, P. Morphological Classification of the Rat Lateral Cerebellar Nuclear Neurons by Principal Component Analysis. J. Comp. Neurol. 2003, 455, 139–155. [Google Scholar] [CrossRef]
- Leto, K.; Carletti, B.; Williams, I.M.; Magrassi, L.; Rossi, F. Different Types of Cerebellar GABAergic Interneurons Originate from a Common Pool of Multipotent Progenitor Cells. J. Neurosci. 2006, 26, 11682–11694. [Google Scholar] [CrossRef]
- Leto, K.; Bartolini, A.; Yanagawa, Y.; Obata, K.; Magrassi, L.; Schilling, K.; Rossi, F. Laminar Fate and Phenotype Specification of Cerebellar GABAergic Interneurons. J. Neurosci. 2009, 29, 7079–7091. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, P.; Parras, C.; Guillemot, F.; Rossi, F.; Wassef, M. Origins and Control of the Differentiation of Inhibitory Interneurons and Glia in the Cerebellum. Dev. Biol. 2009, 328, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje Cell Death in the Cerebellum. Prog. Neurobiol. 2003, 70, 473–507. [Google Scholar] [CrossRef] [PubMed]
- Chedotal, A.; Sotelo, C. Early Development of Olivocerebellar Projections in the Fetal Rat Using CGRP Immunocytochemistry. Eur. J. Neurosci. 1992, 4, 1159–1179. [Google Scholar] [CrossRef]
- Sugihara, I. Microzonal Projection and Climbing Fiber Remodeling in Single Olivocerebellar Axons of Newborn Rats at Postnatal Days 4–7. J. Comp. Neurol. 2005, 487, 93–106. [Google Scholar] [CrossRef]
- Rodriguez, C.I.; Dymecki, S.M. Origin of the Precerebellar System. Neuron 2000, 27, 475–486. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Marcussen, L.; West, J.R. A Single Day of Alcohol Exposure During the Brain Growth Spurt Induces Brain Weight Restriction and Cerebellar Purkinje Cell Loss. Alcohol 1989, 7, 107–114. [Google Scholar] [CrossRef]
- Sim, M.E.; Lyoo, I.K.; Streeter, C.C.; Covell, J.; Sarid-Segal, O.; Ciraulo, D.A.; Kim, M.J.; Kaufman, M.J.; Yurgelun-Todd, D.A.; Renshaw, P.F. Cerebellar Gray Matter Volume Correlates with Duration of Cocaine Use in Cocaine-Dependent Subjects. Neuropsychopharmacology 2007, 32, 2229–2237. [Google Scholar] [CrossRef]
- Manto, M.; Perrotta, G. Toxic-Induced Cerebellar Syndrome: From the Fetal Period to the Elderly. Handb. Clin. Neurol. 2018, 155, 333–352. [Google Scholar]
- Dow-Edwards, D.L.; Benveniste, H.; Behnke, M.; Bandstra, E.S.; Singer, L.T.; Hurd, Y.L.; Stanford, L.R. Neuroimaging of Prenatal Drug Exposure. Neurotoxicol. Teratol. 2006, 28, 386–402. [Google Scholar] [CrossRef]
- Falk, L.; Nordberg, A.; Seiger, Å.; Kjældgaard, A.; Hellström-Lindahl, E. Smoking during Early Pregnancy Affects the Expression Pattern of Both Nicotinic and Muscarinic Acetylcholine Receptors in Human First Trimester Brainstem and Cerebellum. Neuroscience 2005, 132, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Lavezzi, A.M.; Corna, M.F.; Repetti, M.L.; Matturri, L. Cerebellar Purkinje Cell Vulnerability to Prenatal Nicotine Exposure in Sudden Unexplained Perinatal Death. Folia Neuropathol. 2013, 4, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; Khurdayan, V.K.; Goody, R.J.; Nath, A.; Saria, A.; Pauly, J.R. Selective Vulnerability of Cerebellar Granule Neuroblasts and Their Progeny to Drugs with Abuse Liability. Cerebellum 2003, 2, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A.; Altman, J.; Russo, R.J.; Zhang, X. Timetables of Neurogenesis in the Human Brain Based on Experimentally Determined Patterns in the Rat. Neurotoxicology 1993, 14, 83–144. [Google Scholar]
- Riley, E.P.; McGee, C.L. Fetal Alcohol Spectrum Disorders: An Overview with Emphasis on Changes in Brain and Behavior. Exp. Biol. Med. 2005, 230, 357–365. [Google Scholar] [CrossRef]
- Jaatinen, P.; Rintala, J. Mechanisms of Ethanol-Induced Degeneration in the Developing, Mature, and Aging Cerebellum. Cerebellum 2008, 7, 332–347. [Google Scholar] [CrossRef]
- Coviello, C.; Keunen, K.; Kersbergen, K.J.; Groenendaal, F.; Leemans, A.; Peels, B.; Isgum, I.; Viergever, M.A.; de Vries, L.S.; Buonocore, G.; et al. Effects of Early Nutrition and Growth on Brain Volumes, White Matter Microstructure, and Neurodevelopmental Outcome in Preterm Newborns. Pediatr. Res. 2018, 83, 102–110. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Shaikh, A.G. Alcohol Toxicity in the Developing Cerebellum. Diagnostics 2024, 14, 1415. [Google Scholar] [CrossRef]
- Handmaker, N.S.; Rayburn, W.F.; Meng, C.; Bell, J.B.; Rayburn, B.B.; Rappaport, V.J. Impact of Alcohol Exposure After Pregnancy Recognition on Ultrasonographic Fetal Growth Measures. Alcohol. Clin. Exp. Res. 2006, 30, 892–898. [Google Scholar] [CrossRef]
- Astley, S.J.; Aylward, E.H.; Olson, H.C.; Kerns, K.; Brooks, A.; Coggins, T.E.; Davies, J.; Dorn, S.; Gendler, B.; Jirikowic, T.; et al. Magnetic Resonance Imaging Outcomes from a Comprehensive Magnetic Resonance Study of Children with Fetal Alcohol Spectrum Disorders. Alcohol. Clin. Exp. Res. 2009, 33, 1671–1689. [Google Scholar] [CrossRef]
- Autti-Rämö, I.; Autti, T.; Korkman, M.; Kettunen, S.; Salonen, O.; Valanne, L. MRI Findings in Children with School Problems Who Had Been Exposed Prenatally to Alcohol. Dev. Med. Child Neurol. 2002, 44, 98. [Google Scholar] [CrossRef] [PubMed]
- Sowell, E.R.; Jernigan, T.L.; Mattson, S.N.; Riley, E.P.; Sobel, D.F.; Jones, K.L. Abnormal Development of the Cerebellar Vermis in Children Prenatally Exposed to Alcohol: Size Reduction in Lobules I-V. Alcohol. Clin. Exp. Res. 1996, 20, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Archibald, S.L.; Fennema-Notestine, C.; Gamst, A.; Riley, E.P.; Mattson, S.N.; Jernigan, T.L. Brain Dysmorphology in Individuals with Severe Prenatal Alcohol Exposure. Dev. Med. Child. Neurol. 2001, 43, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Moore, E.M.; Lane, B.; Pohl, K.M.; Riley, E.P.; Pfefferbaum, A. Graded Cerebellar Lobular Volume Deficits in Adolescents and Young Adults with Fetal Alcohol Spectrum Disorders (FASD). Cereb. Cortex 2020, 30, 4729–4746. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Meintjes, E.M.; Molteno, C.D.; Spottiswoode, B.S.; Dodge, N.C.; Alhamud, A.A.; Stanton, M.E.; Peterson, B.S.; Jacobson, J.L.; Jacobson, S.W. White Matter Integrity of the Cerebellar Peduncles as a Mediator of Effects of Prenatal Alcohol Exposure on Eyeblink Conditioning. Hum. Brain Mapp. 2015, 36, 2470–2482. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Chong, S.; Tieng, Q.M.; Mardon, K.; Galloway, G.J.; Kurniawan, N.D. Radiological Studies of Fetal Alcohol Spectrum Disorders in Humans and Animal Models: An Updated Comprehensive Review. Magn. Reson. Imaging 2017, 43, 10–26. [Google Scholar] [CrossRef]
- du Plessis, L.; Jacobson, J.L.; Jacobson, S.W.; Hess, A.T.; van der Kouwe, A.; Avison, M.J.; Molteno, C.D.; Stanton, M.E.; Stanley, J.A.; Peterson, B.S.; et al. An In Vivo 1H Magnetic Resonance Spectroscopy Study of the Deep Cerebellar Nuclei in Children with Fetal Alcohol Spectrum Disorders. Alcohol. Clin. Exp. Res. 2014, 38, 1330–1338. [Google Scholar] [CrossRef]
- Diwadkar, V.A.; Meintjes, E.M.; Goradia, D.; Dodge, N.C.; Warton, C.; Molteno, C.D.; Jacobson, S.W.; Jacobson, J.L. Differences in Cortico-striatal-cerebellar Activation during Working Memory in Syndromal and Nonsyndromal Children with Prenatal Alcohol Exposure. Hum. Brain Mapp. 2013, 34, 1931–1945. [Google Scholar] [CrossRef]
- Parnell, S.E.; O’Leary-Moore, S.K.; Godin, E.A.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic Resonance Microscopy Defines Ethanol-Induced Brain Abnormalities in Prenatal Mice: Effects of Acute Insult on Gestational Day 8. Alcohol. Clin. Exp. Res. 2009, 33, 1001–1011. [Google Scholar] [CrossRef]
- Parnell, S.E.; Holloway, H.T.; O’Leary-Moore, S.K.; Dehart, D.B.; Paniaqua, B.; Oguz, I.; Budin, F.; Styner, M.A.; Johnson, G.A.; Sulik, K.K. Magnetic Resonance Microscopy-Based Analyses of the Neuroanatomical Effects of Gestational Day 9 Ethanol Exposure in Mice. Neurotoxicol. Teratol. 2013, 39, 77–83. [Google Scholar] [CrossRef]
- Fish, E.W.; Holloway, H.T.; Rumple, A.; Baker, L.K.; Wieczorek, L.A.; Moy, S.S.; Paniagua, B.; Parnell, S.E. Acute Alcohol Exposure during Neurulation: Behavioral and Brain Structural Consequences in Adolescent C57BL/6J Mice. Behav. Brain Res. 2016, 311, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.W.; Wieczorek, L.A.; Rumple, A.; Suttie, M.; Moy, S.S.; Hammond, P.; Parnell, S.E. The Enduring Impact of Neurulation Stage Alcohol Exposure: A Combined Behavioral and Structural Neuroimaging Study in Adult Male and Female C57BL/6J Mice. Behav. Brain Res. 2018, 338, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Godin, E.A.; O’Leary-Moore, S.K.; Khan, A.A.; Parnell, S.E.; Ament, J.J.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic Resonance Microscopy Defines Ethanol-Induced Brain Abnormalities in Prenatal Mice: Effects of Acute Insult on Gestational Day 7. Alcohol. Clin. Exp. Res. 2010, 34, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Bhalla, R.; Cowin, G.; Stimson, D.H.R.; Song, X.; Chong, S.; Jackson, A.; Trigg, W.J.; Tieng, Q.M.; Mardon, K.; et al. GABAa Receptor Density Alterations Revealed in a Mouse Model of Early Moderate Prenatal Ethanol Exposure Using [18F]AH114726. Nuclear Med. Biol. 2020, 88, 44–51. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Majrashi, M.; Ramesh, S.; Govindarajulu, M.; Bloemer, J.; Fujihashi, A.; Crump, B.R.; Hightower, H.; Bhattacharya, S.; Moore, T.; et al. Assessment of the Cerebellar Neurotoxic Effects of Nicotine in Prenatal Alcohol Exposure in Rats. Life Sci. 2018, 194, 177–184. [Google Scholar] [CrossRef]
- Marcussen, B.L.; Goodlett, C.R.; Mahoney, J.C.; West, J.R. Developing Rat Purkinje Cells Are More Vulnerable to Alcohol-Induced Depletion During Differentiation than During Neurogenesis. Alcohol 1994, 11, 147–156. [Google Scholar] [CrossRef]
- Nirgudkar, P.; Taylor, D.H.; Yanagawa, Y.; Valenzuela, C.F. Ethanol Exposure during Development Reduces GABAergic/Glycinergic Neuron Numbers and Lobule Volumes in the Mouse Cerebellar Vermis. Neurosci. Lett. 2016, 632, 86–91. [Google Scholar] [CrossRef]
- González-Burgos, I.; Alejandre-Gómez, M. Cerebellar Granule Cell and Bergmann Glial Cell Maturation in the Rat Is Disrupted by Pre- and Post-natal Exposure to Moderate Levels of Ethanol. Int. J. Dev. Neurosci. 2005, 23, 383–388. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Eilers, A.T. Alcohol-Induced Purkinje Cell Loss with a Single Binge Exposure in Neonatal Rats: A Stereological Study of Temporal Windows of Vulnerability. Alcohol. Clin. Exp. Res. 1997, 21, 738–744. [Google Scholar] [CrossRef]
- Karaçay, B.; Li, S.; Bonthius, D.J. Maturation-Dependent Alcohol Resistance in the Developing Mouse: Cerebellar Neuronal Loss and Gene Expression during Alcohol-Vulnerable and -Resistant Periods. Alcohol. Clin. Exp. Res. 2008, 32, 1439–1450. [Google Scholar] [CrossRef]
- Hamre, K.M.; West, J.R. The Effects of the Timing of Ethanol Exposure during the Brain Growth Spurt on the Number of Cerebellar Purkinje and Granule Cell Nuclear Profiles. Alcohol. Clin. Exp. Res. 1993, 17, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.A.; Parnell, S.E.; West, J.R.; Chen, W.-J.A.; Parnell, S.E.; Neonatal, J.R.W. Neonatal Alcohol and Nicotine Exposure Limits Brain Growth and Depletes Cerebellar Purkinje Cells. Alcohol 1998, 15, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.M.; Phelan, K.D.; Han, L.; Smith, R.R.; Xie, J.; Douglas, J.C.; Drew, P.D. Protection of Neurons and Microglia against Ethanol in a Mouse Model of Fetal Alcohol Spectrum Disorders by Peroxisome Proliferator-Activated Receptor-γ Agonists. Brain Behav. Immun. 2011, 25, S137–S145. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rowe, J.; Eskue, K.; West, J.R.; Maier, S.E. Alcohol Exposure on Postnatal Day 5 Induces Purkinje Cell Loss and Evidence of Purkinje Cell Degradation in Lobule I of Rat Cerebellum. Alcohol 2008, 42, 295–302. [Google Scholar] [CrossRef]
- Topper, L.A.; Baculis, B.C.; Valenzuela, C.F. Exposure of Neonatal Rats to Alcohol Has Differential Effects on Neuroinflammation and Neuronal Survival in the Cerebellum and Hippocampus. J. Neuroinflamm. 2015, 12, 160. [Google Scholar] [CrossRef]
- Light, K.E.; Hayar, A.M.; Pierce, D.R. Electrophysiological and Immunohistochemical Evidence for an Increase in GABAergic Inputs and HCN Channels in Purkinje Cells That Survive Developmental Ethanol Exposure. Cerebellum 2015, 14, 398–412. [Google Scholar] [CrossRef]
- Pierce, D.R.; Hayar, A.; Williams, D.K.; Light, K.E. Developmental Alterations in Olivary Climbing Fiber Distribution Following Postnatal Ethanol Exposure in the Rat. Neuroscience 2010, 169, 1438–1448. [Google Scholar] [CrossRef]
- Pierce, D.R.; Hayar, A.; Williams, D.K.; Light, K.E. Olivary Climbing Fiber Alterations in PN40 Rat Cerebellum Following Postnatal Ethanol Exposure. Brain Res. 2011, 1378, 54–65. [Google Scholar] [CrossRef]
- Todd, D.; Clapp, M.; Dains, P.; Karacay, B.; Bonthius, D.J. Purkinje Cell-Specific Deletion of CREB Worsens Alcohol-Induced Cerebellar Neuronal Losses and Motor Deficits. Alcohol 2022, 101, 27–35. [Google Scholar] [CrossRef]
- Gursky, Z.H.; Johansson, J.R.; Klintsova, A.Y. Postnatal Alcohol Exposure and Adolescent Exercise Have Opposite Effects on Cerebellar Microglia in Rat. Int. J. Dev. Neurosci. 2020, 80, 558–571. [Google Scholar] [CrossRef]
- Cealie, M.K.Y.; Douglas, J.C.; Swan, H.K.; Vonkaenel, E.D.; McCall, M.N.; Drew, P.D.; Majewska, A.K. Developmental Ethanol Exposure Impacts Purkinje Cells but Not Microglia in the Young Adult Cerebellum. Cells 2024, 13, 386. [Google Scholar] [CrossRef] [PubMed]
- Cealie, M.K.Y.; Douglas, J.C.; Le, L.H.D.; Vonkaenel, E.D.; McCall, M.N.; Drew, P.D.; Majewska, A.K. Developmental Ethanol Exposure Has Minimal Impact on Cerebellar Microglial Dynamics, Morphology, and Interactions with Purkinje Cells during Adolescence. Front. Neurosci. 2023, 17, 1176581. [Google Scholar] [CrossRef] [PubMed]
- Bolbanabad, H.M.; Anvari, E.; Rezai, M.J.; Moayeri, A.; Kaffashian, M.R. Amelioration of Cerebellar Dysfunction in Rats Following Postnatal Ethanol Exposure Using Low-Intensity Pulsed Ultrasound. J. Chem. Neuroanat. 2017, 81, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; Winters, Z.; Karacay, B.; Bousquet, S.L.; Bonthius, D.J. Importance of Genetics in Fetal Alcohol Effects: Null Mutation of the NNOS Gene Worsens Alcohol-Induced Cerebellar Neuronal Losses and Behavioral Deficits. Neurotoxicology 2015, 46, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Gundogan, F.; Tong, M.; Chen, W.C.; Kim, C.; Nguyen, Q.-G.; Yu, R.; de la Monte, S.M. Chronic Prenatal Ethanol Exposure Disrupts WNT Signaling In Adolescent Cerebella. J. Clin. Exp. Pathol. 2013, 3, 144. [Google Scholar] [CrossRef]
- Tong, M.; Ziplow, J.; Chen, W.C.; Nguyen, Q.-G.; Kim, C.; De La Monte, S.M. Motor Function Deficits Following Chronic Prenatal Ethanol Exposure Are Linked to Impairments in Insulin/IGF, Notch and Wnt Signaling in the Cerebellum. J. Diabetes Metab. 2013, 4, 238. [Google Scholar] [CrossRef]
- Holloway, K.N.; Douglas, J.C.; Rafferty, T.M.; Majewska, A.K.; Kane, C.J.M.; Drew, P.D. Ethanol-Induced Cerebellar Transcriptomic Changes in a Postnatal Model of Fetal Alcohol Spectrum Disorders: Focus on Disease Onset. Front. Neurosci. 2023, 17, 1154637. [Google Scholar] [CrossRef]
- Niedzwiedz-Massey, V.M.; Douglas, J.C.; Rafferty, T.; Kane, C.J.M.; Drew, P.D. Ethanol Effects on Cerebellar Myelination in a Postnatal Mouse Model of Fetal Alcohol Spectrum Disorders. Alcohol 2021, 96, 43–53. [Google Scholar] [CrossRef]
- Drew, P.D.; Johnson, J.W.; Douglas, J.C.; Phelan, K.D.; Kane, C.J.M. Pioglitazone Blocks Ethanol Induction of Microglial Activation and Immune Responses in the Hippocampus, Cerebellum, and Cerebral Cortex in a Mouse Model of Fetal Alcohol Spectrum Disorders. Alcohol. Clin. Exp. Res. 2015, 39, 445–454. [Google Scholar] [CrossRef]
- Kane, C.J.M.; Douglas, J.C.; Rafferty, T.; Johnson, J.W.; Niedzwiedz-Massey, V.M.; Phelan, K.D.; Majewska, A.K.; Drew, P.D. Ethanol Modulation of Cerebellar Neuroinflammation in a Postnatal Mouse Model of Fetal Alcohol Spectrum Disorders. J. Neurosci. Res. 2021, 99, 1986–2007. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, H.; Xu, M.; Frank, J.A.; Luo, J. Role of MCP-1 and CCR2 in Ethanol-Induced Neuroinflammation and Neurodegeneration in the Developing Brain. J. Neuroinflamm. 2018, 15, 197. [Google Scholar] [CrossRef] [PubMed]
- Ieraci, A.; Herrera, D.G. Nicotinamide Inhibits Ethanol-Induced Caspase-3 and PARP-1 Over-Activation and Subsequent Neurodegeneration in the Developing Mouse Cerebellum. Cerebellum 2018, 17, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.C.H.; Jain, P.; Michealson, M.A.; Choi, H.; Ellsworth-Kopkowski, A.; Valenzuela, C.F. Recent Breakthroughs in Understanding the Cerebellum’s Role in Fetal Alcohol Spectrum Disorder: A Systematic Review. Alcohol 2024, 119, 37–71. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, M.; Korkeila, J.; Parkkola, R.; Lapinleimu, H.; Haataja, L.; Lehtonen, L. Maternal Smoking during Pregnancy and Regional Brain Volumes in Preterm Infants. J. Pediatr. 2010, 156, 185–190.e1. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Boer, O.D.; Felix, J.F.; Muetzel, R.L.; Franken, I.H.A.; Cecil, C.A.M.; El Marroun, H. Association of Maternal Tobacco Use During Pregnancy with Preadolescent Brain Morphology Among Offspring. JAMA Netw. Open 2022, 5, e2224701. [Google Scholar] [CrossRef] [PubMed]
- Bublitz, M.H.; Stroud, L.R. Maternal Smoking During Pregnancy and Offspring Brain Structure and Function: Review and Agenda for Future Research. Nicotine Tob. Res. 2012, 14, 388–397. [Google Scholar] [CrossRef]
- Roza, S.J.; Verburg, B.O.; Jaddoe, V.W.V.; Hofman, A.; Mackenbach, J.P.; Steegers, E.A.P.; Witteman, J.C.M.; Verhulst, F.C.; Tiemeier, H. Effects of Maternal Smoking in Pregnancy on Prenatal Brain Development. The Generation R Study. Eur. J. Neurosci. 2007, 25, 611–617. [Google Scholar] [CrossRef]
- Bennett, D.S.; Mohamed, F.B.; Carmody, D.P.; Bendersky, M.; Patel, S.; Khorrami, M.; Faro, S.H.; Lewis, M. Response Inhibition among Early Adolescents Prenatally Exposed to Tobacco: An FMRI Study. Neurotoxicol. Teratol. 2009, 31, 283–290. [Google Scholar] [CrossRef]
- Longo, C.A.; Fried, P.A.; Cameron, I.; Smith, A.M. The Long-Term Effects of Prenatal Nicotine Exposure on Response Inhibition: An FMRI Study of Young Adults. Neurotoxicol. Teratol. 2013, 39, 9–18. [Google Scholar] [CrossRef]
- Martin-Ruiz, C.M.; Lee, M.; Perry, R.H.; Baumann, M.; Court, J.A.; Perry, E.K. Molecular Analysis of Nicotinic Receptor Expression in Autism. Mol. Brain Res. 2004, 123, 81–90. [Google Scholar] [CrossRef]
- Lee, M. Nicotinic Receptor Abnormalities in the Cerebellar Cortex in Autism. Brain 2002, 125, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R.; Kellar, K.J. Nicotinic Cholinergic Receptors in the Rat Cerebellum: Multiple Heteromeric Subtypes. J. Neurosci. 2005, 25, 9258–9265. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.; Court, J.A.; Martin-Ruiz, C.M.; Jaros, E.; Perry, R.; Volsen, S.G.; Bose, S.; Evans, N.; Ince, P.; Kuryatov, A.; et al. Immunohistochemical Localisation of Nicotinic Acetylcholine Receptor Subunits in Human Cerebellum. Neuroscience 2002, 113, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, M.O. Association of Smoking During Pregnancy with Compromised Brain Development in Offspring. JAMA Netw. Open 2022, 5, e2224714. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, N.R.; Day, K.D.; Payakachat, N.; Franks, A.M.; McCain, K.R.; Ragland, D. Use and Risk Perception of Electronic Nicotine Delivery Systems and Tobacco in Pregnancy. Women’s Health Issues 2018, 28, 251–257. [Google Scholar] [CrossRef]
- Stroud, L.R.; Papandonatos, G.D.; Borba, K.; Kehoe, T.; Scott-Sheldon, L.A.J. Flavored Electronic Cigarette Use, Preferences, and Perceptions in Pregnant Mothers: A Correspondence Analysis Approach. Addict. Behav. 2019, 91, 21–29. [Google Scholar] [CrossRef]
- O’Leary, K.T.; Leslie, F.M. Developmental Regulation of Nicotinic Acetylcholine Receptor-mediated [3H]Norepinephrine Release from Rat Cerebellum. J. Neurochem. 2003, 84, 952–959. [Google Scholar] [CrossRef]
- Didier, M.; Berman, S.A.; Lindstrom, J.; Bursztajn, S. Characterization of Nicotinic Acetylcholine Receptors Expressed in Primary Cultures of Cerebellar Granule Cells. Mol. Brain Res. 1995, 30, 17–28. [Google Scholar] [CrossRef]
- Tribollet, E.; Bertrand, D.; Marguerat, A.; Raggenbass, M. Comparative Distribution of Nicotinic Receptor Subtypes during Development, Adulthood and Aging: An Autoradiographic Study in the Rat Brain. Neuroscience 2004, 124, 405–420. [Google Scholar] [CrossRef]
- Rossi, D.J.; Hamann, M.; Attwell, D. Multiple Modes of GABAergic Inhibition of Rat Cerebellar Granule Cells. J. Physiol. 2003, 548, 97–110. [Google Scholar] [CrossRef]
- De Filippi, G.; Baldwinson, T.; Sher, E. Evidence for Nicotinic Acetylcholine Receptor Activation in Rat Cerebellar Slices. Pharmacol. Biochem. Behav. 2001, 70, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Caban, K.M.; Seßenhausen, P.; Stöckl, J.B.; Popper, B.; Mayerhofer, A.; Fröhlich, T. Proteome Profile of the Cerebellum from A7 Nicotinic Acetylcholine Receptor Deficient Mice. Proteomics 2024, 24, 2300384. [Google Scholar] [CrossRef] [PubMed]
- de Toro, E.D.; Juíz, J.M.; Smillie, F.I.; Lindstrom, J.; Criado, M. Expression of α 7 Neuronal Nicotinic Receptors during Postnatal Development of the Rat Cerebellum. Dev. Brain Res. 1997, 98, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Role, L.W.; Berg, D.K. Nicotinic Receptors in the Development and Modulation of CNS Synapses. Neuron 1996, 16, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Opanashuk, L.A.; Pauly, J.R.; Hauser, K.F. Effect of Nicotine on Cerebellar Granule Neuron Development. Eur. J. Neurosci. 2001, 13, 48–56. [Google Scholar] [CrossRef]
- Banerjee, S.; Deacon, A.; Suter, M.A.; Aagaard, K.M. Understanding the Placental Biology of Tobacco Smoke, Nicotine, and Marijuana (THC) Exposures During Pregnancy. Clin. Obs. Gynecol. 2022, 65, 347–359. [Google Scholar] [CrossRef]
- Kim, C.W.; Lee, S.M.; Ko, E.B.; Go, R.E.; Jeung, E.B.; Kim, M.S.; Choi, K.C. Inhibitory Effects of Cigarette Smoke Extracts on Neural Differentiation of Mouse Embryonic Stem Cells. Reprod. Toxicol. 2020, 95, 75–85. [Google Scholar] [CrossRef]
- Abdel-Rahman, A.; Dechkovskaia, A.M.; Sutton, J.M.; Chen, W.C.; Guan, X.; Khan, W.A.; Abou-Donia, M.B. Maternal Exposure of Rats to Nicotine via Infusion during Gestation Produces Neurobehavioral Deficits and Elevated Expression of Glial Fibrillary Acidic Protein in the Cerebellum and CA1 Subfield in the Offspring at Puberty. Toxicology 2005, 209, 245–261. [Google Scholar] [CrossRef]
- Abou-Donia, M.B.; Khan, W.A.; Dechkovskaia, A.M.; Goldstein, L.B.; Bullman, S.L.; Abdel-Rahman, A. In Utero Exposure to Nicotine and Chlorpyrifos Alone, and in Combination Produces Persistent Sensorimotor Deficits and Purkinje Neuron Loss in the Cerebellum of Adult Offspring Rats. Arch. Toxicol. 2006, 80, 620–631. [Google Scholar] [CrossRef]
- Al-Amri, I.S.; Kadim, I.T.; Alkindi, A.; Khalaf, S.K.; Hamaed, A.; Al-Yaqoobi, S.S.; Al-Harrasi, A.S.; Al-Hashmi, S.A.; Al-Maasbi, A.A.; Al-Harthy, R.S.; et al. Effects of Prenatal Tobacco Smoke on Cerebellum Histomorphological Changes at Critical Developmental Stages in CD-1 Mice. Int. J. Morphol. 2021, 39, 318–326. [Google Scholar] [CrossRef]
- Chen, W.J.A.; Edwards, R.B. Prenatal Nicotine Exposure Does Not Cause Purkinje Cell Loss in the Developing Rat Cerebellar Vermis. Neurotoxicol. Teratol. 2003, 25, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Z.; Abbott, L.C.; Winzer-Serhan, U.H. Effects of Chronic Neonatal Nicotine Exposure on Nicotinic Acetylcholine Receptor Binding, Cell Death and Morphology in Hippocampus and Cerebellum. Neuroscience 2007, 146, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Southard, M.C.; Adam, S.J.; Cousins, M.M.; Seidler, F.J. A7 Nicotinic Acetylcholine Receptors Targeted by Cholinergic Developmental Neurotoxicants: Nicotine and Chlorpyrifos. Brain Res. Bull. 2004, 64, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Polli, F.S.; Ipsen, T.H.; Caballero-Puntiverio, M.; Østerbøg, T.B.; Aznar, S.; Andreasen, J.T.; Kohlmeier, K.A. Cellular and Molecular Changes in Hippocampal Glutamate Signaling and Alterations in Learning, Attention, and Impulsivity Following Prenatal Nicotine Exposure. Mol. Neurobiol. 2020, 57, 2002–2020. [Google Scholar] [CrossRef] [PubMed]
- Nakauchi, S.; Su, H.; Trang, I.; Sumikawa, K. Long-Term Effects of Early Postnatal Nicotine Exposure on Cholinergic Function in the Mouse Hippocampal CA1 Region. Neurobiol. Learn. Mem. 2021, 181, 107445. [Google Scholar] [CrossRef]
- Roy, T.S.; Sabherwal, U. Effects of Prenatal Nicotine Exposure on the Morphogenesis of Somatosensory Cortex. Neurotoxicol. Teratol. 1994, 16, 411–421. [Google Scholar] [CrossRef]
- Chan, Y.L.; Saad, S.; Pollock, C.; Oliver, B.; Al-Odat, I.; Zaky, A.A.; Jones, N.; Chen, H. Impact of Maternal Cigarette Smoke Exposure on Brain Inflammation and Oxidative Stress in Male Mice Offspring. Sci. Rep. 2016, 6, 25881. [Google Scholar] [CrossRef]
- Wallauer, M.M.; Huf, F.; Tortorelli, L.S.; Rahmeier, F.L.; Carvalho, F.B.; Meurer, R.T.; da Cruz Fernandes, M. Morphological Changes in the Cerebellum as a Result of Ethanol Treatment and Cigarette Smoke Exposure: A Study on Astrogliosis, Apoptosis and Purkinje Cells. Neurosci. Lett. 2018, 672, 70–77. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Maternal E-Cigarette Exposure Results in Cognitive and Epigenetic Alterations in Offspring in a Mouse Model. Chem. Res. Toxicol. 2018, 31, 601–611. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Neurological Effects in the Offspring after Switching from Tobacco Cigarettes to E-Cigarettes during Pregnancy in a Mouse Model. Toxicol. Sci. 2019, 172, 191–200. [Google Scholar] [CrossRef]
- Church, J.S.; Chace-Donahue, F.; Blum, J.L.; Ratner, J.R.; Zelikoff, J.T.; Schwartzer, J.J. Neuroinflammatory and Behavioral Outcomes Measured in Adult Offspring of Mice Exposed Prenatally to E-Cigarette Aerosols. Env. Health Perspect. 2020, 128, 047006. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Jew, C.P.; Lu, H.-C. Lasting Impacts of Prenatal Cannabis Exposure and the Role of Endogenous Cannabinoids in the Developing Brain. Future Neurol. 2011, 6, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, D.E.; Martin, B.R.; Gamagaris, Z.; Miller, N.; Fico, T. Plasma Concentrations of Delta-9-Tetrahydrocannabinol in Dams and Fetuses Following Acute or Multiple Prenatal Dosing in Rats. Life Sci. 1989, 44, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, M.; Wall, M.E. Presence of Δ9-Tetrahydrocannabinol in Human Milk. N. Engl. J. Med. 1982, 307, 819–820. [Google Scholar] [CrossRef]
- Hayer, S.; Mandelbaum, A.D.; Watch, L.; Ryan, K.S.; Hedges, M.A.; Manuzak, J.A.; Easley, C.A.; Schust, D.J.; Lo, J.O. Cannabis and Pregnancy: A Review. Obs. Gynecol. Surv. 2023, 78, 411–428. [Google Scholar] [CrossRef]
- Gunn, J.K.L.; Rosales, C.B.; Center, K.E.; Nuñez, A.; Gibson, S.J.; Christ, C.; Ehiri, J.E. Prenatal Exposure to Cannabis and Maternal and Child Health Outcomes: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e009986. [Google Scholar] [CrossRef]
- Metz, T.D.; Borgelt, L.M. Marijuana Use in Pregnancy and While Breastfeeding. Obstet. Gynecol. 2018, 132, 1198–1210. [Google Scholar] [CrossRef]
- Klebanoff, M.A.; Wilkins, D.G.; Keim, S.A. Marijuana Use during Pregnancy and Preterm Birth: A Prospective Cohort Study. Am. J. Perinatol. 2021, 38, e146–e154. [Google Scholar] [CrossRef]
- Habayeb, O.M.H.; Taylor, A.H.; Bell, S.C.; Taylor, D.J.; Konje, J.C. Expression of the Endocannabinoid System in Human First Trimester Placenta and Its Role in Trophoblast Proliferation. Endocrinology 2008, 149, 5052–5060. [Google Scholar] [CrossRef]
- Paria, B.C.; Dey, S.K. Ligand-Receptor Signaling with Endocannabinoids in Preimplantation Embryo Development and Implantation. Chem. Phys. Lipids 2000, 108, 211–220. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Berrendero, F.; Hernández, M.L.; Ramos, J.A. The Endogenous Cannabinoid System and Brain Development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the Pharmacology and Signal Transduction of the Human Cannabinoid CB1 and CB2 Receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar]
- Takahashi, K.A.; Linden, D.J. Cannabinoid Receptor Modulation of Synapses Received by Cerebellar Purkinje Cells. J. Neurophysiol. 2000, 83, 1167–1180. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 Receptors Regulate Neuronal Energy Metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes That Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.R.; Myoga, M.H.; McDaniels, K.R.; Marsicano, G.; Lutz, B.; Mackie, K.; Regehr, W.G. Presynaptic CB1 Receptors Regulate Synaptic Plasticity at Cerebellar Parallel Fiber Synapses. J. Neurophysiol. 2011, 105, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.; Johnson, M.; Melvin, L.; de Costa, B.; Rice, K. Characterization and Localization of Cannabinoid Receptors in Rat Brain: A Quantitative In Vitro Autoradiographic Study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Groen, B.G.S.; Lynn, A.B.; De Costa, B.R.; Richfield, E.K. Neuronal Localization of Cannabinoid Receptors and Second Messengers in Mutant Mouse Cerebellum. Brain Res. 1991, 552, 301–310. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid Receptor Localization in Brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef]
- Grewen, K.; Salzwedel, A.P.; Gao, W. Functional Connectivity Disruption in Neonates with Prenatal Marijuana Exposure. Front. Hum. Neurosci. 2015, 9, 601. [Google Scholar] [CrossRef]
- Richardson, K.A.; Hester, A.K.; McLemore, G.L. Prenatal Cannabis Exposure—The “First Hit” to the Endocannabinoid System. Neurotoxicol. Teratol. 2016, 58, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Calvigioni, D.; Hurd, Y.L.; Harkany, T.; Keimpema, E. Neuronal Substrates and Functional Consequences of Prenatal Cannabis Exposure. Eur. Child Adolesc. Psychiatry 2014, 23, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alonso, J.; Aguado, T.; Wu, C.-S.; Palazuelos, J.; Hofmann, C.; Garcez, P.; Guillemot, F.; Lu, H.-C.; Lutz, B.; Guzmán, M.; et al. The CB1 Cannabinoid Receptor Drives Corticospinal Motor Neuron Differentiation through the Ctip2/Satb2 Transcriptional Regulation Axis. J. Neurosci. 2012, 32, 16651–16665. [Google Scholar] [CrossRef] [PubMed]
- Huizink, A.C. Prenatal Cannabis Exposure and Infant Outcomes: Overview of Studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 52, 45–52. [Google Scholar] [CrossRef]
- Fried, P.A.; Watkinson, B.; Gray, R. Differential Effects on Cognitive Functioning in 9- to 12-Year Olds Prenatally Exposed to Cigarettes and Marihuana. Neurotoxicol. Teratol. 1998, 20, 293–306. [Google Scholar] [CrossRef]
- Motamedi, S.; Amleshi, R.S.; Javar, B.A.; Shams, P.; Kohlmeier, K.A.; Shabani, M. Cannabis during Pregnancy: A Way to Transfer an Impairment to Later Life. Birth Defects Res. 2023, 115, 1327–1344. [Google Scholar] [CrossRef]
- Thomason, M.E.; Palopoli, A.C.; Jariwala, N.N.; Werchan, D.M.; Chen, A.; Adhikari, S.; Espinoza-Heredia, C.; Brito, N.H.; Trentacosta, C.J. Miswiring the Brain: Human Prenatal Δ9-Tetrahydrocannabinol Use Associated with Altered Fetal Hippocampal Brain Network Connectivity. Dev. Cogn. Neurosci. 2021, 51, 101000. [Google Scholar] [CrossRef]
- Smith, A.M.; Fried, P.A.; Hogan, M.J.; Cameron, I. Effects of Prenatal Marijuana on Response Inhibition: An FMRI Study of Young Adults. Neurotoxicol. Teratol. 2004, 26, 533–542. [Google Scholar] [CrossRef]
- Bossong, M.G.; Jansma, J.M.; van Hell, H.H.; Jager, G.; Oudman, E.; Saliasi, E.; Kahn, R.S.; Ramsey, N.F. Effects of Δ9-Tetrahydrocannabinol on Human Working Memory Function. Biol. Psychiatry 2012, 71, 693–699. [Google Scholar] [CrossRef]
- Jutras-Aswad, D.; DiNieri, J.A.; Harkany, T.; Hurd, Y.L. Neurobiological Consequences of Maternal Cannabis on Human Fetal Development and Its Neuropsychiatric Outcome. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 395–412. [Google Scholar] [CrossRef]
- Martinez, L.R.; Black, K.C.; Webb, B.T.; Bell, A.; Baygani, S.K.; Mier, T.J.; Dominguez, L.; Mackie, K.; Kalinovsky, A. Components of Endocannabinoid Signaling System Are Expressed in the Perinatal Mouse Cerebellum and Required for Its Normal Development. eNeuro 2020, 7, ENEURO.0471-19.2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhu, J.; Wager-Miller, J.; Wang, S.; O’Leary, D.; Monory, K.; Lutz, B.; Mackie, K.; Lu, H. Requirement of Cannabinoid CB1 Receptors in Cortical Pyramidal Neurons for Appropriate Development of Corticothalamic and Thalamocortical Projections. Eur. J. Neurosci. 2010, 32, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid Signaling Controls Pyramidal Cell Specification and Long-Range Axon Patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765. [Google Scholar] [CrossRef] [PubMed]
- Suárez, I.; Bodega, G.; Rubio, M.; Fernández-Ruiz, J.J.; Ramos, J.A.; Fernández, B. Prenatal Cannabinoid Exposure Down- Regulates Glutamate Transporter Expressions (GLAST and EAAC1) in the Rat Cerebellum. Dev. Neurosci. 2004, 26, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Pinky, P.D.; Majrashi, M.; Fujihashi, A.; Bloemer, J.; Govindarajulu, M.; Ramesh, S.; Reed, M.N.; Moore, T.; Suppiramaniam, V.; Dhanasekaran, M. Effects of Prenatal Synthetic Cannabinoid Exposure on the Cerebellum of Adolescent Rat Offspring. Heliyon 2021, 7, e06730. [Google Scholar] [CrossRef]
- van Amsterdam, J.; Talhout, R.; Vleeming, W.; Opperhuizen, A. Contribution of Monoamine Oxidase (MAO) Inhibition to Tobacco and Alcohol Addiction. Life Sci. 2006, 79, 1969–1973. [Google Scholar] [CrossRef]
- Day, N.L.; Goldschmidt, L.; Thomas, C.A. Prenatal Marijuana Exposure Contributes to the Prediction of Marijuana Use at Age 14. Addiction 2006, 101, 1313–1322. [Google Scholar] [CrossRef]
- Benevenuto, S.G.; Domenico, M.D.; Yariwake, V.Y.; Dias, C.T.; Mendes-da-Silva, C.; Alves, N.D.O.; Caumo, S.E.D.S.; Vasconcellos, P.; Morais, D.R.; Cardoso, M.S.; et al. Veras Prenatal Exposure to Cannabis Smoke Induces Early and Lasting Damage to the Brain. Neurochem. Int. 2022, 160, 105406. [Google Scholar] [CrossRef]
- Shabani, M.; Hosseinmardi, N.; Haghani, M.; Shaibani, V.; Janahmadi, M. Maternal Exposure to the CB1 Cannabinoid Agonist WIN 55212–2 Produces Robust Changes in Motor Function and Intrinsic Electrophysiological Properties of Cerebellar Purkinje Neurons in Rat Offspring. Neuroscience 2011, 172, 139–152. [Google Scholar] [CrossRef]
- Honein, M.A.; Boyle, C.; Redfield, R.R. Public Health Surveillance of Prenatal Opioid Exposure in Mothers and Infants. Pediatrics 2019, 143, e20183801. [Google Scholar] [CrossRef]
- Yeoh, S.L.; Eastwood, J.; Wright, I.M.; Morton, R.; Melhuish, E.; Ward, M.; Oei, J.L. Cognitive and Motor Outcomes of Children with Prenatal Opioid Exposure A Systematic Review and Meta-Analysis. JAMA Netw. Open 2019, 2, e197025. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Clark, A.L.; Kiss, A.; Hahn, J.W.; Wesselschmidt, R.; Coscia, C.J.; Belcheva, M.M. μ- and κ-Opioids Induce the Differentiation of Embryonic Stem Cells to Neural Progenitors. J. Biol. Chem. 2006, 281, 33749–33760. [Google Scholar] [CrossRef] [PubMed]
- Schadrack, J.; Willoch, F.; Platzer, S.; Bartenstein, P.; Mahal, B.; Dworzak, D.; Wester, H.J.; Zieglgänsberger, W.; Tölle, T.R. Opioid Receptors in the Human Cerebellum: Evidence from [11C] Diprenorphine PET, MRNA Expression and Autoradiography. Neuroreport 1999, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Platzer, S.; Winkler, A.; Schadrack, J.; Dworzak, D.; Tölle, T.R.; Zieglgänsberger, W.; Spanagel, R. Autoradiographic Distribution of μ-, δ- and Κ1-Opioid Stimulated [35S]Guanylyl-5’-O-(γ-Thio)-Triphosphate Binding in Human Frontal Cortex and Cerebellum. Neurosci. Lett. 2000, 283, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Hiller, J.M.; Fan, L.Q. Laminar Distribution of the Multiple Opioid Receptors in the Human Cerebral Cortex. Neurochem. Res. 1996, 21, 1333–1345. [Google Scholar] [CrossRef]
- Kinney, H.C.; White, W.F. Opioid Receptors Localize to the External Granular Cell Layer of the Developing Human Cerebellum. Neuroscience 1991, 45, 13–21. [Google Scholar] [CrossRef]
- McPherson, C.; Haslam, M.; Pineda, R.; Rogers, C.; Neil, J.J.; Inder, T.E. Brain Injury and Development in Preterm Infants Exposed to Fentanyl. Ann. Pharmacother. 2015, 49, 1291–1297. [Google Scholar] [CrossRef]
- Yuan, Q.; Rubic, M.; Seah, J.; Rae, C.; Wright, I.M.R.; Kaltenbach, K.; Feller, J.M.; Abdel-Latif, M.E.; Chu, C.; Oei, J.L.; et al. Do Maternal Opioids Reduce Neonatal Regional Brain Volumes? A Pilot Study. J. Perinatol. 2014, 34, 909–913. [Google Scholar] [CrossRef]
- Sirnes, E.; Oltedal, L.; Bartsch, H.; Eide, G.E.; Elgen, I.B.; Aukland, S.M. Brain Morphology in School-Aged Children with Prenatal Opioid Exposure: A Structural MRI Study. Early Hum. Dev. 2017, 106, 33–39. [Google Scholar] [CrossRef]
- Nygaard, E.; Slinning, K.; Moe, V.; Due-Tønnessen, P.; Fjell, A.; Walhovd, K.B. Neuroanatomical Characteristics of Youths with Prenatal Opioid and Poly-Drug Exposure. Neurotoxicol. Teratol. 2018, 68, 13–26. [Google Scholar] [CrossRef]
- Walhovd, K.B.; Moe, V.; Slinning, K.; Due-Tønnessen, P.; Bjørnerud, A.; Dale, A.M.; van der Kouwe, A.; Quinn, B.T.; Kosofsky, B.; Greve, D.; et al. Volumetric Cerebral Characteristics of Children Exposed to Opiates and Other Substances in Utero. Neuroimage 2007, 36, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hsu, M.S.; Pintar, J.E. Developmental Expression of the μ, κ and δ Opioid Receptor MRNAs in Mouse. J. Neurosci. 1998, 18, 2538–2549. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; Houdi, A.A.; Turbek, C.S.; Elde, R.P.; Maxson, W. Opioids Intrinsically Inhibit the Genesis of Mouse Cerebellar Granule Neuron Precursors In Vitro: Differential Impact of μ and δ Receptor Activation on Proliferation and Neurite Elongation. Eur. J. Neurosci. 2000, 12, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Gibo, D.M.; McLaughlin, P.J. Zeta (ξ), a Growth-Related Opioid Receptor in Developing Rat Cerebellum: Identification and Characterization. Brain Res. 1991, 551, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Verderame, M.F.; McLaughlin, P.J. The Biology of the Opioid Growth Factor Receptor (OGFr). Brain Res. Rev. 2002, 38, 351–376. [Google Scholar] [CrossRef]
- Abeyta, A.; Dettmer, T.S.; Barnes, A.; Vega, D.; Carta, M.; Gallegos, N.; Raymond-Stintz, M.; Savage, D.D.; Valenzuela, C.F.; Saland, L.C. Delta Opioid Receptor Localization in the Rat Cerebellum. Brain Res. 2002, 931, 100–105. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Methadone and Brain Development. Experientia 1977, 33, 1486–1487. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Morphine and Brain Growth Retardation in the Rat. Pharmacology 1977, 15, 276–282. [Google Scholar] [CrossRef]
- Lorber, B.A.; Freitag, S.K.; Bartolome, J.V. Effects of Beta-Endorphin on DNA Synthesis in Brain Regions of Preweanling Rats. Brain Res. 1990, 531, 329–332. [Google Scholar] [CrossRef]
- Gibson, J.M.; Chu, T.; Zeng, W.; Wethall, A.C.; Kong, M.; Mellen, N.; Devlin Phinney, L.A.; Cai, J. Perinatal Methadone Exposure Attenuates Myelination and Induces Oligodendrocyte Apoptosis in Neonatal Rat Brain. Exp. Biol. Med. 2022, 247, 1067–1079. [Google Scholar] [CrossRef]
- Hauser, K.F.; Gurwell, J.A.; Turbek, C.S. Morphine Inhibits Purkinje Cell Survival and Dendritic Differentiation in Organotypic Cultures of the Mouse Cerebellum. Exp. Neurol. 1994, 130, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; McLaughlin, P.J.; Zagon, I.S. Endogenous Opioid Systems and the Regulation of Dendritic Growth and Spine Formation. J. Comp. Neurol. 1989, 281, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; McLaughlin, P.J.; Zagon, I.S. Endogenous Opioids Regulate Dendritic Growth and Spine Formation in Developing Rat Brain. Brain Res. 1987, 416, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of Stress throughout the Lifespan on the Brain, Behaviour and Cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef]
- Glover, V.; O’Donnell, K.J.; O’Connor, T.G.; Fisher, J. Prenatal Maternal Stress, Fetal Programming, and Mechanisms Underlying Later Psychopathology—A Global Perspective. Dev. Psychopathol. 2018, 30, 843–854. [Google Scholar] [CrossRef]
- Adamson, B.; Letourneau, N.; Lebel, C. Prenatal Maternal Anxiety and Children’s Brain Structure and Function: A Systematic Review of Neuroimaging Studies. J. Affect. Disord. 2018, 241, 117–126. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.-N.; Chen, Y.-P.; Dong, Y.-P.; Shuai, H.-L.; Xiao, X.-M.; Reichetzeder, C.; Hocher, B. Late Gestational Maternal Serum Cortisol Is Inversely Associated with Fetal Brain Growth. Neurosci. Biobehav. Rev. 2012, 36, 1085–1092. [Google Scholar] [CrossRef]
- Eberle, C.; Fasig, T.; Brüseke, F.; Stichling, S. Impact of Maternal Prenatal Stress by Glucocorticoids on Metabolic and Cardiovascular Outcomes in Their Offspring: A Systematic Scoping Review. PLoS ONE 2021, 16, e0245386. [Google Scholar] [CrossRef]
- Dunkel Schetter, C.; Tanner, L. Anxiety, Depression and Stress in Pregnancy. Curr. Opin. Psychiatry 2012, 25, 141–148. [Google Scholar] [CrossRef]
- Buss, C.; Davis, E.P.; Muftuler, L.T.; Head, K.; Sandman, C.A. High Pregnancy Anxiety during Mid-Gestation Is Associated with Decreased Gray Matter Density in 6–9-Year-Old Children. Psychoneuroendocrinology 2010, 35, 141–153. [Google Scholar] [CrossRef]
- van den Heuvel, M.I.; Hect, J.L.; Smarr, B.L.; Qawasmeh, T.; Kriegsfeld, L.J.; Barcelona, J.; Hijazi, K.E.; Thomason, M.E. Maternal Stress during Pregnancy Alters Fetal Cortico-Cerebellar Connectivity in Utero and Increases Child Sleep Problems after Birth. Sci. Rep. 2021, 11, 2228. [Google Scholar] [CrossRef] [PubMed]
- Ulupinar, E.; Yucel, F. Prenatal Stress Reduces Interneuronal Connectivity in the Rat Cerebellar Granular Layer. Neurotoxicol. Teratol. 2005, 27, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Ulupinar, E.; Yucel, F.; Ortug, G. The Effects of Prenatal Stress on the Purkinje Cell Neurogenesis. Neurotoxicol. Teratol. 2006, 28, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Pascual, R.; Ebner, D.; Araneda, R.; Urqueta, M.J.; Bustamante, C. Maternal Stress Induces Long-Lasting Purkinje Cell Developmental Impairments in Mouse Offspring. Eur. J. Pediatr. 2010, 169, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Pascual, R.; Valencia, M.; Bustamante, C. Purkinje Cell Dendritic Atrophy Induced by Prenatal Stress Is Mitigated by Early Environmental Enrichment. Neuropediatrics 2015, 46, 37–43. [Google Scholar] [CrossRef]
- Pascual, R.; Santander, O.; Cuevas, I.; Valencia, M. Prenatal Glucocorticoid Administration Persistently Increased the Immunohistochemical Expression of Type-1 Metabotropic Glutamate Receptor and Purkinje Cell Dendritic Growth in the Cerebellar Cortex of the Rat. Rom. J. Morphol. Embryol. 2017, 58, 67–72. [Google Scholar]
- Rivas-Manzano, P.; Ramírez-Escoto, M.M.; la Rosa-Rugerio, D.; Rugerio-Vargas, C.; Ortiz-Hernández, R.; Torres-Ramírez, N. Argentic Staining Reveals Changes in Cerebellar Tissue Organisation by Prenatal Glucocorticoid Administration in Rats. Histol. Histopathol. 2021, 36, 195–205. [Google Scholar] [CrossRef]
- Wilber, A.A.; Lin, G.L.; Wellman, C.L. Neonatal Corticosterone Administration Impairs Adult Eyeblink Conditioning and Decreases Glucocorticoid Receptor Expression in the Cerebellar Interpositus Nucleus. Neuroscience 2011, 177, 56–65. [Google Scholar] [CrossRef]
- Wilber, A.A.; Wellman, C.L. Neonatal Maternal Separation Alters the Development of Glucocorticoid Receptor Expression in the Interpositus Nucleus of the Cerebellum. Int. J. Dev. Neurosci. 2009, 27, 649–654. [Google Scholar] [CrossRef]
- Schneider, E.R.; Civillico, E.F.; Wang, S.S.-H. Calcium-Based Dendritic Excitability and Its Regulation in the Deep Cerebellar Nuclei. J. Neurophysiol. 2013, 109, 2282–2292. [Google Scholar] [CrossRef]
- Miki, T.; Yokoyama, T.; Kusaka, T.; Suzuki, S.; Ohta, K.; Warita, K.; Wang, Z.-Y.; Ueki, M.; Sumitani, K.; Bellinger, F.P.; et al. Early Postnatal Repeated Maternal Deprivation Causes a Transient Increase in OMpg and BDNF in Rat Cerebellum Suggesting Precocious Myelination. J. Neurol. Sci. 2014, 336, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Roque, A.; Lajud, N.; Valdez, J.J.; Torner, L. Early-Life Stress Increases Granule Cell Density in the Cerebellum of Male Rats. Brain Res. 2019, 1723, 146358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-X.; Levine, S.; Dent, G.; Zhan, Y.; Xing, G.; Okimoto, D.; Gordon, M.K.; Post, R.M.; Smith, M.A. Maternal Deprivation Increases Cell Death in the Infant Rat Brain. Dev. Brain Res. 2002, 133, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Llorente, R.; Gallardo, M.L.; Berzal, A.L.; Prada, C.; Garcia-Segura, L.M.; Viveros, M. Early Maternal Deprivation in Rats Induces Gender-dependent Effects on Developing Hippocampal and Cerebellar Cells. Int. J. Dev. Neurosci. 2009, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Enthoven, L.; Van Der Mark, M.; Levine, S.; De Kloet, E.R.; Oitzl, M.S. The Postnatal Development of the Hypothalamic-Pituitary-Adrenal Axis in the Mouse. Int. J. Dev. Neurosci. 2003, 21, 125–132. [Google Scholar] [CrossRef]
- Sapolsky’, R.M.; Meaney’, M.J. Maturation of the Adrenocortical Stress Response: Neuroendocrine Control Mechanisms and the Stress Hyporesponsive Period. Brain Res. Rev. 1986, 11, 65–76. [Google Scholar] [CrossRef]
- Schmidt, M.V. Stress-Hyporesponsive Period. In Stress: Physiology, Biochemistry, and Pathology Handbook of Stress Series, Volume 3; Elsevier: Amsterdam, The Netherlands, 2019; pp. 49–56. ISBN 9780128131466. [Google Scholar]
- Noguchi, K.K.; Walls, K.C.; Wozniak, D.F.; Olney, J.W.; Roth, K.A.; Farber, N.B. Acute Neonatal Glucocorticoid Exposure Produces Selective and Rapid Cerebellar Neural Progenitor Cell Apoptotic Death. Cell Death Differ. 2008, 15, 1582–1592. [Google Scholar] [CrossRef]
- Warland, J.; Dorrian, J.; Morrison, J.L.; O’Brien, L.M. Maternal Sleep during Pregnancy and Poor Fetal Outcomes: A Scoping Review of the Literature with Meta-Analysis. Sleep Med. Rev. 2018, 41, 197–219. [Google Scholar] [CrossRef]
- Wu, F.; Sun, L.; Chen, J.; Du, Y.; Fan, Z.; Cao, Z.; Liu, H.; Lei, X.; Zhang, F. Sleep Quality during Pregnancy and Fetal Growth: A Prospective Cohort Study. J. Sleep Res. 2024, e14233. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Li, W.; Zhu, Y.; Ma, R.; Wang, Y.; Zhang, Y.; Zhu, D.; Zhu, P. Association of Maternal Short Sleep Duration with Neurodevelopmental Delay in Offspring: A Prospective Cohort Study. J. Clin. Endocrinol. Metab. 2024, dgae569. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Aswathy, B.S.; Kumar, V.M.; Gulia, K.K. Sleep Deprivation during Late Pregnancy Produces Hyperactivity and Increased Risk-Taking Behavior in Offspring. Brain Res. 2015, 1596, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Gulia, K.K.; Patel, N.; Kumar, V.M. Increased Ultrasonic Vocalizations and Risk-Taking in Rat Pups of Sleep-Deprived Dams. Physiol. Behav. 2015, 139, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xie, X.; Fan, Y.; Zhang, J.; Jiang, W.; Wu, X.; Yan, S.; Chen, Y.; Peng, C.; You, Z. Phenotypic Dysregulation of Microglial Activation in Young Offspring Rats with Maternal Sleep Deprivation-Induced Cognitive Impairment. Sci. Rep. 2015, 5, 9513. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Peng, C.; Wu, X.; Chen, Y.; Wang, C.; You, Z. Maternal Sleep Deprivation Inhibits Hippocampal Neurogenesis Associated with Inflammatory Response in Young Offspring Rats. Neurobiol. Dis. 2014, 68, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, W.; Tan, T.; He, W.; Dong, Z.; Wang, Y.T.; Han, H. Maternal Sleep Deprivation at Different Stages of Pregnancy Impairs the Emotional and Cognitive Functions, and Suppresses Hippocampal Long-Term Potentiation in the Offspring Rats. Mol. Brain 2016, 9, 17. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, T.; Yu, J.; Lei, X. Sleep Deprivation Makes the Young Brain Resemble the Elderly Brain: A Large-Scale Brain Networks Study. Brain Connect. 2017, 7, 58–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Y.; Yang, Y.; Li, J.; Xin, W.; Huang, Y.; Shao, Y.; Zhang, X. Alterations in Cerebellar Functional Connectivity Are Correlated with Decreased Psychomotor Vigilance Following Total Sleep Deprivation. Front. Neurosci. 2019, 13, 134. [Google Scholar] [CrossRef]
- Benarroch, E. What Is the Involvement of the Cerebellum During Sleep? Neurology 2023, 100, 572–577. [Google Scholar] [CrossRef]
- Canto, C.B.; Onuki, Y.; Bruinsma, B.; van der Werf, Y.D.; De Zeeuw, C.I. The Sleeping Cerebellum. Trends Neurosci. 2017, 40, 309–323. [Google Scholar] [CrossRef]
- Blumberg, M.; Dooley, J.; Sokoloff, G. The Developing Brain Revealed during Sleep. Curr. Opin. Physiol. 2020, 15, 14–22. [Google Scholar] [CrossRef]
- Canto, C.B.; Bauer, S.; Hoogland, T.M.; Hoedemaker, H.H.; Geelen, C.; Loyola, S.; Miaja, P.; De Zeeuw, C.I. Sleep-State Dependent Cerebellar Processing in Adult Mice. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tagliazucchi, E.; Laufs, H. Decoding Wakefulness Levels from Typical FMRI Resting-State Data Reveals Reliable Drifts between Wakefulness and Sleep. Neuron 2014, 82, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, L.; Yang, G.; Gan, W.-B. REM Sleep Selectively Prunes and Maintains New Synapses in Development and Learning. Nat. Neurosci. 2017, 20, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Dooley, J.C.; Sokoloff, G.; Blumberg, M.S. Movements during Sleep Reveal the Developmental Emergence of a Cerebellar-Dependent Internal Model in Motor Thalamus. Curr. Biol. 2021, 31, 5501–5511.e5. [Google Scholar] [CrossRef]
- Mukherjee, D.; Sokoloff, G.; Blumberg, M.S. Corollary Discharge in Precerebellar Nuclei of Sleeping Infant Rats. Elife 2018, 7, e38213. [Google Scholar] [CrossRef]
- Crapse, T.B.; Sommer, M.A. Corollary Discharge across the Animal Kingdom. Nat. Rev. Neurosci. 2008, 9, 587–600. [Google Scholar] [CrossRef]
- Hu, Y.; Korovaichuk, A.; Astiz, M.; Schroeder, H.; Islam, R.; Barrenetxea, J.; Fischer, A.; Oster, H.; Bringmann, H. Functional Divergence of Mammalian TFAP2a and TFAP2b Transcription Factors for Bidirectional Sleep Control. Genetics 2020, 216, 735–752. [Google Scholar] [CrossRef]
- Hu, Y.; Bringmann, H. Tfap2b Acts in GABAergic Neurons to Control Sleep in Mice. Sci. Rep. 2023, 13, 8026. [Google Scholar] [CrossRef]
- Pardo, G.V.E.; Goularte, J.F.; Hoefel, A.L.; de Castro, A.L.; Kucharski, L.C.; da Rosa Araujo, A.S.; Lucion, A.B. Effects of Sleep Restriction during Pregnancy on the Mother and Fetuses in Rats. Physiol. Behav. 2016, 155, 66–76. [Google Scholar] [CrossRef]
- Vollmer, B.; Edmonds, C.J. School Age Neurological and Cognitive Outcomes of Fetal Growth Retardation or Small for Gestational Age Birth Weight. Front. Endocrinol. 2019, 10, 186. [Google Scholar] [CrossRef]
- Hartkopf, J.; Schleger, F.; Keune, J.; Wiechers, C.; Pauluschke-Froehlich, J.; Weiss, M.; Conzelmann, A.; Brucker, S.; Preissl, H.; Kiefer-Schmidt, I. Impact of Intrauterine Growth Restriction on Cognitive and Motor Development at 2 Years of Age. Front. Physiol. 2018, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Chu, A. Intrauterine Growth Restriction: Hungry for an Answer. Physiology 2016, 31, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fu, W.; Liu, J. Neurodevelopment in Children with Intrauterine Growth Restriction: Adverse Effects and Interventions. J. Matern.-Fetal Neonatal Med. 2016, 29, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Huppi, P.S.; Mallard, C. The Consequences of Fetal Growth Restriction on Brain Structure and Neurodevelopmental Outcome. J. Physiol. 2016, 594, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Olivier, P.; Baud, O.; Bouslama, M.; Evrard, P.; Gressens, P.; Verney, C. Moderate Growth Restriction: Deleterious and Protective Effects on White Matter Damage. Neurobiol. Dis. 2007, 26, 253–263. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain Injury in Premature Infants: A Complex Amalgam of Destructive and Developmental Disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Eixarch, E.; Muñoz-Moreno, E.; Bargallo, N.; Batalle, D.; Gratacos, E. Motor and Cortico-Striatal-Thalamic Connectivity Alterations in Intrauterine Growth Restriction. Am. J. Obs. Gynecol. 2016, 214, 725.e1–725.e9. [Google Scholar] [CrossRef]
- Fischi-Gomez, E.; Muñoz-Moreno, E.; Vasung, L.; Griffa, A.; Borradori-Tolsa, C.; Monnier, M.; Lazeyras, F.; Thiran, J.-P.; Hüppi, P.S. Brain Network Characterization of High-Risk Preterm-Born School-Age Children. Neuroimage Clin. 2016, 11, 195–209. [Google Scholar] [CrossRef]
- Chang, J.L.; Bashir, M.; Santiago, C.; Farrow, K.; Fung, C.; Brown, A.S.; Dettman, R.W.; Dizon, M.L.V. Intrauterine Growth Restriction and Hyperoxia as a Cause of White Matter Injury. Dev. Neurosci. 2018, 40, 344–357. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Fattakhov, N.; Buddington, R.K.; Chizhikov, V. V Intrauterine Growth Restriction Compromises Cerebellar Development by Affecting Radial Migration of Granule Cells via the JamC/Pard3a Molecular Pathway. Exp. Neurol. 2021, 336, 113537. [Google Scholar] [CrossRef]
- McDougall, A.R.A.; Wiradjaja, V.; Azhan, A.; Li, A.; Hale, N.; Wlodek, M.E.; Hooper, S.B.; Wallace, M.J.; Tolcos, M. Intrauterine Growth Restriction Alters the Postnatal Development of the Rat Cerebellum. Dev. Neurosci. 2017, 39, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Tolcos, M.; McDougall, A.; Shields, A.; Chung, Y.; O’Dowd, R.; Turnley, A.; Wallace, M.; Rees, S. Intrauterine Growth Restriction Affects Cerebellar Granule Cells in the Developing Guinea Pig Brain. Dev. Neurosci. 2018, 40, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Bonatto, F.; Polydoro, M.; Andrades, M.E.; da Frota Júnior, M.L.; Dal-Pizzol, F.; Rotta, L.N.; Souza, D.O.; Perry, M.L.; Moreira, J.C.F. Effects of Maternal Protein Malnutrition on Oxidative Markers in the Young Rat Cortex and Cerebellum. Neurosci. Lett. 2006, 406, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Chanez, C.; Rabin, O.; Heroux, M.; Giguere, J.F. Cerebral Amino Acid Changes in an Animal Model of Intrauterine Growth Retardation. Metab. Brain Dis. 1993, 8, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Tita, A.T.N.; Andrews, W.W. Diagnosis and Management of Clinical Chorioamnionitis. Clin. Perinatol. 2010, 37, 339–354. [Google Scholar] [CrossRef]
- Ylijoki, M.; Lehtonen, L.; Lind, A.; Ekholm, E.; Lapinleimu, H.; Kujari, H.; Haataja, L. Chorioamnionitis and Five-Year Neurodevelopmental Outcome in Preterm Infants. Neonatology 2016, 110, 286–295. [Google Scholar] [CrossRef]
- Hendson, L.; Russell, L.; Robertson, C.M.T.; Liang, Y.; Chen, Y.; Abdalla, A.; Lacaze-Masmonteil, T. Neonatal and Neurodevelopmental Outcomes of Very Low Birth Weight Infants with Histologic Chorioamnionitis. J. Pediatr. 2011, 158, 397–402. [Google Scholar] [CrossRef]
- Nasef, N.; Shabaan, A.E.; Schurr, P.; Iaboni, D.; Choudhury, J.; Church, P.; Dunn, M.S. Effect of Clinical and Histological Chorioamnionitis on the Outcome of Preterm Infants. Am. J. Perinatol. 2013, 30, 59–68. [Google Scholar] [CrossRef]
- Pappas, A.; Kendrick, D.E.; Shankaran, S.; Stoll, B.J.; Bell, E.F.; Laptook, A.R.; Walsh, M.C.; Das, A.; Hale, E.C.; Newman, N.S.; et al. Chorioamnionitis and Early Childhood Outcomes among Extremely Low-Gestational-Age Neonates. JAMA Pediatr. 2014, 168, 137–147. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Sullivan, N.R.; Soul, J.S.; Robertson, R.L.J.; Moore, M.; Ringer, S.A.; Volpe, J.J.; du Plessis, A.J. Positive Screening for Autism in Ex-Preterm Infants: Prevalence and Risk Factors. Pediatrics 2008, 121, 758–765. [Google Scholar] [CrossRef]
- Jain, V.G.; Willis, K.A.; Jobe, A.; Ambalavanan, N. Chorioamnionitis and Neonatal Outcomes. Pediatr. Res. 2022, 91, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Normann, E.; Lacaze-Masmonteil, T.; Eaton, F.; Schwendimann, L.; Gressens, P.; Thébaud, B. A Novel Mouse Model of Ureaplasma-Induced Perinatal Inflammation: Effects on Lung and Brain Injury. Pediatr. Res. 2009, 65, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Van Steenwinckel, J.; Fleiss, B.; Scheuer, T.; Bührer, C.; Faivre, V.; Lemoine, S.; Blugeon, C.; Schwendimann, L.; Csaba, Z.; et al. A Unique Cerebellar Pattern of Microglia Activation in a Mouse Model of Encephalopathy of Prematurity. Glia 2022, 70, 1699–1719. [Google Scholar] [CrossRef] [PubMed]
- Gudsnuk, K.M.A.; Champagne, F.A. Epigenetic Effects of Early Developmental Experiences. Clin. Perinatol. 2011, 38, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Broersen, R.; Canto, C.B.; De Zeeuw, C.I. Cerebellar Nuclei: Associative Motor Learning in Zebrafish. Curr. Biol. 2023, 33, R867–R870. [Google Scholar] [CrossRef]
- Canto, C.B.; Broersen, R.; De Zeeuw, C.I. Intrinsic Excitement in Cerebellar Nuclei Neurons during Learning. Proc. Natl. Acad. Sci. USA 2018, 115, 9824–9826. [Google Scholar] [CrossRef]
- Witter, L.; Canto, C.B.; Hoogland, T.M.; de Gruijl, J.R.; De Zeeuw, C.I. Strength and Timing of Motor Responses Mediated by Rebound Firing in the Cerebellar Nuclei after Purkinje Cell Activation. Front. Neural. Circuits 2013, 7, 133. [Google Scholar] [CrossRef]
- Hoogland, T.M.; De Gruijl, J.R.; Witter, L.; Canto, C.B.; De Zeeuw, C.I. Role of Synchronous Activation of Cerebellar Purkinje Cell Ensembles in Multi-Joint Movement Control. Curr. Biol. 2015, 25, 1157–1165. [Google Scholar] [CrossRef]
- Lange, W. Regional Differences in the Distribution of Golgi Cells in the Cerebellar Cortex of Man and Some Other Mammals. Cell Tissue Res. 1974, 153, 219–226. [Google Scholar] [CrossRef]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling Transformations of Neurodevelopmental Sequences across Mammalian Species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Haldipur, P.; Dang, D.; Millen, K.J. Embryology. Handb. Clin. Neurol. 2018, 154, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Wojcinski, A.; Morabito, M.; Lawton, A.K.; Stephen, D.N.; Joyner, A.L. Genetic Deletion of Genes in the Cerebellar Rhombic Lip Lineage Can Stimulate Compensation through Adaptive Reprogramming of Ventricular Zone-Derived Progenitors. Neural. Dev. 2019, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.E.; Sillitoe, R.V. Interactions Between Purkinje Cells and Granule Cells Coordinate the Development of Functional Cerebellar Circuits. Neuroscience 2021, 462, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.; Tong, X.; Tan, A.; De Paiva, V.N.; Presicce, P.; Kannan, P.S.; Williams, K.; Damianos, A.; Newsam, M.T.; Benny, M.; et al. Single-Nucleus RNA-Seq Reveals Disrupted Cell Maturation by Chorioamnionitis in the Preterm Cerebellum of Nonhuman Primates. bioRxiv 2022. [Google Scholar] [CrossRef]
- Newman, J.; Tong, X.; Tan, A.; Yeasky, T.; De Paiva, V.N.; Presicce, P.; Kannan, P.S.; Williams, K.; Damianos, A.; Tamase Newsam, M.; et al. Chorioamnionitis Accelerates Granule Cell and Oligodendrocyte Maturation in the Cerebellum of Preterm Nonhuman Primates. J. Neuroinflamm. 2024, 21, 16. [Google Scholar] [CrossRef]
- Norman, A.L.; Crocker, N.; Mattson, S.N.; Riley, E.P. Neuroimaging and Fetal Alcohol Spectrum Disorders. Dev. Disabil. Res. Rev. 2009, 15, 209–217. [Google Scholar] [CrossRef]
- Kalinovsky, A.; Boukhtouche, F.; Blazeski, R.; Bornmann, C.; Suzuki, N.; Mason, C.A.; Scheiffele, P. Development of Axon-Target Specificity of Ponto-Cerebellar Afferents. PLoS Biol. 2011, 9, e1001013. [Google Scholar] [CrossRef]
- Ashwell, K.W.S.; Zhang, L.-L. Ontogeny of Afferents to the Fetal Rat Cerebellum. Cells Tissues Organs 1992, 145, 17–23. [Google Scholar] [CrossRef]
- Stoodley, C.J. Distinct Regions of the Cerebellum Show Gray Matter Decreases in Autism, ADHD, and Developmental Dyslexia. Front. Syst. Neurosci. 2014, 8, 92. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Schmahmann, J.D. Functional Topography of the Human Cerebellum. Handb. Clin. Neurol. 2018, 154, 59–70. [Google Scholar] [CrossRef]
- Stoodley, C.J. The Cerebellum and Cognition: Evidence from Functional Imaging Studies. Cerebellum 2012, 11, 352–365. [Google Scholar] [CrossRef]
- D’Mello, A.M.; Stoodley, C.J. Cerebro-Cerebellar Circuits in Autism Spectrum Disorder. Front. Neurosci. 2015, 9, 408. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, C.J. The Cerebellum and Neurodevelopmental Disorders. Cerebellum 2016, 15, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Limperopoulos, C.; Chilingaryan, G.; Guizard, N.; Robertson, R.L.; Du Plessis, A.J. Cerebellar Injury in the Premature Infant Is Associated with Impaired Growth of Specific Cerebral Regions. Pediatr. Res. 2010, 68, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Li, Z.Q.; Sun, Y.; Wang, T.; Wan, C.L.; Li, X.W.; Zhao, X.Z.; Feng, G.Y.; Li, S.; Clair, D.S.; et al. The Role of Pro-Inflammatory Factors in Mediating the Effects on the Fetus of Prenatal Undernutrition: Implications for Schizophrenia. Schizophr. Res. 2008, 99, 48–55. [Google Scholar] [CrossRef]
- Walker, C.K.; Krakowiak, P.; Baker, A.; Hansen, R.L.; Ozonoff, S.; Hertz-Picciotto, I. Preeclampsia, Placental Insufficiency, and Autism Spectrum Disorder or Developmental Delay. JAMA Pediatr. 2015, 169, 154. [Google Scholar] [CrossRef]


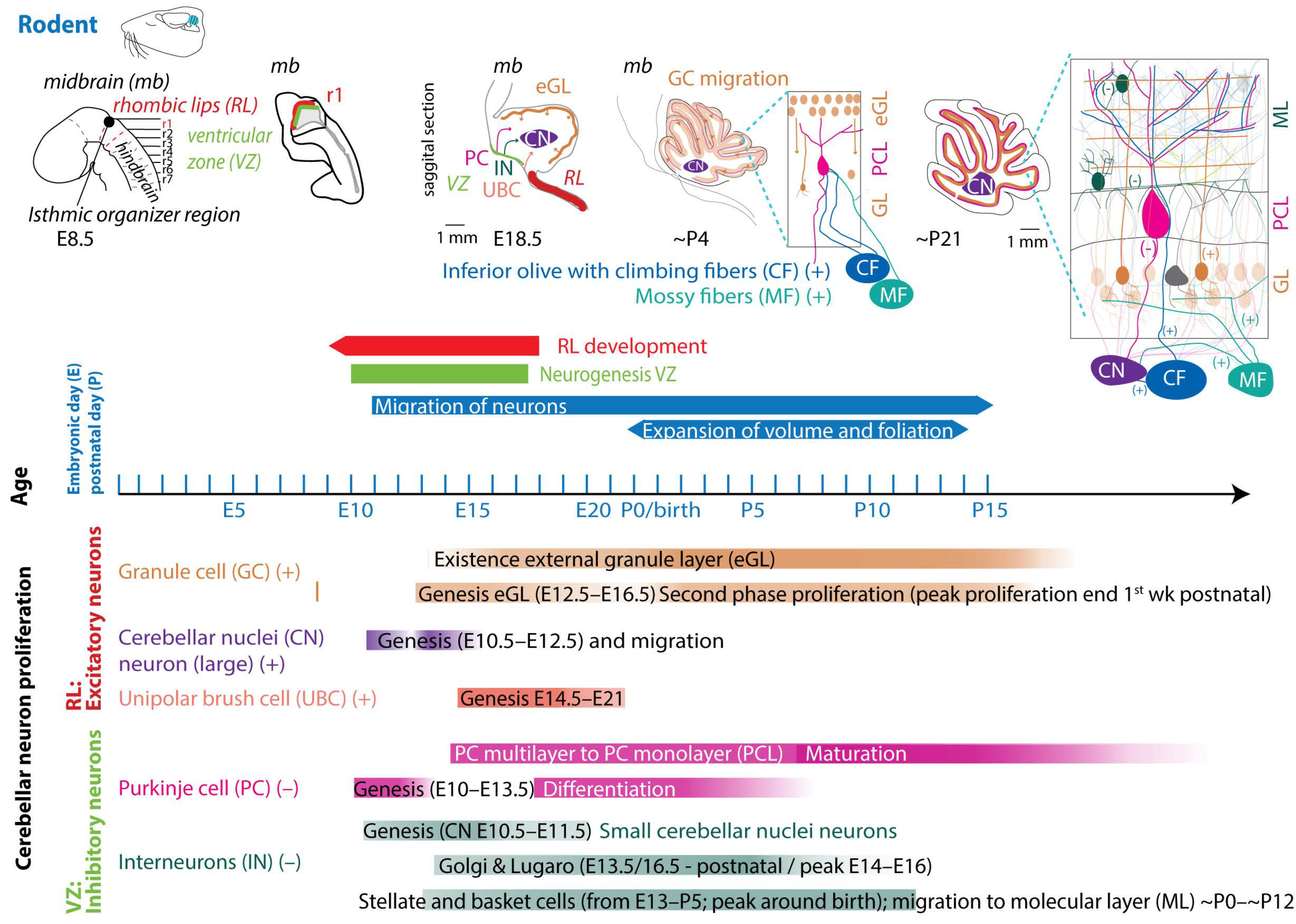
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westerhuis, J.A.W.; Dudink, J.; Wijnands, B.E.C.A.; De Zeeuw, C.I.; Canto, C.B. Impact of Intrauterine Insults on Fetal and Postnatal Cerebellar Development in Humans and Rodents. Cells 2024, 13, 1911. https://doi.org/10.3390/cells13221911
Westerhuis JAW, Dudink J, Wijnands BECA, De Zeeuw CI, Canto CB. Impact of Intrauterine Insults on Fetal and Postnatal Cerebellar Development in Humans and Rodents. Cells. 2024; 13(22):1911. https://doi.org/10.3390/cells13221911
Chicago/Turabian StyleWesterhuis, Judith A. W., Jeroen Dudink, Bente E. C. A. Wijnands, Chris I. De Zeeuw, and Cathrin B. Canto. 2024. "Impact of Intrauterine Insults on Fetal and Postnatal Cerebellar Development in Humans and Rodents" Cells 13, no. 22: 1911. https://doi.org/10.3390/cells13221911
APA StyleWesterhuis, J. A. W., Dudink, J., Wijnands, B. E. C. A., De Zeeuw, C. I., & Canto, C. B. (2024). Impact of Intrauterine Insults on Fetal and Postnatal Cerebellar Development in Humans and Rodents. Cells, 13(22), 1911. https://doi.org/10.3390/cells13221911