
The Formyl Peptid Receptor Ligand Ac2-26 Improves the Integrity of the Blood−Brain Barrier in the Course of Pneumococcal Meningitis


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Induction of Experimental Pneumococcal Meningitis
2.4. Immunohistochemistry or Immunofluorescence
2.5. Quantification of the Optical Density
2.6. Quantification of Fluorescence Intensity
2.7. Statistical Analyses
3. Results
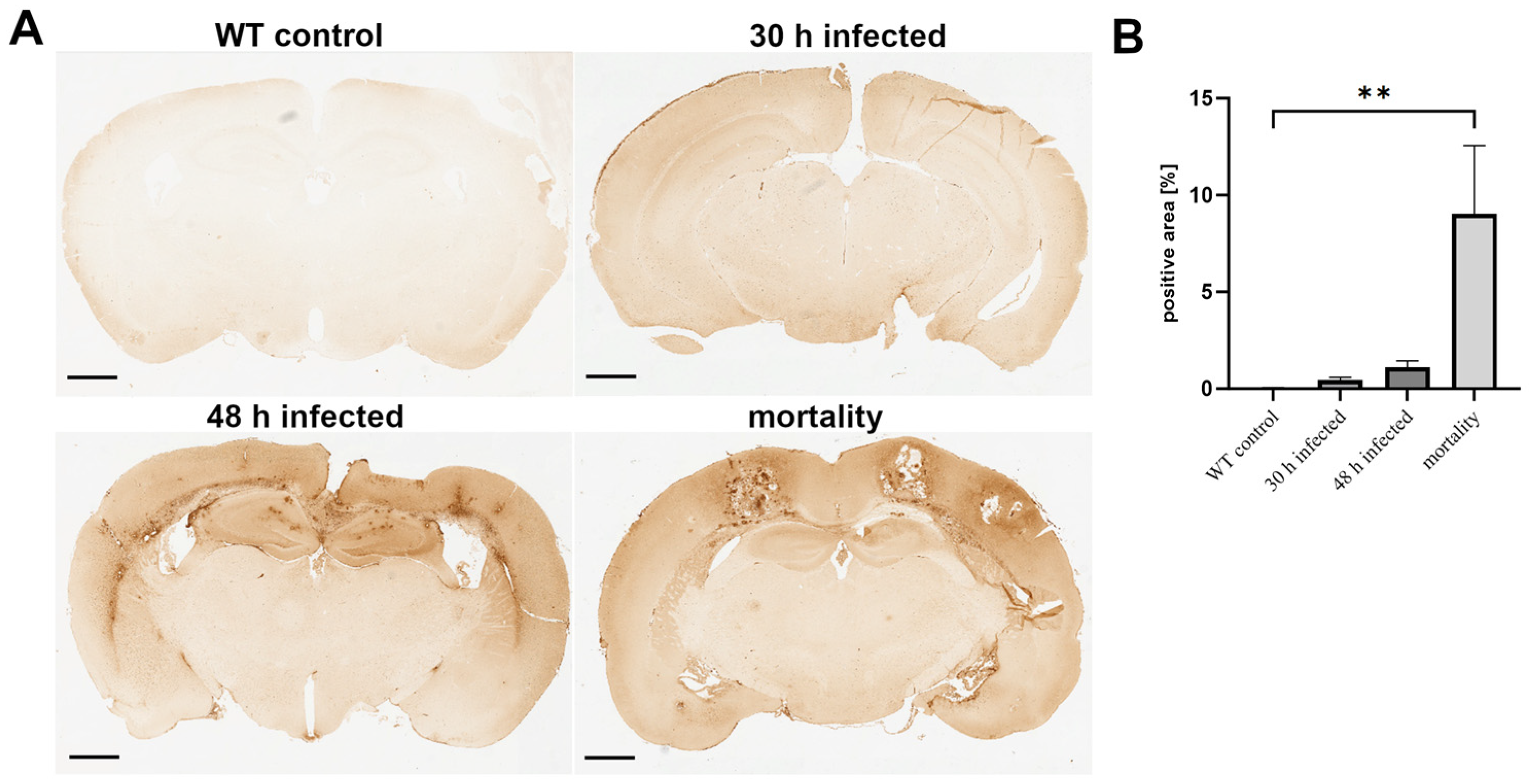
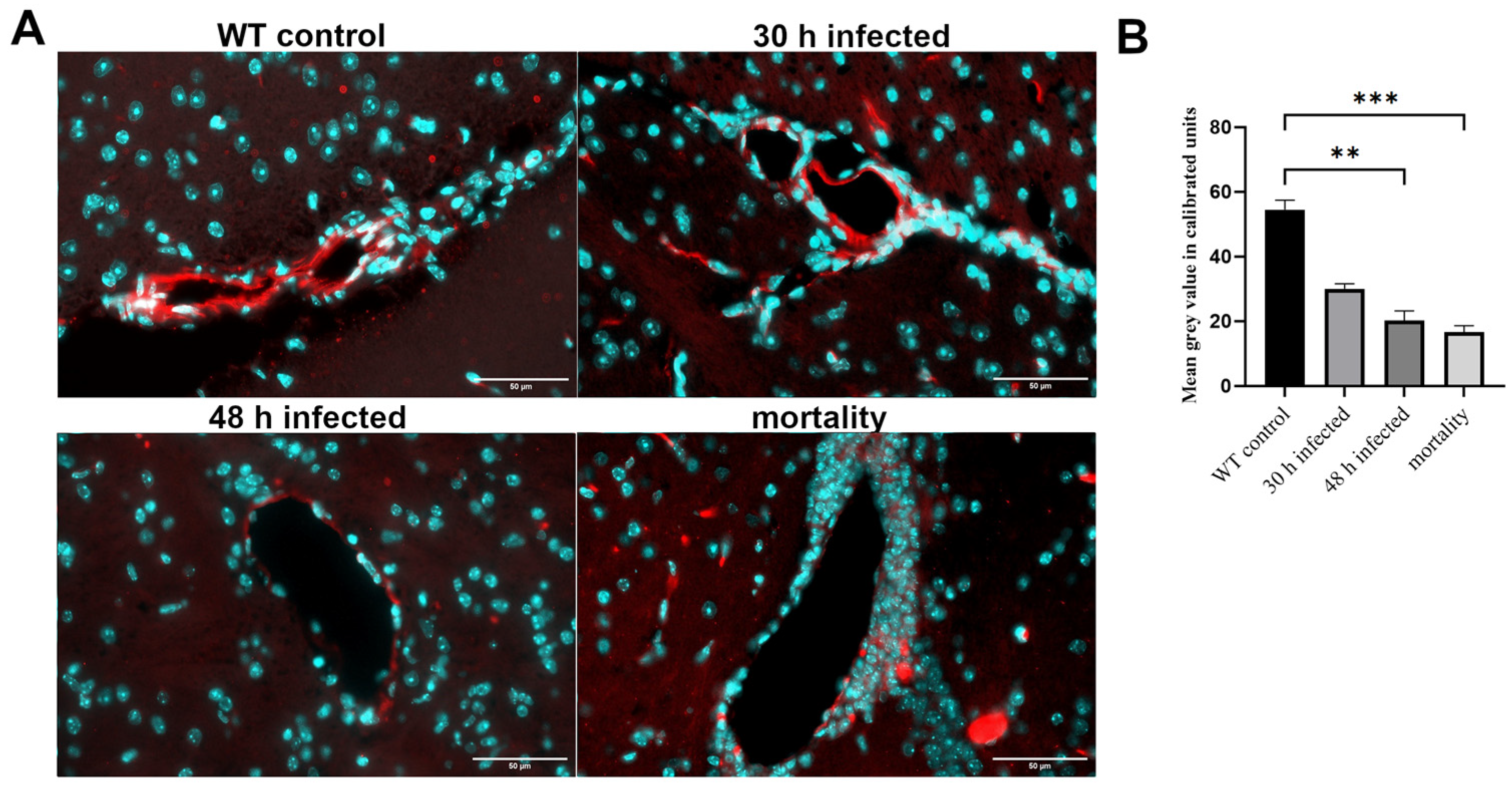
3.1. Acute Bacterial Meningitis Induces Breakdown of the Blood−Brain Barrier
3.2. Lack of FPR1 Changes Permeability of BBB in Uninfected Controls
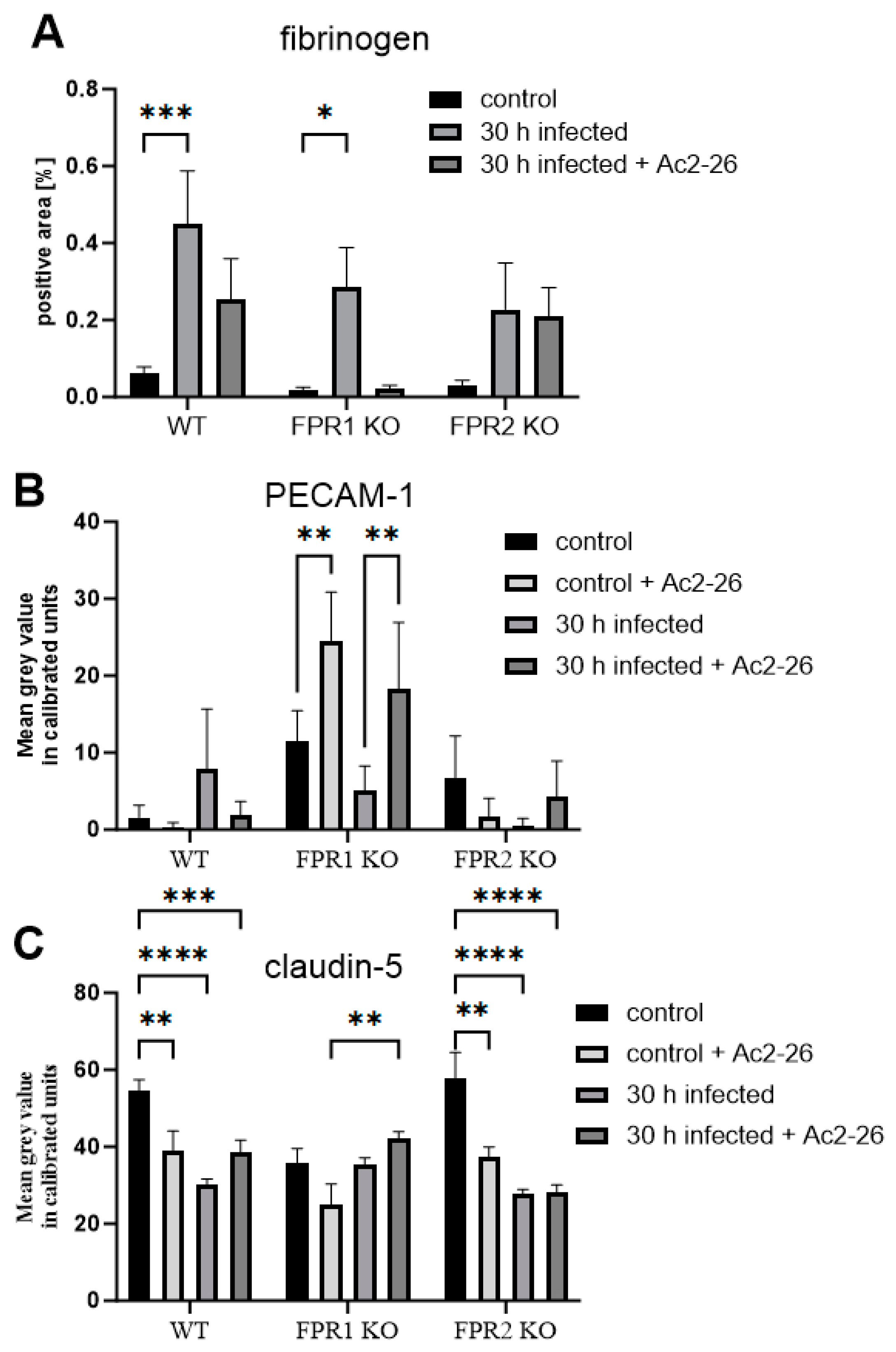
3.3. Ac2-26 Ameliorates BBB Permeability Loss During Pneumococcal Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heckenberg, S.G.B.; Brouwer, M.C.; van de Beek, D. Bacterial meningitis. Handb. Clin. Neurol. 2014, 121, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.J.; Brouwer, M.C.; van de Beek, D. Neurological sequelae of bacterial meningitis. J. Infect. 2016, 73, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Heimann, B.; Djukic, M.; Mazurek, C.; Fels, C.; Wallesch, C.-W.; Nau, R. Neuropsychological sequelae of bacterial and viral meningitis. Brain 2006, 129, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Gil, E.; Wall, E.; Noursadeghi, M.; Brown, J.S. Streptococcus pneumoniae meningitis and the CNS barriers. Front. Cell. Infect. Microbiol. 2022, 12, 1106596. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef]
- Gao, J.L.; Chen, H.; Filie, J.D.; Kozak, C.A.; Murphy, P.M. Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics 1998, 51, 270–276. [Google Scholar] [CrossRef]
- Bihler, K.; Kress, E.; Esser, S.; Nyamoya, S.; Tauber, S.C.; Clarner, T.; Stope, M.B.; Pufe, T.; Brandenburg, L.-O. Formyl Peptide Receptor 1-Mediated Glial Cell Activation in a Mouse Model of Cuprizone-Induced Demyelination. J. Mol. Neurosci. 2017, 62, 232–243. [Google Scholar] [CrossRef]
- Lacy, M.; Jones, J.; Whittemore, S.R.; Haviland, D.L.; Wetsel, R.A.; Barnum, S.R. Expression of the receptors for the C5a anaphylatoxin, interleukin-8 and FMLP by human astrocytes and microglia. J. Neuroimmunol. 1995, 61, 71–78. [Google Scholar] [CrossRef]
- Oldekamp, S.; Pscheidl, S.; Kress, E.; Soehnlein, O.; Jansen, S.; Pufe, T.; Wang, J.M.; Tauber, S.C.; Brandenburg, L.-O. Lack of formyl peptide receptor 1 and 2 leads to more severe inflammation and higher mortality in mice with of pneumococcal meningitis. Immunology 2014, 143, 447–461. [Google Scholar] [CrossRef]
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Raabe, C.A.; Gröper, J.; Rescher, U. Biased perspectives on formyl peptide receptors. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.-O.; Konrad, M.; Wruck, C.; Koch, T.; Pufe, T.; Lucius, R. Involvement of formyl-peptide-receptor-like-1 and phospholipase D in the internalization and signal transduction of amyloid beta 1-42 in glial cells. Neuroscience 2008, 156, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Schröder, N.; Schaffrath, A.; Welter, J.A.; Putzka, T.; Griep, A.; Ziegler, P.; Brandt, E.; Samer, S.; Heneka, M.T.; Kaddatz, H.; et al. Inhibition of formyl peptide receptors improves the outcome in a mouse model of Alzheimer disease. J. Neuroinflamm. 2020, 17, 131. [Google Scholar] [CrossRef] [PubMed]
- Rüger, M.; Kipp, E.; Schubert, N.; Schröder, N.; Pufe, T.; Stope, M.B.; Kipp, M.; Blume, C.; Tauber, S.C.; Brandenburg, L.-O. The formyl peptide receptor agonist Ac2-26 alleviates neuroinflammation in a mouse model of pneumococcal meningitis. J. Neuroinflamm. 2020, 17, 325. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct Signaling Cascades Elicited by Different Formyl Peptide Receptor 2 (FPR2) Agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar] [CrossRef]
- Gavins, F.N.E.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef]
- Leoni, G.; Alam, A.; Neumann, P.-A.; Lambeth, J.D.; Cheng, G.; McCoy, J.; Hilgarth, R.S.; Kundu, K.; Murthy, N.; Kusters, D.; et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J. Clin. Investig. 2012, 123, 443–454. [Google Scholar] [CrossRef]
- Machado, M.G.; Tavares, L.P.; Souza, G.V.S.; Queiroz-Junior, C.M.; Ascenção, F.R.; Lopes, M.E.; Garcia, C.C.; Menezes, G.B.; Perretti, M.; Russo, R.C.; et al. The Annexin A1/FPR2 pathway controls the inflammatory response and bacterial dissemination in experimental pneumococcal pneumonia. FASEB J. 2020, 34, 2749–2764. [Google Scholar] [CrossRef]
- Kao, W.; Gu, R.; Jia, Y.; Wei, X.; Fan, H.; Harris, J.; Zhang, Z.; Quinn, J.; Morand, E.F.; Yang, Y.H. A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis. Br. J. Pharmacol. 2014, 171, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Le, Y.; Liu, Y.; Gong, W.; Ying, G.; Huang, J.; Yoshimura, T.; Tessarollo, L.; Wang, J.M. A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J. Immunol. 2010, 184, 3331–3335. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Aust, V.; Kress, E.; Abraham, S.; Schröder, N.; Kipp, M.; Stope, M.B.; Pufe, T.; Tauber, S.C.; Brandenburg, L.-O. Lack of chemokine (C-C motif) ligand 3 leads to decreased survival and reduced immune response after bacterial meningitis. Cytokine 2018, 111, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Merres, J.; Höss, J.; Albrecht, L.-J.; Kress, E.; Soehnlein, O.; Jansen, S.; Pufe, T.; Tauber, S.C.; Brandenburg, L.-O. Role of the Cathelicidin-Related Antimicrobial Peptide in Inflammation and Mortality in a Mouse Model of Bacterial Meningitis. J. Innate Immun. 2013, 6, 205–218. [Google Scholar] [CrossRef]
- Roseborough, A.D.; Zhu, Y.; Zhao, L.; Laviolette, S.R.; Pasternak, S.H.; Whitehead, S.N. Fibrinogen primes the microglial NLRP3 inflammasome and propagates pro-inflammatory signaling via extracellular vesicles: Implications for blood-brain barrier dysfunction. Neurobiol. Dis. 2023, 177, 106001. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Iovino, F.; Engelen-Lee, J.-Y.; Brouwer, M.; van de Beek, D.; van der Ende, A.; Valls Seron, M.; Mellroth, P.; Muschiol, S.; Bergstrand, J.; Widengren, J.; et al. pIgR and PECAM-1 bind to pneumococcal adhesins RrgA and PspC mediating bacterial brain invasion. J. Exp. Med. 2017, 214, 1619–1630. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Campbell, M. Tight junction modulation at the blood-brain barrier: Current and future perspectives. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183298. [Google Scholar] [CrossRef]
- Farmen, K.; Tofiño-Vian, M.; Iovino, F. Neuronal Damage and Neuroinflammation, a Bridge Between Bacterial Meningitis and Neurodegenerative Diseases. Front. Cell. Neurosci. 2021, 15, 680858. [Google Scholar] [CrossRef]
- Yang, R.; Wang, J.; Wang, F.; Zhang, H.; Tan, C.; Chen, H.; Wang, X. Blood-Brain Barrier Integrity Damage in Bacterial Meningitis: The Underlying Link, Mechanisms, and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 2852. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Walsh, D.M.; O’Malley, T.; Hudson, N.; Crosbie, D.E.; Loftus, T.; Sheehan, F.; McDaid, J.; Humphries, M.M.; Callanan, J.J.; et al. Autoregulated paracellular clearance of amyloid-β across the blood-brain barrier. Sci. Adv. 2015, 1, e1500472. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef]
- Gilpin, T.E.; Walter, F.R.; Herbath, M.; Sandor, M.; Fabry, Z. Mycobacterium bovis Bacillus Calmette-Guérin-Infected Dendritic Cells Induce TNF-α-Dependent Cell Cluster Formation That Promotes Bacterial Dissemination through an In Vitro Model of the Blood-Brain Barrier. J. Immunol. 2021, 207, 1065–1077. [Google Scholar] [CrossRef]
- Kim, B.J.; Hancock, B.M.; Bermudez, A.; Del Cid, N.; Reyes, E.; van Sorge, N.M.; Lauth, X.; Smurthwaite, C.A.; Hilton, B.J.; Stotland, A.; et al. Bacterial induction of Snail1 contributes to blood-brain barrier disruption. J. Clin. Investig. 2015, 125, 2473–2483. [Google Scholar] [CrossRef]
- Muller, W.A. Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. 2011, 6, 323–344. [Google Scholar] [CrossRef]
- Iovino, F.; Molema, G.; Bijlsma, J.J.E. Platelet endothelial cell adhesion molecule-1, a putative receptor for the adhesion of Streptococcus pneumoniae to the vascular endothelium of the blood-brain barrier. Infect. Immun. 2014, 82, 3555–3566. [Google Scholar] [CrossRef]
- Zysk, G.; Schneider-Wald, B.K.; Hwang, J.H.; Bejo, L.; Kim, K.S.; Mitchell, T.J.; Hakenbeck, R.; Heinz, H.-P. Pneumolysin Is the Main Inducer of Cytotoxicity to Brain Microvascular Endothelial Cells Caused by Streptococcus pneumoniae. Infect. Immun. 2001, 69, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; Dalziel, C.E. The biology of pneumolysin. Subcell. Biochem. 2014, 80, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Hupp, S.; Heimeroth, V.; Wippel, C.; Förtsch, C.; Ma, J.; Mitchell, T.J.; Iliev, A.I. Astrocytic tissue remodeling by the meningitis neurotoxin pneumolysin facilitates pathogen tissue penetration and produces interstitial brain edema. Glia 2012, 60, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Hupp, S.; Grandgirard, D.; Mitchell, T.J.; Leib, S.L.; Hathaway, L.J.; Iliev, A.I. Pneumolysin and the bacterial capsule of Streptococcus pneumoniae cooperatively inhibit taxis and motility of microglia. J. Neuroinflamm. 2019, 16, 105. [Google Scholar] [CrossRef]
- Wall, E.C.; Gordon, S.B.; Hussain, S.; Goonetilleke, U.R.S.; Gritzfeld, J.; Scarborough, M.; Kadioglu, A. Persistence of pneumolysin in the cerebrospinal fluid of patients with pneumococcal meningitis is associated with mortality. Clin. Infect. Dis. 2012, 54, 701–705. [Google Scholar] [CrossRef]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J. Clin. Investig. 2002, 109, 19–27. [Google Scholar] [CrossRef]
- Braun, J.S.; Hoffmann, O.; Schickhaus, M.; Freyer, D.; Dagand, E.; Bermpohl, D.; Mitchell, T.J.; Bechmann, I.; Weber, J.R. Pneumolysin causes neuronal cell death through mitochondrial damage. Infect. Immun. 2007, 75, 4245–4254. [Google Scholar] [CrossRef]
- Kim, K.S. Pathogenesis of bacterial meningitis: From bacteraemia to neuronal injury. Nat. Rev. Neurosci. 2003, 4, 376–385. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Hayhoe, R.P.G.; Kamal, A.M.; Solito, E.; Flower, R.J.; Cooper, D.; Perretti, M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: Indication of distinct receptor involvement. Blood 2006, 107, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Riehemann, K.; Gerke, V. A novel ligand of the formyl peptide receptor: Annexin I regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 2000, 5, 831–840. [Google Scholar] [CrossRef]
- Lim, L.H.K.; Solito, E.; Russo-Marie, F.; Flower, R.J.; Perretti, M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: Effect of lipocortin 1. Proc. Natl. Acad. Sci. USA 1998, 95, 14535–14539. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Gao, S.; Zheng, Y.; Liu, P.; Zhai, Y.; Huang, W.; Jiang, H.; Lv, Q.; Kong, D.; Jiang, Y. Annexin A1 Attenuates Neutrophil Migration and IL-6 Expression through Fpr2 in a Mouse Model of Streptococcus suis-Induced Meningitis. Infect. Immun. 2021, 89, e00680-20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2016, 136, 826–836. [Google Scholar] [CrossRef]
- Gimenes, A.D.; Andrade, T.R.M.; Mello, C.B.; Ramos, L.; Gil, C.D.; Oliani, S.M. Beneficial effect of annexin A1 in a model of experimental allergic conjunctivitis. Exp. Eye Res. 2015, 134, 24–32. [Google Scholar] [CrossRef]
- Guido, B.C.; Zanatelli, M.; Tavares-de-Lima, W.; Oliani, S.M.; Damazo, A.S. Annexin-A1 peptide down-regulates the leukocyte recruitment and up-regulates interleukin-10 release into lung after intestinal ischemia-reperfusion in mice. J. Inflamm. 2013, 10, 10. [Google Scholar] [CrossRef]
- Girol, A.P.; Mimura, K.K.O.; Drewes, C.C.; Bolonheis, S.M.; Solito, E.; Farsky, S.H.P.; Gil, C.D.; Oliani, S.M. Anti-inflammatory mechanisms of the annexin A1 protein and its mimetic peptide Ac2-26 in models of ocular inflammation in vivo and in vitro. J. Immunol. 2013, 190, 5689–5701. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deutloff, J.; Pöhner, I.; Rößler, J.; Kipp, M.; Tauber, S.C.; Brandenburg, L.-O. The Formyl Peptid Receptor Ligand Ac2-26 Improves the Integrity of the Blood−Brain Barrier in the Course of Pneumococcal Meningitis. Cells 2024, 13, 2104. https://doi.org/10.3390/cells13242104
Deutloff J, Pöhner I, Rößler J, Kipp M, Tauber SC, Brandenburg L-O. The Formyl Peptid Receptor Ligand Ac2-26 Improves the Integrity of the Blood−Brain Barrier in the Course of Pneumococcal Meningitis. Cells. 2024; 13(24):2104. https://doi.org/10.3390/cells13242104
Chicago/Turabian StyleDeutloff, Johannes, Irina Pöhner, Johann Rößler, Markus Kipp, Simone C. Tauber, and Lars-Ove Brandenburg. 2024. "The Formyl Peptid Receptor Ligand Ac2-26 Improves the Integrity of the Blood−Brain Barrier in the Course of Pneumococcal Meningitis" Cells 13, no. 24: 2104. https://doi.org/10.3390/cells13242104
APA StyleDeutloff, J., Pöhner, I., Rößler, J., Kipp, M., Tauber, S. C., & Brandenburg, L.-O. (2024). The Formyl Peptid Receptor Ligand Ac2-26 Improves the Integrity of the Blood−Brain Barrier in the Course of Pneumococcal Meningitis. Cells, 13(24), 2104. https://doi.org/10.3390/cells13242104