
A New Synthesized Dicarboxylated Oxy-Heparin Efficiently Attenuates Tumor Growth and Metastasis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Compound XII
2.2. Heparanase Enzymatic Activity
2.2.1. Fondaparinux (Arixtra)
2.2.2. FRET Analysis
2.2.3. 35Sulfate-Labeled ECM
2.3. Cells
2.4. Cancer Models
2.4.1. Pancreatic Ductal Adenocarcinoma (PDAC)
2.4.2. PDX Model
2.4.3. Breast Carcinoma
2.4.4. Mesothelioma
2.4.5. Myeloma
2.4.6. IVIS Imaging
2.5. Western Blot Analysis
2.6. Heparanase Uptake and Processing
2.7. Pharmacokinetics
2.8. Statistics
3. Results
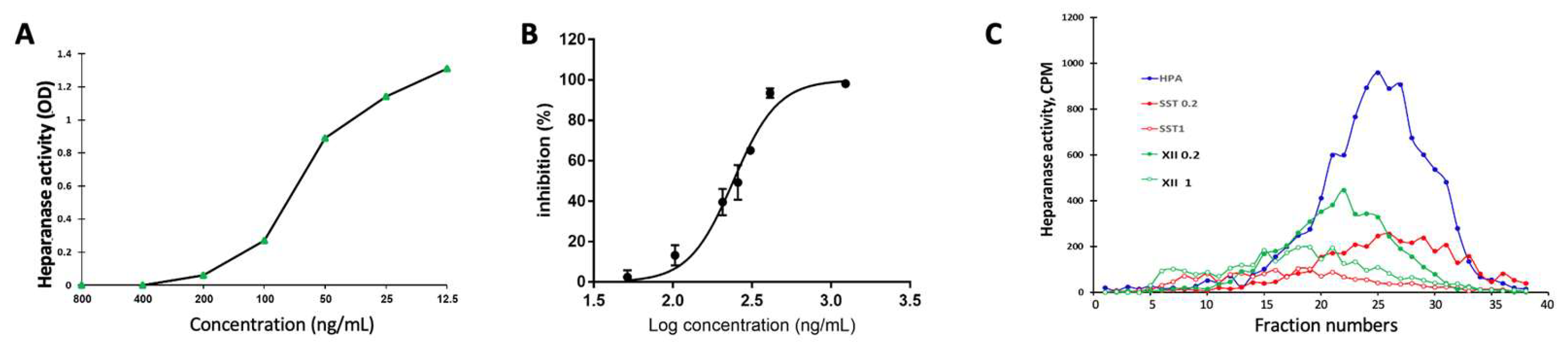
3.1. Preparation of Dicarboxylated Oxy-Heparin (DcoxyH = compound XII)
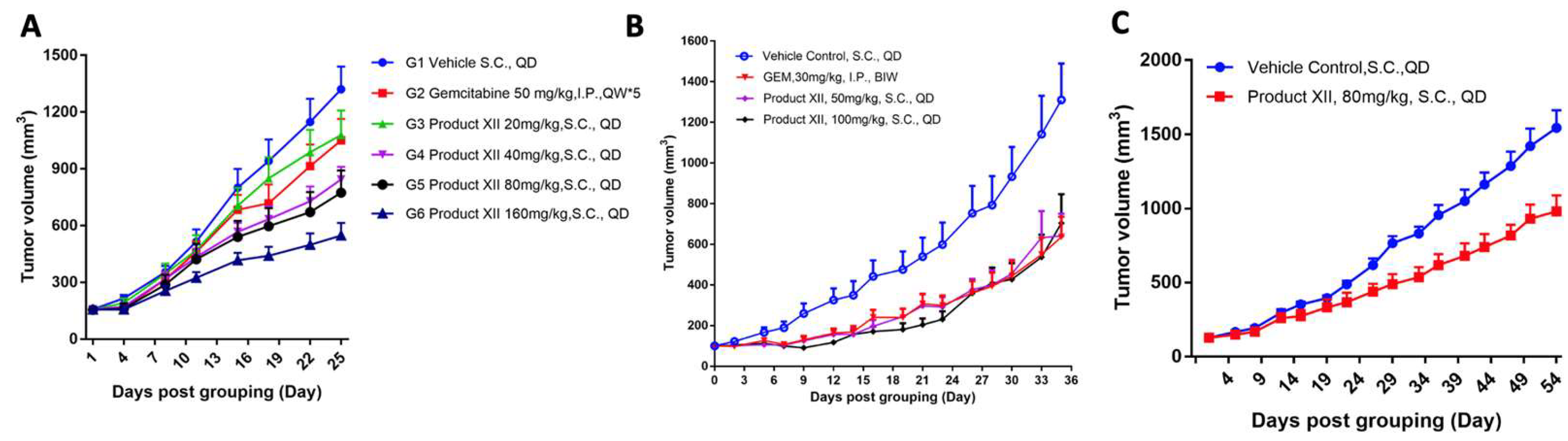
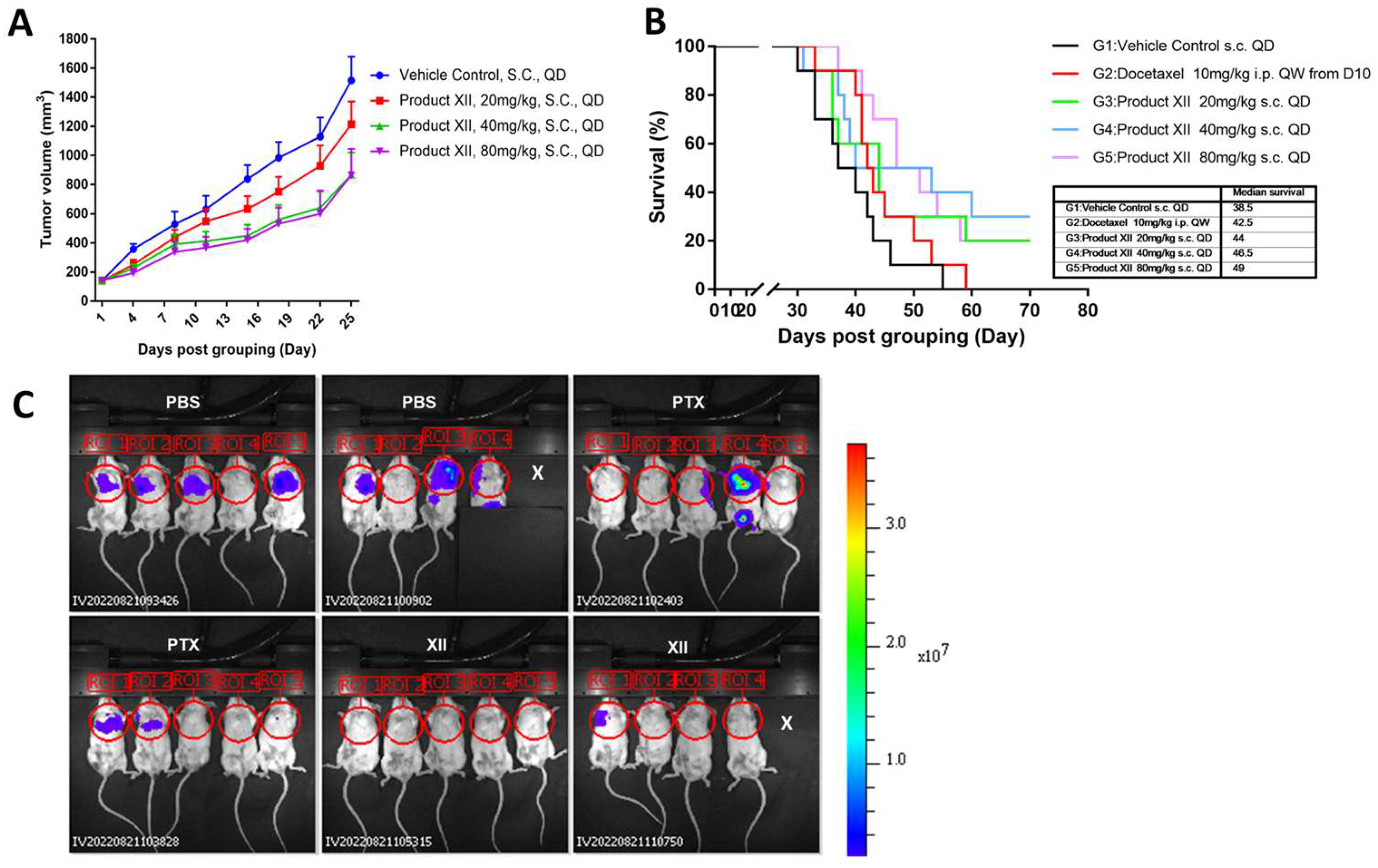
3.2. Pancreatic Cancer
3.3. Breast Cancer
3.4. Breast Cancer Metastasis
3.5. Mesothelioma
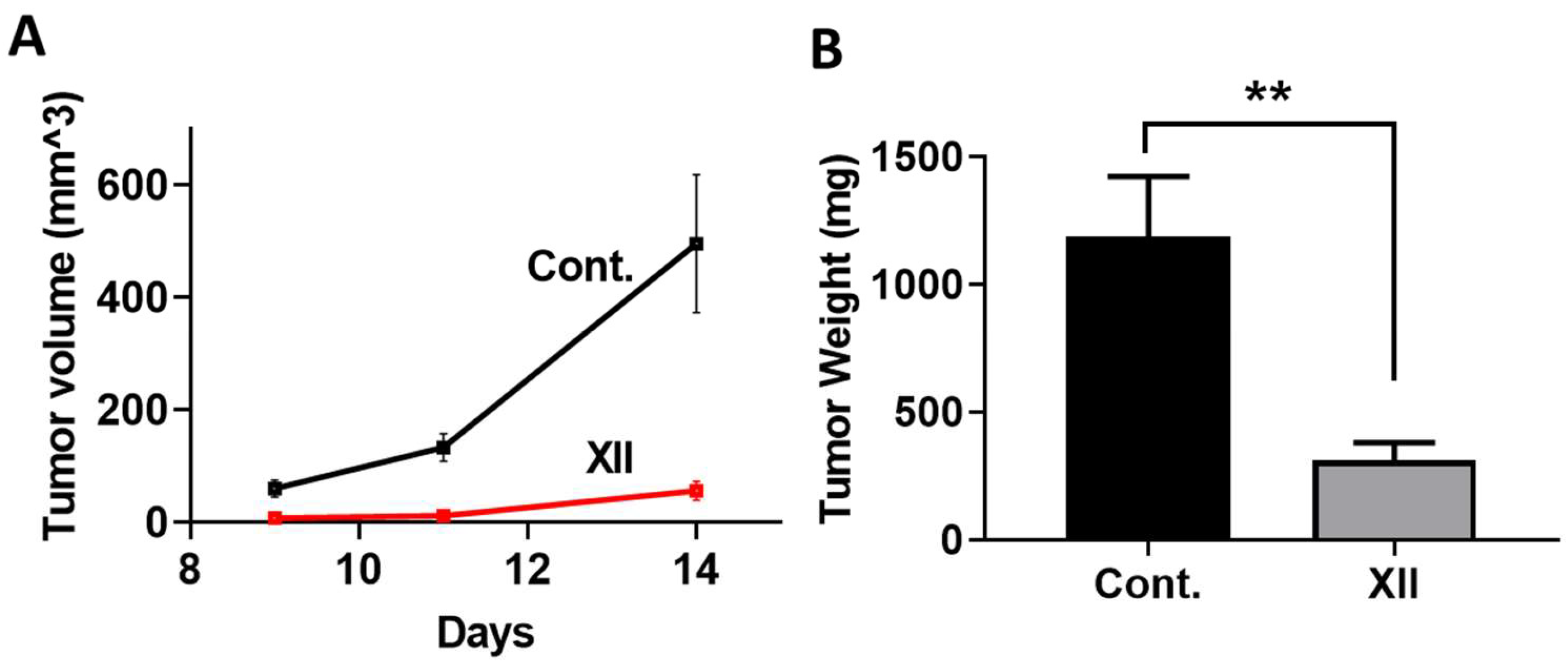
3.6. Mouse Myeloma
3.7. Heparanase Levels in PDAC Tumors
3.8. Heparanase Uptake and Processing
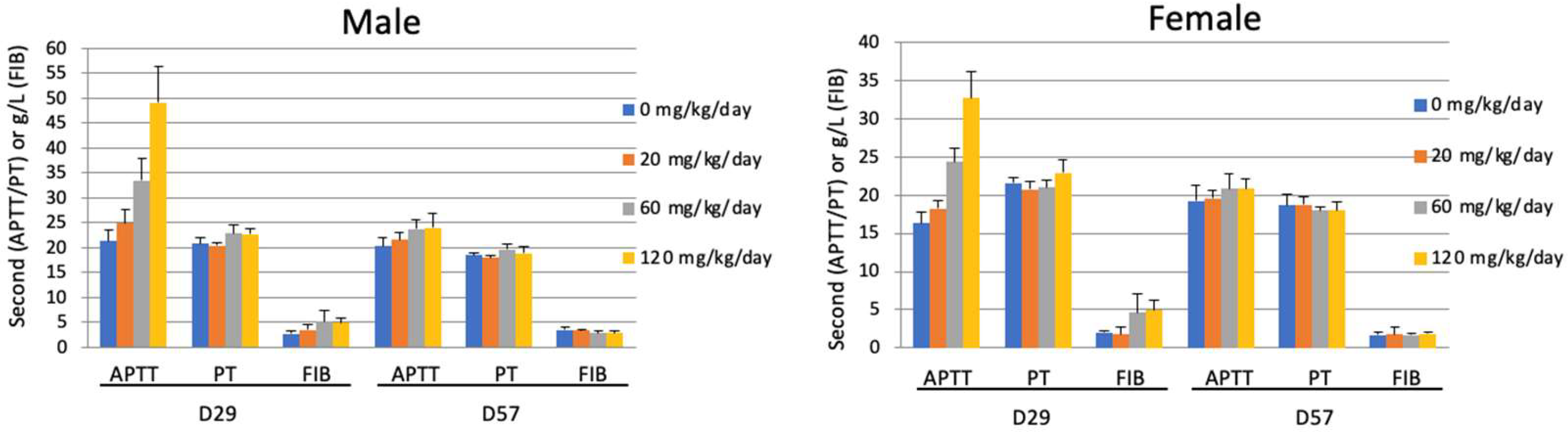
3.9. Effect of Compound XII on Coagulation Parameters
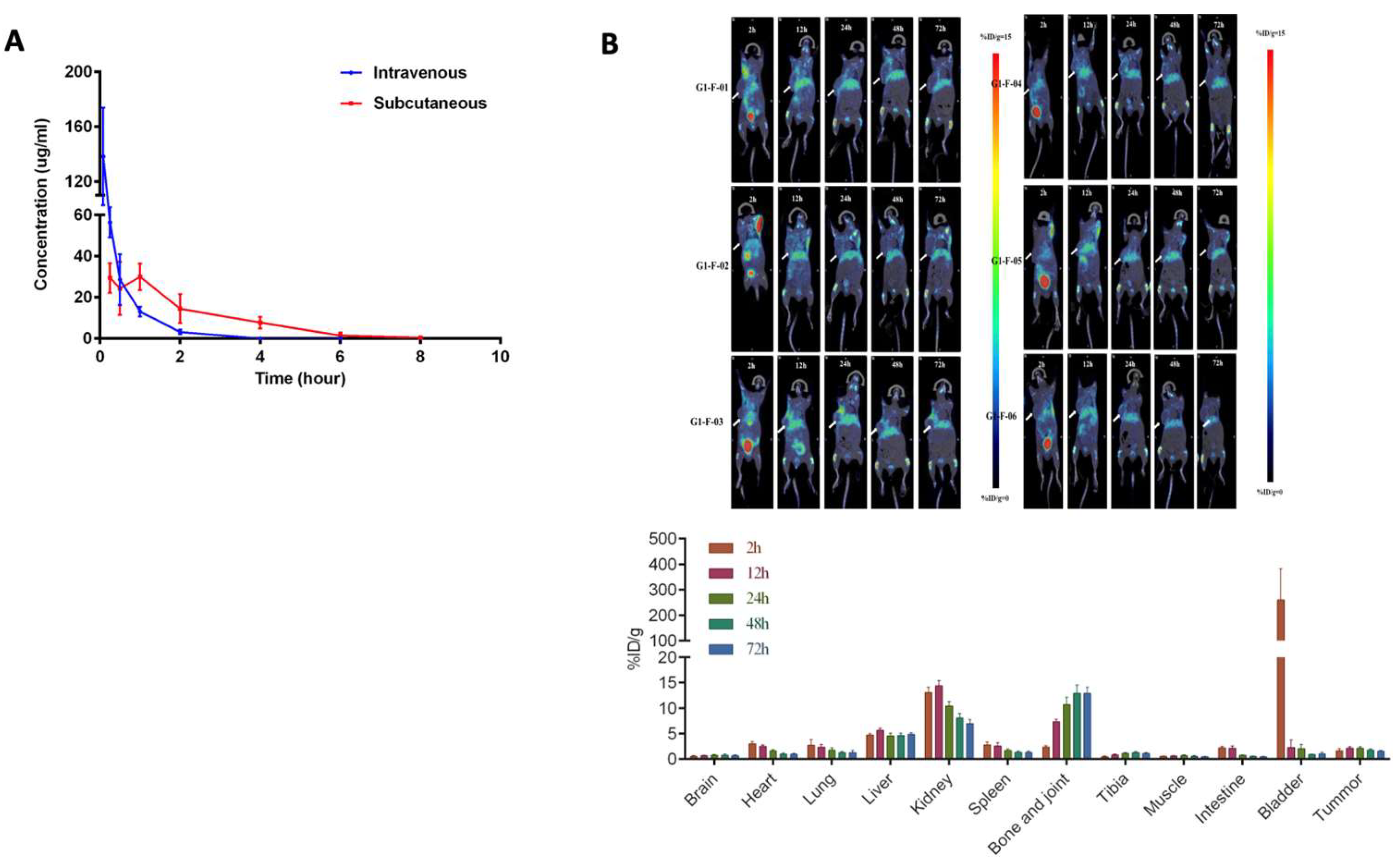
3.10. Pharmacokinetics of Compound XII
4. Discussion
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaskin, S.M.; Soares Da Costa, T.P.; Hulett, M.D. Heparanase: Cloning, Function and Regulation. Adv. Exp. Med. Biol. 2020, 1221, 189–229. [Google Scholar] [CrossRef]
- Ilan, N.; Bhattacharya, U.; Barash, U.; Boyango, I.; Yanku, Y.; Gross-Cohen, M.; Vlodavsky, I. Heparanase-The message comes in different flavors. Adv. Exp. Med. Biol. 2020, 1221, 253–283. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Bandari, S.K.; Vlodavsky, I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019, 75–76, 160–169. [Google Scholar] [CrossRef]
- Agelidis, A.; Suryawanshi, R.K.; Patil, C.D.; Campeau, A.; Gonzalez, D.J.; Shukla, D. Dissociation of DNA damage sensing by endoglycosidase HPSE. iScience 2021, 24, 102242. [Google Scholar] [CrossRef] [PubMed]
- Jayatilleke, K.M.; Hulett, M.D. Heparanase and the hallmarks of cancer. J. Transl. Med. 2020, 18, 453. [Google Scholar] [CrossRef] [PubMed]
- Mayfosh, A.J.; Nguyen, T.K.; Hulett, M.D. The Heparanase Regulatory Network in Health and Disease. Int. J. Mol. Sci. 2021, 22, 1096. [Google Scholar] [CrossRef] [PubMed]
- Khanna, M.; Parish, C.R. Heparanase: Historical Aspects and Future Perspectives. Adv. Exp. Med. Biol. 2020, 1221, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef]
- Purushothaman, A.; Hurst, D.R.; Pisano, C.; Mizumoto, S.; Sugahara, K.; Sanderson, R.D. Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. J. Biol. Chem. 2011, 286, 30377–30383. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updates 2016, 29, 54–75. [Google Scholar] [CrossRef]
- Arvatz, G.; Shafat, I.; Levy-Adam, F.; Ilan, N.; Vlodavsky, I. The heparanase system and tumor metastasis: Is heparanase the seed and soil? Cancer Metastasis Rev. 2011, 30, 253–268. [Google Scholar] [CrossRef]
- Ramani, V.C.; Zhan, F.; He, J.; Barbieri, P.; Noseda, A.; Tricot, G.; Sanderson, R.D. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget 2016, 7, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Shteingauz, A.; Boyango, I.; Naroditsky, I.; Hammond, E.; Gruber, M.; Doweck, I.; Ilan, N.; Vlodavsky, I. Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer Res. 2015, 75, 3946–3957. [Google Scholar] [CrossRef] [PubMed]
- Barash, U.; Lapidot, M.; Zohar, Y.; Loomis, C.; Moreira, A.; Feld, S.; Goparaju, C.; Yang, H.; Hammond, E.; Zhang, G.; et al. Involvement of Heparanase in the Pathogenesis of Mesothelioma: Basic Aspects and Clinical Applications. J. Natl. Cancer Inst. 2018, 110, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M.; Naomoto, Y.; Ohkawa, T.; Uetsuka, H.; Shirakawa, Y.; Uno, F.; Fujiwara, T.; Gunduz, M.; Nagatsuka, H.; Nakajima, M.; et al. Heparanase expression correlates with invasion and poor prognosis in gastric cancers. Lab. Investig. 2003, 83, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Xiong, A.; Spyrou, A.; Wicher, G.; Marinescu, V.D.; Edqvist, P.D.; Zhang, L.; Essand, M.; Dimberg, A.; Smits, A.; et al. Heparanase promotes glioma progression and is inversely correlated with patient survival. Mol. Cancer Res. 2016, 14, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, G.; Nian, J.; Yu, M.; Chen, S.; Zhang, Y.; Yang, G.; Yang, L.; Cheng, P.; Yan, C.; et al. Elevated heparanase expression is associated with poor prognosis in breast cancer: A study based on systematic review and TCGA data. Oncotarget 2017, 27, 43521–43535. [Google Scholar] [CrossRef]
- Galli, M.; Chatterjee, M.; Grasso, M.; Specchia, G.; Magen, H.; Einsele, H.; Celeghini, I.; Barbieri, P.; Paoletti, D.; Pace, S.; et al. Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica 2018, 103, e469–e472. [Google Scholar] [CrossRef]
- Lemech, C.; Dredge, K.; Bampton, D.; Hammond, E.; Clouston, A.; Waterhouse, N.J.; Stanley, A.C.; Leveque-El Mouttie, L.; Chojnowski, G.M.; Haydon, A.; et al. Phase Ib open-label, multicenter study of pixatimod, an activator of TLR9, in combination with nivolumab in subjects with microsatellite-stable metastatic colorectal cancer, metastatic pancreatic ductal adenocarcinoma and other solid tumors. J. Immunother. Cancer 2023, 11, e006136. [Google Scholar] [CrossRef]
- Liu, C.J.; Chang, J.; Lee, P.H.; Lin, D.Y.; Wu, C.C.; Jeng, L.B.; Lin, Y.J.; Mok, K.T.; Lee, W.C.; Yeh, H.Z.; et al. Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World J. Gastroenterol. 2014, 20, 11384–11393. [Google Scholar] [CrossRef]
- Noseda, A.; Barbieri, P. Roneparstat: Development, Preclinical and Clinical Studies. Adv. Exp. Med. Biol. 2020, 1221, 523–538. [Google Scholar] [CrossRef]
- Giannini, G.; Battistuzzi, G.; Rivara, S. The Control of Heparanase Through the Use of Small Molecules. Adv. Exp. Med. Biol. 2020, 1221, 567–603. [Google Scholar] [CrossRef]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef]
- Cassinelli, G.; Torri, G.; Naggi, A. Non-Anticoagulant Heparins as Heparanase Inhibitors. Adv. Exp. Med. Biol. 2020, 1221, 493–522. [Google Scholar] [CrossRef]
- Hammond, E.; Li, C.P.; Ferro, V. Development of a colorimetric assay for heparanase activity suitable for kinetic analysis and inhibitor screening. Anal. Biochem. 2011, 396, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, K.; Okamoto, H.; Numata, Y.; Takemoto, H. A simple and rapid assay for heparanase activity using homogeneous time-resolved fluorescence. J. Pharm. Biomed. Anal. 2006, 41, 912–917. [Google Scholar] [CrossRef]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arter. Thromb. Vascular Biol. 2013, 33, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 1999, 5, 793–802. [Google Scholar] [CrossRef]
- Gingis-Velitski, S.; Zetser, A.; Kaplan, V.; Ben-Zaken, O.; Cohen, E.; Levy-Adam, F.; Bashenko, Y.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J. Biol. Chem. 2004, 279, 44084–44092. [Google Scholar] [CrossRef]
- Levy-Adam, F.; Feld, S.; Cohen-Kaplan, V.; Shteingauz, A.; Gross, M.; Arvatz, G.; Naroditsky, I.; Ilan, N.; Doweck, I.; Vlodavsky, I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J. Biol. Chem. 2010, 285, 28010–28019. [Google Scholar] [CrossRef]
- Katz, A.; Barash, U.; Boyango, I.; Feld, S.; Zohar, Y.; Hammond, E.; Ilan, N.; Kremer, R.; Vlodavsky, I. Patient derived xenografts (PDX) predict an effective heparanase-based therapy for lung cancer. Oncotarget 2018, 9, 19294–19306. [Google Scholar] [CrossRef]
- Ferguson, V.L.; Simske, S.J.; Ayers, R.A.; Bateman, T.A.; Wang, H.T.; Bendele, A.; Rich, B.; Collins, D.; Scherrer, J.; Sennello, R.; et al. Effect of MPC-11 myeloma and MPC-11 + IL-1 receptor antagonist treatment on mouse bone properties. Bone 2002, 30, 109–116. [Google Scholar] [CrossRef]
- Alekseeva, A.; Mazzini, G.; Giannini, G.; Naggi, A. Structural features of heparanase-inhibiting non-anticoagulant heparin derivative Roneparstat. Carbohydr. Polym. 2017, 156, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Rohloff, J.; Zinke, J.; Schoppmeyer, K.; Tannapfel, A.; Witzigmann, H.; Mossner, J.; Wittekind, C.; Caca, K. Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. Br. J. Cancer 2002, 86, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Maly, B.; Simon, I.; Meirovitz, A.; Pikarsky, E.; Zcharia, E.; Peretz, T.; Vlodavsky, I.; Elkin, M. Tamoxifen induces heparanase expression in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2007, 13, 4069–4077. [Google Scholar] [CrossRef] [PubMed]
- Hermano, E.; Goldberg, R.; Rubinstein, A.M.; Sonnenblick, A.; Maly, B.; Nahmias, D.; Li, J.P.; Bakker, M.A.H.; van der Vlag, J.; Vlodavsky, I.; et al. Heparanase Accelerates Obesity-Associated Breast Cancer Progression. Cancer Res. 2019, 79, 5342–5354. [Google Scholar] [CrossRef]
- Kelly, T.; Suva, L.J.; Huang, Y.; Macleod, V.; Miao, H.Q.; Walker, R.C.; Sanderson, R.D. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res. 2005, 65, 5778–5784. [Google Scholar] [CrossRef]
- Maxhimer, J.B.; Pesce, C.E.; Stewart, R.A.; Gattuso, P.; Prinz, R.A.; Xu, X. Ductal carcinoma in situ of the breast and heparanase-1 expression: A molecular explanation for more aggressive subtypes. J. Am. Coll. Surg. 2005, 200, 328–335. [Google Scholar] [CrossRef]
- Zahavi, T.; Salmon-Divon, M.; Salgado, R.; Elkin, M.; Hermano, E.; Rubinstein, A.M.; Francis, P.A.; Di Leo, A.; Viale, G.; de Azambuja, E.; et al. Heparanase: A potential marker of worse prognosis in estrogen receptor-positive breast cancer. NPJ Breast Cancer 2021, 7, 67. [Google Scholar] [CrossRef]
- Hammond, E.; Dredge, K. Heparanase Inhibition by Pixatimod (PG545): Basic Aspects and Future Perspectives. Adv. Exp. Med. Biol. 2020, 1221, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Zcharia, E.; Jia, J.; Zhang, X.; Baraz, L.; Lindahl, U.; Peretz, T.; Vlodavsky, I.; Li, J.P. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE 2009, 4, e5181. [Google Scholar] [CrossRef]
- de Boer, C.; Armstrong, Z.; Lit, V.A.J.; Barash, U.; Ruijgrok, G.; Boyango, I.; Weitzenberg, M.M.; Schroder, S.P.; Sarris, A.J.C.; Meeuwenoord, N.J.; et al. Mechanism-based heparanase inhibitors reduce cancer metastasis in vivo. Proc. Natl. Acad. Sci. USA 2022, 119, e2203167119. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.A.; Rolph, R.; Cutress, R.I.; Copson, E.R. A Review of Modifiable Risk Factors in Young Women for the Prevention of Breast Cancer. Breast Cancer 2021, 13, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef]
- Carbone, M.; Ly, B.H.; Dodson, R.F.; Pagano, I.; Morris, P.T.; Dogan, U.A.; Gazdar, A.F.; Pass, H.I.; Yang, H. Malignant mesothelioma: Facts, myths, and hypotheses. J. Cell Physiol. 2012, 227, 44–58. [Google Scholar] [CrossRef]
- Sekido, Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis 2013, 34, 1413–1419. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Barash, U.; Ilan, N.; Farhoud, M.; Zhang, X.; Vlodavsky, I.; Li, J.-P. A New Synthesized Dicarboxylated Oxy-Heparin Efficiently Attenuates Tumor Growth and Metastasis. Cells 2024, 13, 211. https://doi.org/10.3390/cells13030211
Li L, Barash U, Ilan N, Farhoud M, Zhang X, Vlodavsky I, Li J-P. A New Synthesized Dicarboxylated Oxy-Heparin Efficiently Attenuates Tumor Growth and Metastasis. Cells. 2024; 13(3):211. https://doi.org/10.3390/cells13030211
Chicago/Turabian StyleLi, Li, Uri Barash, Neta Ilan, Malik Farhoud, Xiao Zhang, Israel Vlodavsky, and Jin-Ping Li. 2024. "A New Synthesized Dicarboxylated Oxy-Heparin Efficiently Attenuates Tumor Growth and Metastasis" Cells 13, no. 3: 211. https://doi.org/10.3390/cells13030211