
Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data Reveals Memory-like NK Cell Subset Associated with Mycobacterium tuberculosis Latency

 ,
,  ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Data Collection
2.3. Data Processing and Differential Gene Expression Analysis
2.4. Reference-Based Decomposition
2.5. Peripheral Immune Cell and NK Cell Reference Map
2.6. Preparation of PBMCs
2.7. Staining of Surface Antigens for Flow Cytometry
2.8. Statistical Analysis
3. Results
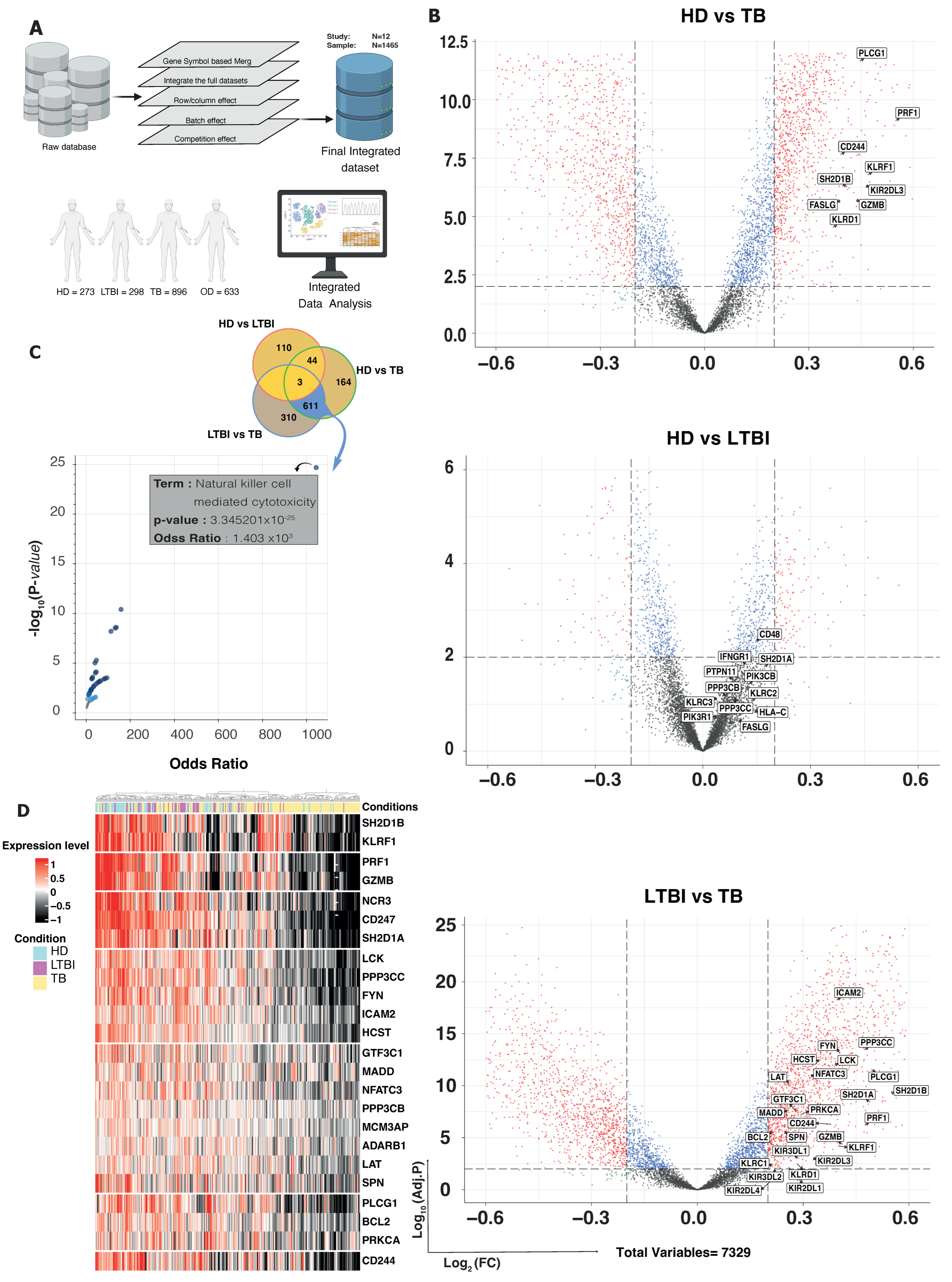
3.1. DEG and GSEA Analysis Reveal Specific Enrichment of NK-Mediated Immune Responses in LTBI Subjects
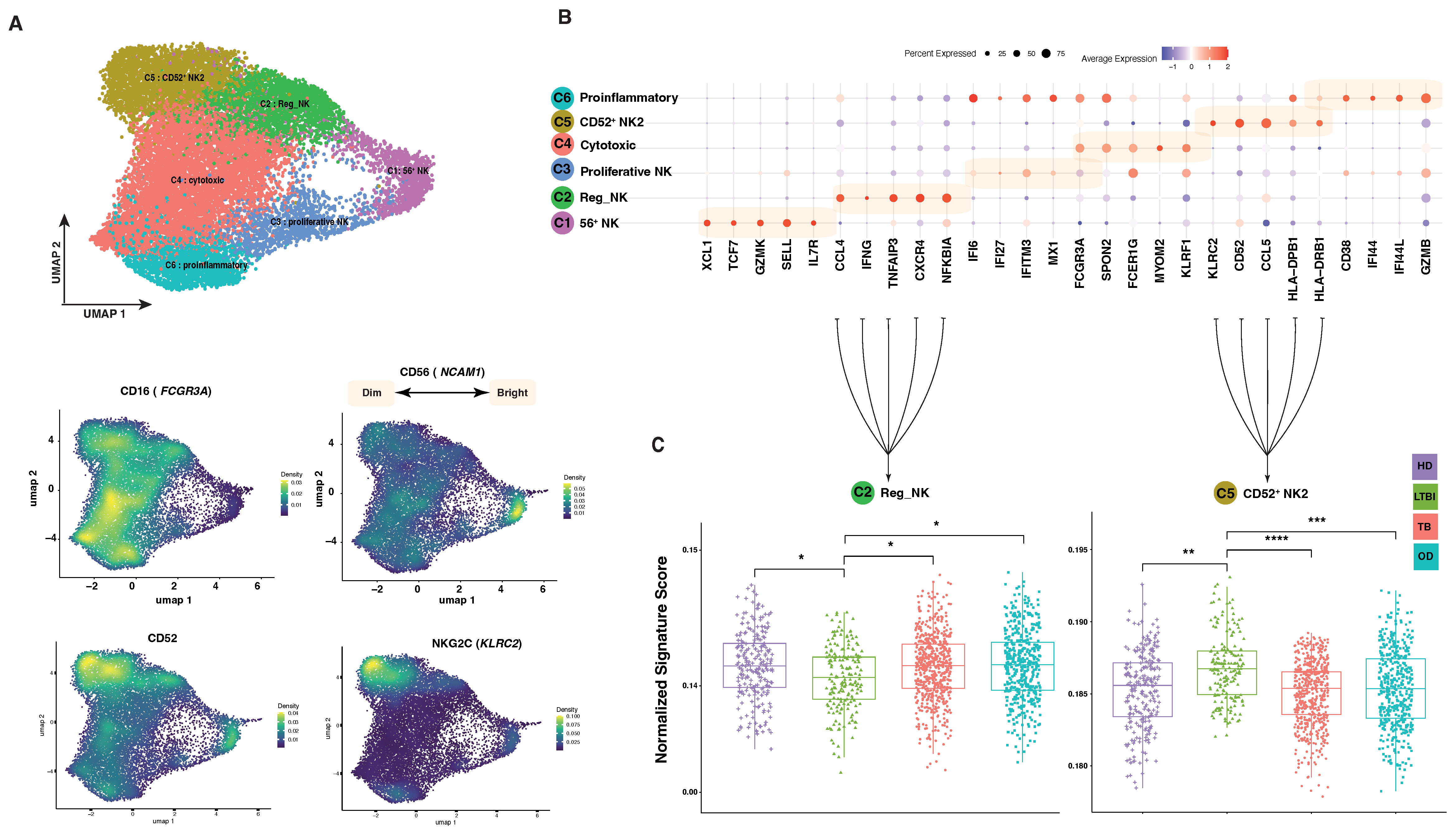
3.2. Computational Exploration into Immune Cell Composition in Different TB Conditions
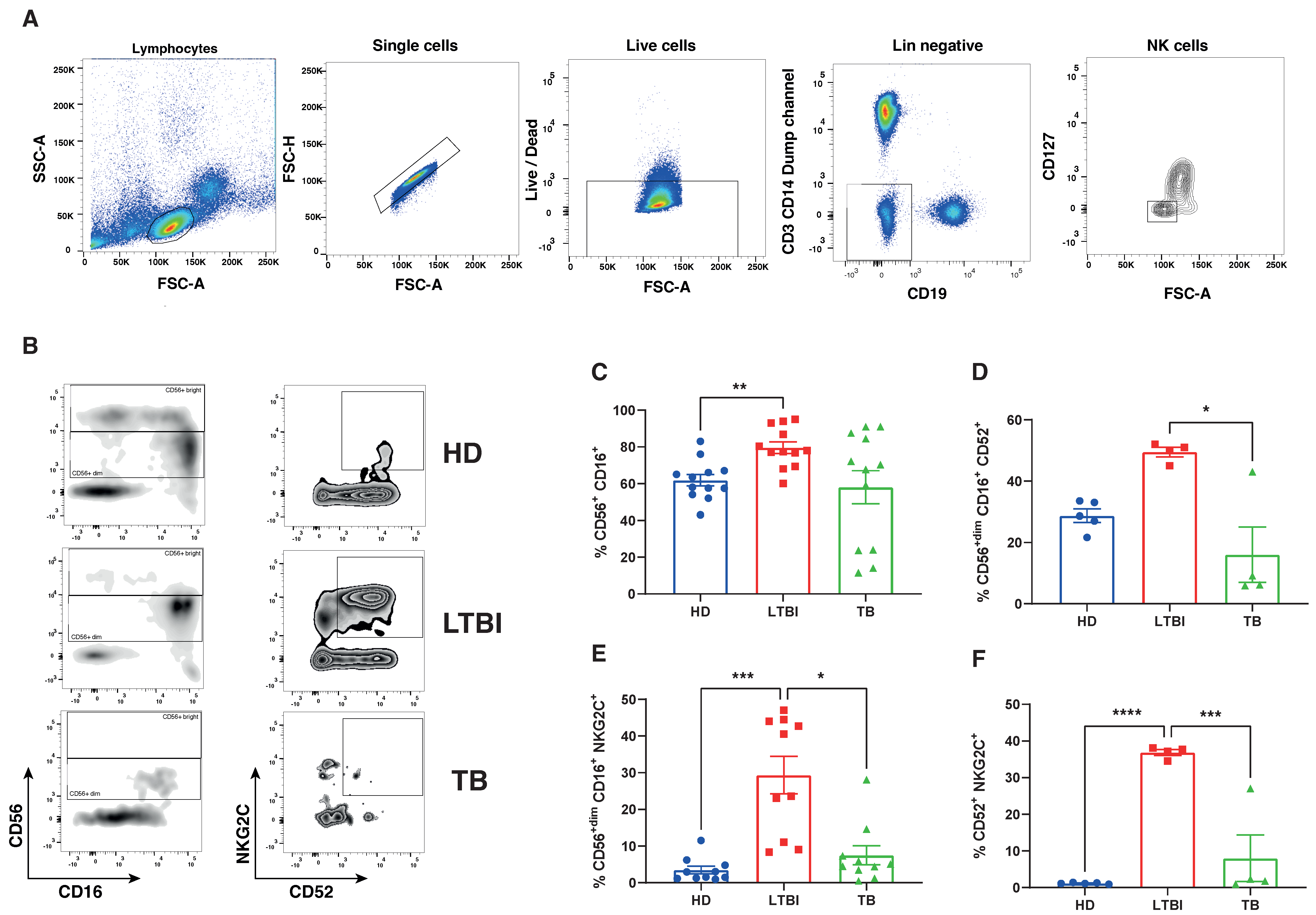
3.3. Comparison of NK Cell Transcriptional Landscape in Peripheral Blood of Different TB Conditions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quatrini, L.; Della Chiesa, M.; Sivori, S.; Mingari, M.C.; Pende, D.; Moretta, L. Human NK cells, their receptors and function. Eur. J. Immunol. 2021, 51, 1566–1579. [Google Scholar] [CrossRef]
- Dhiman, R.; Periasamy, S.; Barnes, P.F.; Jaiswal, A.G.; Paidipally, P.; Barnes, A.B.; Tvinnereim, A.; Vankayalapati, R. NK1.1+ Cells and IL-22 Regulate Vaccine-Induced Protective Immunity against Challenge with Mycobacterium tuberculosis. J. Immunol. 2012, 189, 897–905. [Google Scholar] [CrossRef]
- Roy, S.; Barnes, P.F.; Garg, A.; Wu, S.; Cosman, D.; Vankayalapati, R. NK Cells Lyse T Regulatory Cells That Expand in Response to an Intracellular Pathogen1. J. Immunol. 2008, 180, 1729–1736. [Google Scholar] [CrossRef]
- Zhang, R.; Zheng, X.; Li, B.; Wei, H.; Tian, Z. Human NK Cells Positively Regulate γδ T Cells in Response to Mycobacterium tuberculosis1. J. Immunol. 2006, 176, 2610–2616. [Google Scholar] [CrossRef]
- Vankayalapati, R.; Klucar, P.; Wizel, B.; Weis, S.E.; Samten, B.; Safi, H.; Shams, H.; Barnes, P.F. NK Cells Regulate CD8+ T Cell Effector Function in Response to an Intracellular Pathogen 1. J. Immunol. 2004, 172, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Venkatasubramanian, S.; Cheekatla, S.; Paidipally, P.; Tripathi, D.; Welch, E.; Tvinnereim, A.R.; Nurieva, R.; Vankayalapati, R. IL-21-dependent expansion of memory-like NK cells enhances protective immune responses against Mycobacterium tuberculosis. Mucosal Immunol. 2017, 10, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, C.; Germano, S.; Dutta, B.; Ritz, N.; Curtis, N. The Contribution of Non-Conventional T Cells and NK Cells in the Mycobacterial-Specific IFNγ Response in Bacille Calmette-Guérin (BCG)-Immunized Infants. PLoS ONE 2013, 8, e77334. [Google Scholar] [CrossRef] [PubMed]
- Portevin, D.; Via, L.E.; Eum, S.; Young, D. Natural killer cells are recruited during pulmonary tuberculosis and their ex vivo responses to mycobacteria vary between healthy human donors in association with KIR haplotype. Cell. Microbiol. 2012, 14, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Yu, S.; Yang, B.; Lao, S.; Li, B.; Wu, C. Memory-Like Antigen-Specific Human NK Cells from TB Pleural Fluids Produced IL-22 in Response to IL-15 or Mycobacterium tuberculosis Antigens. PLoS ONE 2016, 11, e0151721. [Google Scholar] [CrossRef] [PubMed]
- Brillantes, M.; Beaulieu, A.M. Memory and Memory-like NK Cell Responses to Microbial Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 102. [Google Scholar] [CrossRef]
- Roy Chowdhury, R.; Vallania, F.; Yang, Q.; Lopez Angel, C.J.; Darboe, F.; Penn-Nicholson, A.; Rozot, V. Author Correction: A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature 2018, 564, E5. [Google Scholar] [CrossRef]
- Esaulova, E.; Das, S.; Singh, D.K.; Choreño-Parra, J.A.; Swain, A.; Arthur, L.; Rangel-Moreno, J.; Ahmed, M.; Singh, B.; Gupta, A.; et al. The immune landscape in tuberculosis reveals populations linked to disease and latency. Cell Host Microbe 2021, 29, 165–178.e168. [Google Scholar] [CrossRef] [PubMed]
- Choreño-Parra, J.A.; Jiménez-Álvarez, L.A.; Maldonado-Díaz, E.D.; Cárdenas, G.; Fernández-Lopez, L.A.; Soto-Hernandez, J.L.; Muñoz-Torrico, M.; Ramírez-Martínez, G.; Cruz-Lagunas, A.; Vega-López, A.; et al. Phenotype of Peripheral NK Cells in Latent, Active, and Meningeal Tuberculosis. J. Immunol. Res. 2021, 2021, 5517856. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.H.; Ahmed, A.M.; Choudhuri, S.; Sen, A.; Hazra, A.; Pal, N.K.; Bhattacharya, B.; Bahar, B. Alteration of serum inflammatory cytokines in active pulmonary tuberculosis following anti-tuberculosis drug therapy. Mol. Immunol. 2014, 62, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lan, H.; Shi, H.; Wu, P.; Zhou, Y. Evaluating the diversity of circulating natural killer cells between active tuberculosis and latent tuberculosis infection. Tuberculosis 2022, 135, 102221. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Dai, Y.; Wang, Y.; Yang, Q.; Guo, J.; Wei, C.; Chen, W.; Huang, H.; Zhu, J.; Zhang, C.; et al. Single-cell transcriptomics of blood reveals a natural killer cell subset depletion in tuberculosis. eBioMedicine 2020, 53, 102686. [Google Scholar] [CrossRef]
- Prada-Medina, C.A.; Fukutani, K.F.; Pavan Kumar, N.; Gil-Santana, L.; Babu, S.; Lichtenstein, F.; West, K.; Sivakumar, S.; Menon, P.A.; Viswanathan, V.; et al. Systems Immunology of Diabetes-Tuberculosis Comorbidity Reveals Signatures of Disease Complications. Sci. Rep. 2017, 7, 1999. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef]
- Wickham, H.; Wickham, H. Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Yong, Y.K.; Tan, H.Y.; Saeidi, A.; Wong, W.F.; Vignesh, R.; Velu, V.; Eri, R.; Larsson, M.; Shankar, E.M. Immune Biomarkers for Diagnosis and Treatment Monitoring of Tuberculosis: Current Developments and Future Prospects. Front. Microbiol. 2019, 10, 2789. [Google Scholar] [CrossRef]
- Guo, C.; Wu, M.; Huang, B.; Zhao, R.; Jin, L.; Fu, B.; Wang, P.; Wang, D.; Zheng, M.; Fang, J.; et al. Single-cell transcriptomics reveal a unique memory-like NK cell subset that accumulates with ageing and correlates with disease severity in COVID-19. Genome Med. 2022, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Krämer, B.; Knoll, R.; Bonaguro, L.; ToVinh, M.; Raabe, J.; Astaburuaga-García, R.; Schulte-Schrepping, J.; Kaiser, K.M.; Rieke, G.J.; Bischoff, J.; et al. Early IFN-α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity 2021, 54, 2650–2669.e2614. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Siebert, J.R.; Burns, R.; Gerbec, Z.J.; Bonacci, B.; Rymaszewski, A.; Rau, M.; Riese, M.J.; Rao, S.; Carlson, K.-S.; et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 2019, 10, 3931. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.; Badami, G.D.; Azgomi, M.S.; Dieli, F.; La Manna, M.P.; Caccamo, N. Role of hematopoietic cells in Mycobacterium tuberculosis infection. Tuberculosis 2021, 130, 102109. [Google Scholar] [CrossRef] [PubMed]
- Maertzdorf, J.; Repsilber, D.; Parida, S.K.; Stanley, K.; Roberts, T.; Black, G.; Walzl, G.; Kaufmann, S.H.E. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun. 2011, 12, 15–22. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Vergès, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood J. Am. Soc. Hematol. 2012, 119, 2665–2674. [Google Scholar] [CrossRef]
- Béziat, V.; Dalgard, O.; Asselah, T.; Halfon, P.; Bedossa, P.; Boudifa, A.; Hervier, B.; Theodorou, I.; Martinot, M.; Debré, P. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur. J. Immunol. 2012, 42, 447–457. [Google Scholar] [CrossRef]
- Björkström, N.K.; Lindgren, T.; Stoltz, M.; Fauriat, C.; Braun, M.; Evander, M.; Michaëlsson, J.; Malmberg, K.-J.; Klingström, J.; Ahlm, C. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 2011, 208, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Petitdemange, C.; Becquart, P.; Wauquier, N.; Béziat, V.; Debré, P.; Leroy, E.M.; Vieillard, V. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog. 2011, 7, e1002268. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.; Cooley, S.; Verneris, M.R.; Curtsinger, J.; Luo, X.; Waller, E.K.; Anasetti, C.; Weisdorf, D.; Miller, J.S. Human cytomegalovirus (CMV)-induced memory-like NKG2C+ NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol. 2012, 189, 5082–5088. [Google Scholar] [CrossRef] [PubMed]
- Rölle, A.; Pollmann, J.; Ewen, E.-M.; Le, V.T.K.; Halenius, A.; Hengel, H.; Cerwenka, A. IL-12–producing monocytes and HLA-E control HCMV-driven NKG2C+ NK cell expansion. J. Clin. Investig. 2014, 124, 5305–5316. [Google Scholar] [CrossRef] [PubMed]
- Gumá, M.n.; Angulo, A.; Vilches, C.; Gómez-Lozano, N.; Malats, N.R.; López-Botet, M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Garand, M.; Goodier, M.; Owolabi, O.; Donkor, S.; Kampmann, B.; Sutherland, J.S. Functional and Phenotypic Changes of Natural Killer Cells in Whole Blood during Mycobacterium tuberculosis Infection and Disease. Front. Immunol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Vales-Gomez, M.; Reyburn, H.T.; Erskine, R.A.; Lopez-Botet, M.; Strominger, J.L. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J. 1999, 18, 4250–4260. [Google Scholar] [CrossRef] [PubMed]
- Björkström, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Björklund, A.T.; Flodström-Tullberg, M.; Michaëlsson, J.; Rottenberg, M.E. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood J. Am. Soc. Hematol. 2010, 116, 3853–3864. [Google Scholar] [CrossRef] [PubMed]
- Rölle, A.; Brodin, P. Immune adaptation to environmental influence: The case of NK cells and HCMV. Trends Immunol. 2016, 37, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Taras, E.; Chiuppesi, F.; Wagner, J.E.; Blazar, B.R.; Brunstein, C.; Luo, X.; Diamond, D.J.; Cooley, S.; Weisdorf, D.J. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight 2019, 4, e125553. [Google Scholar] [CrossRef]
- Wang, T.; Dang, N.; Tang, G.; Li, Z.; Li, X.; Shi, B.; Xu, Z.; Li, L.; Yang, X.; Xu, C.; et al. Integrating bulk and single-cell RNA sequencing reveals cellular heterogeneity and immune infiltration in hepatocellular carcinoma. Mol. Oncol. 2022, 16, 2195–2213. [Google Scholar] [CrossRef]
- Feng, Q.; Huang, Z.; Song, L.; Wang, L.; Lu, H.; Wu, L. Combining bulk and single-cell RNA-sequencing data to develop an NK cell-related prognostic signature for hepatocellular carcinoma based on an integrated machine learning framework. Eur. J. Med. Res. 2023, 28, 306. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shekarkar Azgomi, M.; Badami, G.D.; Lo Pizzo, M.; Tamburini, B.; Dieli, C.; La Manna, M.P.; Dieli, F.; Caccamo, N. Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data Reveals Memory-like NK Cell Subset Associated with Mycobacterium tuberculosis Latency. Cells 2024, 13, 293. https://doi.org/10.3390/cells13040293
Shekarkar Azgomi M, Badami GD, Lo Pizzo M, Tamburini B, Dieli C, La Manna MP, Dieli F, Caccamo N. Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data Reveals Memory-like NK Cell Subset Associated with Mycobacterium tuberculosis Latency. Cells. 2024; 13(4):293. https://doi.org/10.3390/cells13040293
Chicago/Turabian StyleShekarkar Azgomi, Mojtaba, Giusto Davide Badami, Marianna Lo Pizzo, Bartolo Tamburini, Costanza Dieli, Marco Pio La Manna, Francesco Dieli, and Nadia Caccamo. 2024. "Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data Reveals Memory-like NK Cell Subset Associated with Mycobacterium tuberculosis Latency" Cells 13, no. 4: 293. https://doi.org/10.3390/cells13040293
APA StyleShekarkar Azgomi, M., Badami, G. D., Lo Pizzo, M., Tamburini, B., Dieli, C., La Manna, M. P., Dieli, F., & Caccamo, N. (2024). Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data Reveals Memory-like NK Cell Subset Associated with Mycobacterium tuberculosis Latency. Cells, 13(4), 293. https://doi.org/10.3390/cells13040293