

Taming Microglia in Alzheimer’s Disease: Exploring Potential Implications of Choline Alphoscerate via α7 nAChR Modulation


 , ,
, ,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs and Chemicals
2.2. Preparation of Aβ1–42 Oligomers
2.3. Cell Cultures
2.4. Cell Viability Test
2.5. Western Blot Analysis
2.6. Fluorescent Immunocytochemistry
2.7. [Ca2+]i Measurements
2.8. Patch-Clamp Electrophysiology
2.9. Statistical Evaluation
3. Results
3.1. Alpha-GPC Counteracts Aβ-Induced Toxicity in BV2 Microglial Cells
3.2. α-GPC Blunts the Aβ-Induced Inflammatory Phenotype in BV2 Microglial Cells
3.3. α-GPC Treatment Modulates α7 nAchR Expression in BV2 Cells
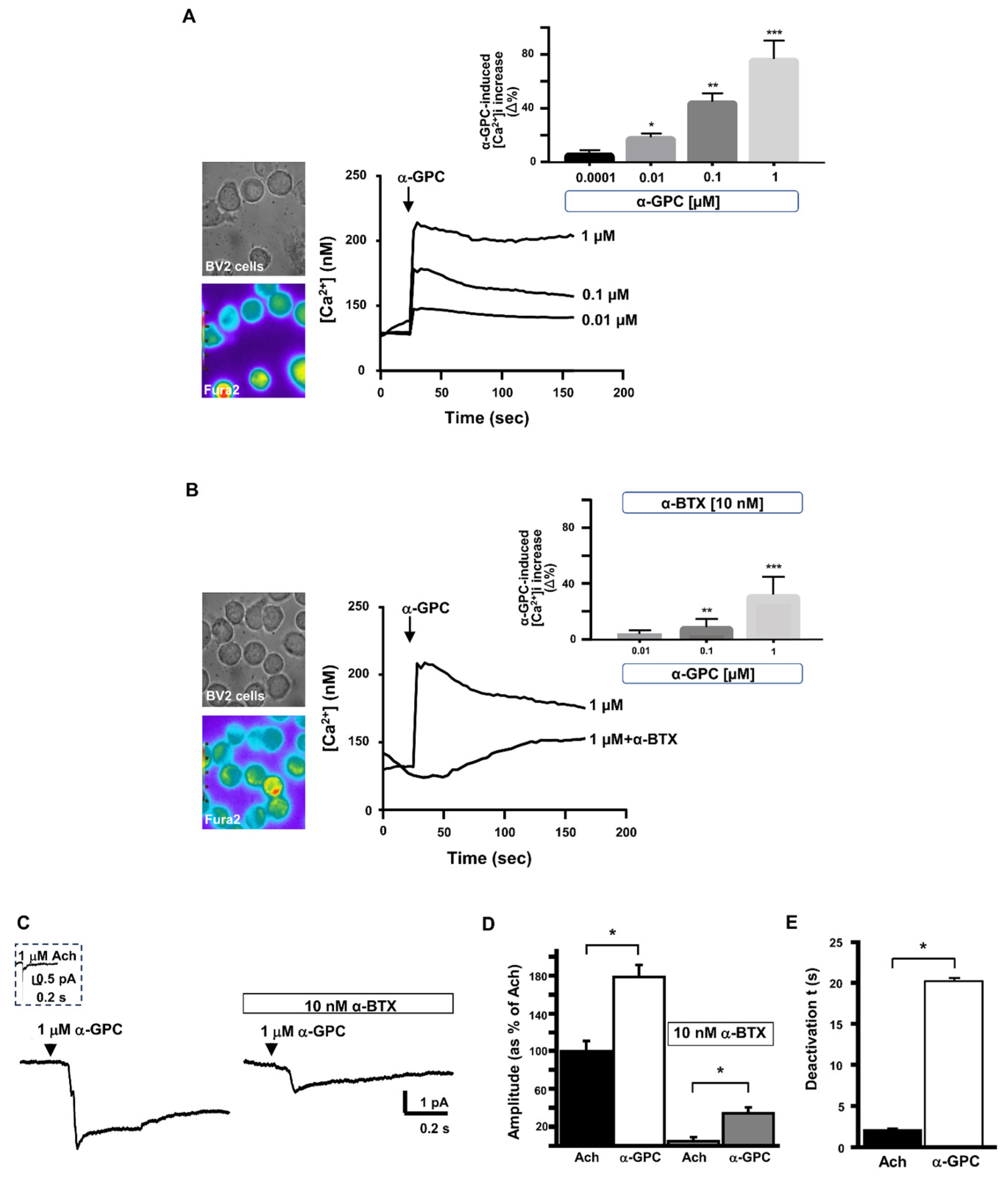
3.4. Alpha-Bungarotoxin Prevents the Effect of α-GPC on α7-nAChR-Mediated [Ca2+]i Increase and Inward Currents in Microglial Cells
3.5. Alpha-Bungarotoxin Reverts the Effects of α-GPC on Anti-Inflammatory Signaling Molecules
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Martinez, J.L.; Zammit, M.D.; West, N.R.; Christian, B.T.; Bhattacharyya, A. Basal Forebrain Cholinergic Neurons: Linking Down Syndrome and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 703876. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, E.; Ananth, M.; Talmage, D.A.; Role, L. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Traini, E.; Bramanti, V.; Amenta, F. Choline Alphoscerate (Alpha-Glyceryl-Phosphoryl-Choline) an Old Choline- Containing Phospholipid with a Still Interesting Profile as Cognition Enhancing Agent. Curr. Alzheimer Res. 2013, 10, 1070–1079. [Google Scholar] [CrossRef]
- Kansakar, U.; Trimarco, V.; Mone, P.; Varzideh, F.; Lombardi, A.; Santulli, G. Choline Supplements: An Update. Front. Endocrinol. 2023, 14, 1148166. [Google Scholar] [CrossRef]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic Acetylcholine Receptors: From Basic Science to Therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef] [PubMed]
- Galvin, V.C.; Arnsten, A.F.T.; Wang, M. Involvement of Nicotinic Receptors in Working Memory Function. In Behavioral Pharmacology of the Cholinergic System; Shoaib, M., Wallace, T.L., Eds.; Current Topics in Behavioral Neurosciences; Springer International Publishing: Cham, Switzerland, 2020; Volume 45, pp. 89–99. ISBN 978-3-030-56012-6. [Google Scholar]
- Ma, K.-G.; Qian, Y.-H. Alpha 7 Nicotinic Acetylcholine Receptor and Its Effects on Alzheimer’s Disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef]
- Orr-Urtreger, A.; Broide, R.S.; Kasten, M.R.; Dang, H.; Dani, J.A.; Beaudet, A.L.; Patrick, J.W. Mice Homozygous for the L250T Mutation in the A7 Nicotinic Acetylcholine Receptor Show Increased Neuronal Apoptosis and Die Within 1 Day of Birth. J. Neurochem. 2000, 74, 2154–2166. [Google Scholar] [CrossRef]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef]
- Burgaletto, C.; Munafò, A.; Di Benedetto, G.; De Francisci, C.; Caraci, F.; Di Mauro, R.; Bucolo, C.; Bernardini, R.; Cantarella, G. The Immune System on the TRAIL of Alzheimer’s Disease. J. Neuroinflamm. 2020, 17, 298. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Fa, M.; Orozco, I.J.; Francis, Y.I.; Saeed, F.; Gong, Y.; Arancio, O. Preparation of Oligomeric Beta-Amyloid 1-42 and Induction of Synaptic Plasticity Impairment on Hippocampal Slices. J. Vis. Exp. 2010, 41, e1884. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Boscia, F.; Gala, R.; Pannaccione, A.; Secondo, A.; Scorziello, A.; Di Renzo, G.; Annunziato, L. NCX1 Expression and Functional Activity Increase in Microglia Invading the Infarct Core. Stroke 2009, 40, 3608–3617. [Google Scholar] [CrossRef]
- Pan, X.; Zhu, Y.; Lin, N.; Zhang, J.; Ye, Q.; Huang, H.; Chen, X. Microglial Phagocytosis Induced by Fibrillar β-Amyloid Is Attenuated by Oligomeric β-Amyloid: Implications for Alzheimer’s Disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kimura, S.; Takada, N.; Takemura, M.; Iwamoto, M.; Hisaoka-Nakashima, K.; Nakata, Y.; Morioka, N. Stimulation of Toll-like Receptor 4 Downregulates the Expression of A7 Nicotinic Acetylcholine Receptors via Histone Deacetylase in Rodent Microglia. Neurochem. Int. 2020, 138, 104751. [Google Scholar] [CrossRef]
- Suzuki, T.; Hide, I.; Matsubara, A.; Hama, C.; Harada, K.; Miyano, K.; Andrä, M.; Matsubayashi, H.; Sakai, N.; Kohsaka, S.; et al. Microglial A7 Nicotinic Acetylcholine Receptors Drive a Phospholipase C/IP 3 Pathway and Modulate the Cell Activation toward a Neuroprotective Role. J. Neurosci. Res. 2006, 83, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.; Khan, S.J.; Butt, H.A.T.; Zaidi, S.A.T.; Na, V. From Symptomatic Treatment to Disease Modification: A Turning Point in Alzheimer’s Disease Management. Cureus 2023, 15, e47251. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Tomassoni, D.; Nittari, G.; Traini, E.; Amenta, F. Effects of Choline Containing Phospholipids on the Neurovascular Unit: A Review. Front. Cell Neurosci. 2022, 16, 988759. [Google Scholar] [CrossRef]
- Sagaro, G.G.; Traini, E.; Amenta, F. Activity of Choline Alphoscerate on Adult-Onset Cognitive Dysfunctions: A Systematic Review and Meta-Analysis. JAD 2023, 92, 59–70. [Google Scholar] [CrossRef]
- Burgaletto, C.; Di Benedetto, G.; Munafò, A.; Bernardini, R.; Cantarella, G. Beneficial Effects of Choline Alphoscerate on Amyloid-β Neurotoxicity in an In Vitro Model of Alzheimer’s Disease. CAR 2021, 18, 298–309. [Google Scholar] [CrossRef]
- Catanesi, M.; d’Angelo, M.; Antonosante, A.; Castelli, V.; Alfonsetti, M.; Benedetti, E.; Desideri, G.; Ferri, C.; Cimini, A. Neuroprotective Potential of Choline Alfoscerate against Β-amyloid Injury: Involvement of Neurotrophic Signals. Cell Biol. Int. 2020, 44, 1734–1744. [Google Scholar] [CrossRef]
- Tayebati, S.; Amenta, F.; Tomassoni, D. Cerebrovascular and Blood-Brain Barrier Morphology in Spontaneously Hypertensive Rats: Effect of Treatment with Choline Alphoscerate. CNSNDDT 2015, 14, 421–429. [Google Scholar] [CrossRef]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, X.-W.; Wang, J.-Z. The Dual Roles of Cytokines in Alzheimer’s Disease: Update on Interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef]
- Plantone, D.; Pardini, M.; Righi, D.; Manco, C.; Colombo, B.M.; De Stefano, N. The Role of TNF-α in Alzheimer’s Disease: A Narrative Review. Cells 2023, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, C.A.; das Graças Carvalho, M.; de Sousa, L.P.; Caramelli, P.; Gomes, K.B. Alzheimer’s Disease and Cytokine IL-10 Gene Polymorphisms: Is There an Association? Arq. Neuropsiquiatr. 2017, 75, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus Nerve Stimulation Attenuates the Systemic Inflammatory Response to Endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic Acetylcholine Receptor Alpha7 Subunit Is an Essential Regulator of Inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Piovesana, R.; Salazar Intriago, M.S.; Dini, L.; Tata, A.M. Cholinergic Modulation of Neuroinflammation: Focus on A7 Nicotinic Receptor. Int. J. Mol. Sci. 2021, 22, 4912. [Google Scholar] [CrossRef]
- Gallowitsch-Puerta, M.; Tracey, K.J. Immunologic Role of the Cholinergic Anti-Inflammatory Pathway and the Nicotinic Acetylcholine A7 Receptor. Ann. N. Y. Acad. Sci. 2005, 1062, 209–219. [Google Scholar] [CrossRef]
- Czura, C.J.; Tracey, K.J. Autonomic Neural Regulation of Immunity. J. Intern. Med. 2005, 257, 156–166. [Google Scholar] [CrossRef]
- Tracey, K.J. Physiology and Immunology of the Cholinergic Antiinflammatory Pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 Nicotinic Acetylcholine Receptor-Specific Agonist DMXBA (GTS-21) Attenuates Aβ Accumulation through Suppression of Neuronal γ-Secretase Activity and Promotion of Microglial Amyloid-β Phagocytosis and Ameliorates Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar] [CrossRef]
- Medeiros, R.; Castello, N.A.; Cheng, D.; Kitazawa, M.; Baglietto-Vargas, D.; Green, K.N.; Esbenshade, T.A.; Bitner, R.S.; Decker, M.W.; LaFerla, F.M. A7 Nicotinic Receptor Agonist Enhances Cognition in Aged 3xTg-AD Mice with Robust Plaques and Tangles. Am. J. Pathol. 2014, 184, 520–529. [Google Scholar] [CrossRef]
- Fucile, S.; Renzi, M.; Lax, P.; Eusebi, F. Fractional Ca2+ Current through Human Neuronal A7 Nicotinic Acetylcholine Receptors. Cell Calcium 2003, 34, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Yakel, J.L. Nicotinic Acetylcholine Receptor-Mediated Calcium Signaling in the Nervous System. Acta Pharmacol. Sin. 2009, 30, 673–680. [Google Scholar] [CrossRef]
- Fucile, S. Ca2+ Permeability of Nicotinic Acetylcholine Receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leiser, S.C.; Bowlby, M.R.; Comery, T.A.; Dunlop, J. A Cog in Cognition: How the A7 Nicotinic Acetylcholine Receptor Is Geared towards Improving Cognitive Deficits. Pharmacol. Ther. 2009, 122, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.; Adamson, L.; Neill, J.C. Partial Agonism at the A7 Nicotinic Acetylcholine Receptor Improves Attention, Impulsive Action and Vigilance in Low Attentive Rats. Eur. Neuropsychopharmacol. 2017, 27, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh Receptors as Therapeutic Targets in CNS Disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic Modulation of Microglial Activation by A7 Nicotinic Receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef]
- De Jesus Moreno Moreno, M. Cognitive Improvement in Mild to Moderate Alzheimer’s Dementia after Treatment with the Acetylcholine Precursor Choline Alfoscerate: A Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Clin. Ther. 2003, 25, 178–193. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantone, A.F.; Burgaletto, C.; Di Benedetto, G.; Pannaccione, A.; Secondo, A.; Bellanca, C.M.; Augello, E.; Munafò, A.; Tarro, P.; Bernardini, R.; et al. Taming Microglia in Alzheimer’s Disease: Exploring Potential Implications of Choline Alphoscerate via α7 nAChR Modulation. Cells 2024, 13, 309. https://doi.org/10.3390/cells13040309
Cantone AF, Burgaletto C, Di Benedetto G, Pannaccione A, Secondo A, Bellanca CM, Augello E, Munafò A, Tarro P, Bernardini R, et al. Taming Microglia in Alzheimer’s Disease: Exploring Potential Implications of Choline Alphoscerate via α7 nAChR Modulation. Cells. 2024; 13(4):309. https://doi.org/10.3390/cells13040309
Chicago/Turabian StyleCantone, Anna Flavia, Chiara Burgaletto, Giulia Di Benedetto, Anna Pannaccione, Agnese Secondo, Carlo Maria Bellanca, Egle Augello, Antonio Munafò, Paola Tarro, Renato Bernardini, and et al. 2024. "Taming Microglia in Alzheimer’s Disease: Exploring Potential Implications of Choline Alphoscerate via α7 nAChR Modulation" Cells 13, no. 4: 309. https://doi.org/10.3390/cells13040309
APA StyleCantone, A. F., Burgaletto, C., Di Benedetto, G., Pannaccione, A., Secondo, A., Bellanca, C. M., Augello, E., Munafò, A., Tarro, P., Bernardini, R., & Cantarella, G. (2024). Taming Microglia in Alzheimer’s Disease: Exploring Potential Implications of Choline Alphoscerate via α7 nAChR Modulation. Cells, 13(4), 309. https://doi.org/10.3390/cells13040309