
Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Glioblastoma: Insights and Biological Characteristics of the Most Aggressive CNS Tumor
3. Characteristics and Functions of AXL Receptor Tyrosine Kinase
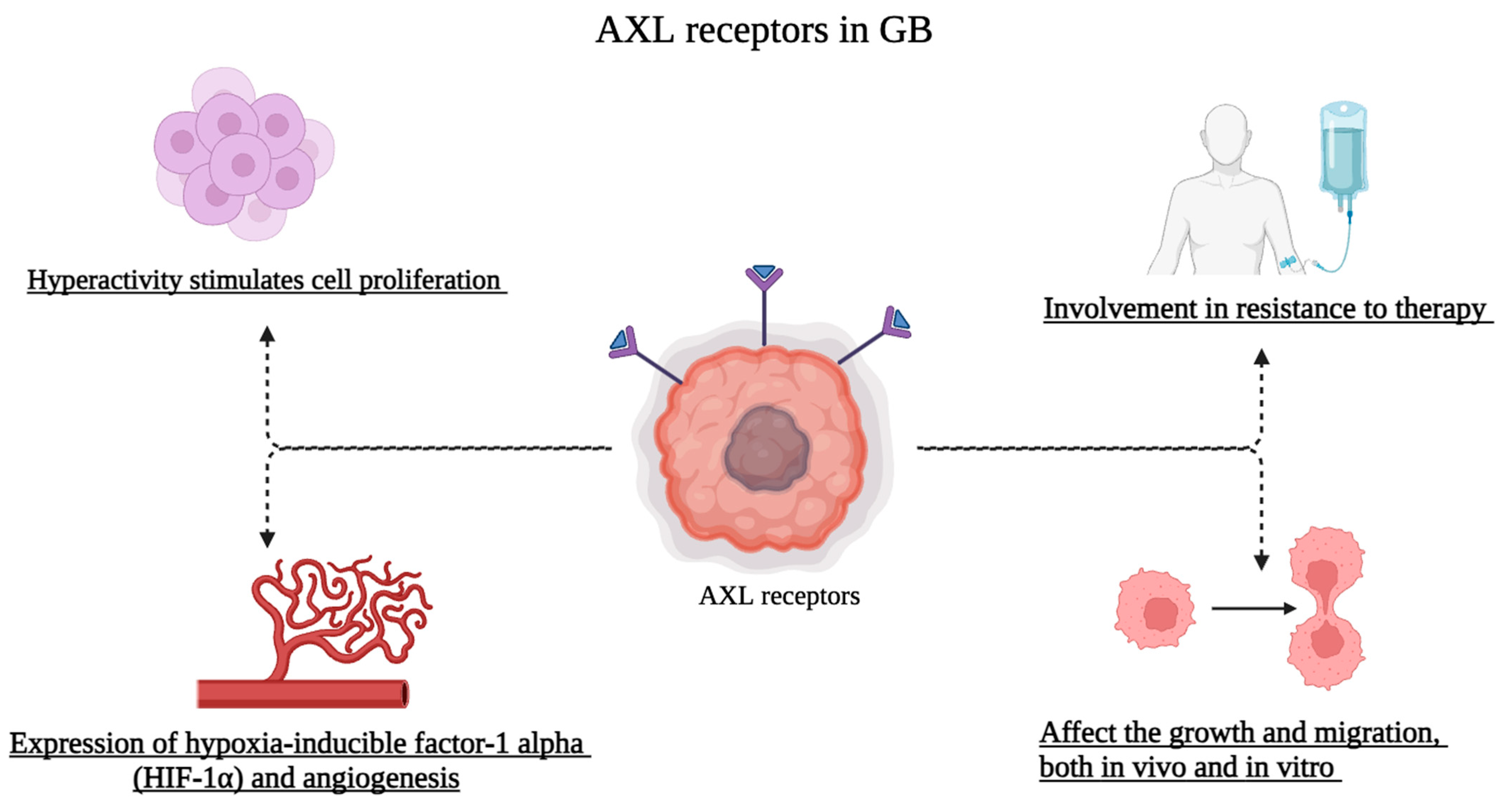
4. Role of AXL Receptors in Glioblastoma
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Repici, A.; Ardizzone, A.; Filippone, A.; Colarossi, C.; Mare, M.; Raciti, G.; Mannino, D.; Cuzzocrea, S.; Paterniti, I.; Esposito, E. Interleukin-21 Influences Glioblastoma Course: Biological Mechanisms and Therapeutic Potential. Cells 2023, 12, 2284. [Google Scholar] [CrossRef]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Siminska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Ardizzone, A.; Scuderi, S.A.; Giuffrida, D.; Colarossi, C.; Puglisi, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers 2020, 12, 3825. [Google Scholar] [CrossRef]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Gilard, V.; Tebani, A.; Dabaj, I.; Laquerriere, A.; Fontanilles, M.; Derrey, S.; Marret, S.; Bekri, S. Diagnosis and Management of Glioblastoma: A Comprehensive Perspective. J. Pers. Med. 2021, 11, 258. [Google Scholar] [CrossRef]
- Metcalfe, S.E.; Grant, R. Biopsy versus resection for malignant glioma. Cochrane Database Syst. Rev. 2001, 3, CD002034. [Google Scholar] [CrossRef]
- Ardizzone, A.; Calabrese, G.; Campolo, M.; Filippone, A.; Giuffrida, D.; Esposito, F.; Colarossi, C.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of miRNA-19a in Cancer Diagnosis and Poor Prognosis. Int. J. Mol. Sci. 2021, 22, 4697. [Google Scholar] [CrossRef]
- Cantrell, J.N.; Waddle, M.R.; Rotman, M.; Peterson, J.L.; Ruiz-Garcia, H.; Heckman, M.G.; Quinones-Hinojosa, A.; Rosenfeld, S.S.; Brown, P.D.; Trifiletti, D.M. Progress Toward Long-Term Survivors of Glioblastoma. Mayo Clin. Proc. 2019, 94, 1278–1286. [Google Scholar] [CrossRef]
- Ardizzone, A.; Bova, V.; Casili, G.; Repici, A.; Lanza, M.; Giuffrida, R.; Colarossi, C.; Mare, M.; Cuzzocrea, S.; Esposito, E.; et al. Role of Basic Fibroblast Growth Factor in Cancer: Biological Activity, Targeted Therapies, and Prognostic Value. Cells 2023, 12, 1002. [Google Scholar] [CrossRef]
- Goyette, M.A.; Cote, J.F. AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers 2022, 14, 466. [Google Scholar] [CrossRef]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef]
- Lozneanu, L.; Pinciroli, P.; Ciobanu, D.A.; Carcangiu, M.L.; Canevari, S.; Tomassetti, A.; Caruntu, I.D. Computational and Immunohistochemical Analyses Highlight AXL as a Potential Prognostic Marker for Ovarian Cancer Patients. Anticancer Res. 2016, 36, 4155–4163. [Google Scholar]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [CrossRef]
- Yu, H.; Liu, R.; Ma, B.; Li, X.; Yen, H.Y.; Zhou, Y.; Krasnoperov, V.; Xia, Z.; Zhang, X.; Bove, A.M.; et al. Axl receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br. J. Cancer 2015, 113, 616–625. [Google Scholar] [CrossRef]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Ait-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Bhalla, S.; Fattah, F.J.; Ahn, C.; Williams, J.; Macchiaroli, A.; Padro, J.; Pogue, M.; Dowell, J.E.; Putnam, W.C.; McCracken, N.; et al. Phase 1 trial of bemcentinib (BGB324), a first-in-class, selective AXL inhibitor, with docetaxel in patients with previously treated advanced non-small cell lung cancer. Lung Cancer 2023, 182, 107291. [Google Scholar] [CrossRef]
- Ibrahim, A.M.; Gray, Z.; Gomes, A.M.; Myers, L.; Behbod, F.; Machado, H.L. Gas6 expression is reduced in advanced breast cancers. NPJ Precis. Oncol. 2020, 4, 9. [Google Scholar] [CrossRef]
- Kirane, A.; Ludwig, K.F.; Sorrelle, N.; Haaland, G.; Sandal, T.; Ranaweera, R.; Toombs, J.E.; Wang, M.; Dineen, S.P.; Micklem, D.; et al. Warfarin Blocks Gas6-Mediated Axl Activation Required for Pancreatic Cancer Epithelial Plasticity and Metastasis. Cancer Res. 2015, 75, 3699–3705. [Google Scholar] [CrossRef]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S.; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef]
- Wei, J.; Sun, H.; Zhang, A.; Wu, X.; Li, Y.; Liu, J.; Duan, Y.; Xiao, F.; Wang, H.; Lv, M.; et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell Immunol. 2018, 331, 49–58. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Casili, G.; Ardizzone, A.; Forte, S.; Colarossi, L.; Sava, S.; Paterniti, I.; Esposito, E.; Cuzzocrea, S.; Campolo, M. KYP-2047, an Inhibitor of Prolyl-Oligopeptidase, Reduces GlioBlastoma Proliferation through Angiogenesis and Apoptosis Modulation. Cancers 2021, 13, 3444. [Google Scholar] [CrossRef]
- Rodriguez-Camacho, A.; Flores-Vazquez, J.G.; Moscardini-Martelli, J.; Torres-Rios, J.A.; Olmos-Guzman, A.; Ortiz-Arce, C.S.; Cid-Sanchez, D.R.; Perez, S.R.; Macias-Gonzalez, M.D.S.; Hernandez-Sanchez, L.C.; et al. Glioblastoma Treatment: State-of-the-Art and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7207. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Thier, K.; Calabek, B.; Tinchon, A.; Grisold, W.; Oberndorfer, S. The Last 10 Days of Patients with Glioblastoma: Assessment of Clinical Signs and Symptoms as well as Treatment. Am. J. Hosp. Palliat. Care 2016, 33, 985–988. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2-8. [Google Scholar] [CrossRef]
- Johnson, D.R.; Fogh, S.E.; Giannini, C.; Kaufmann, T.J.; Raghunathan, A.; Theodosopoulos, P.V.; Clarke, J.L. Case-Based Review: Newly diagnosed glioblastoma. Neurooncol. Pract. 2015, 2, 106–121. [Google Scholar] [CrossRef]
- Ellor, S.V.; Pagano-Young, T.A.; Avgeropoulos, N.G. Glioblastoma: Background, standard treatment paradigms, and supportive care considerations. J. Law Med. Ethics 2014, 42, 171–182. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef]
- Haque, A.; Banik, N.L.; Ray, S.K. Molecular alterations in glioblastoma: Potential targets for immunotherapy. Prog. Mol. Biol. Transl. Sci. 2011, 98, 187–234. [Google Scholar] [CrossRef]
- Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6385. [Google Scholar] [CrossRef]
- Norden, A.D.; Wen, P.Y. Glioma therapy in adults. Neurologist 2006, 12, 279–292. [Google Scholar] [CrossRef]
- Poppe, J.P.; Machegger, L.; Steinbacher, J.; Stefanits, H.; Eisschiel, S.; Gruber, A.; Demetz, M.; Ladisich, B.; Kraus, T.F.J.; Weis, S.; et al. Surgeon experience in glioblastoma surgery of the elderly-a multicenter, retrospective cohort study. J. Neurooncol. 2023, 161, 563–572. [Google Scholar] [CrossRef]
- Tatsuzaki, H.; Urie, M.M.; Linggood, R. Comparative treatment planning: Proton vs. X-ray beams against glioblastoma multiforme. Int. J. Radiat Oncol. Biol. Phys. 1992, 22, 265–273. [Google Scholar] [CrossRef]
- Karati, D.; Mahadik, K.R.; Trivedi, P.; Kumar, D. Alkylating Agents, the Road Less Traversed, Changing Anticancer Therapy. Anticancer Agents Med. Chem. 2022, 22, 1478–1495. [Google Scholar] [CrossRef]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology 2018, 20, 1566–1572. [Google Scholar] [CrossRef]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef]
- Rocha Pinheiro, S.L.; Lemos, F.F.B.; Marques, H.S.; Silva Luz, M.; de Oliveira Silva, L.G.; Faria Souza Mendes Dos Santos, C.; da Costa Evangelista, K.; Calmon, M.S.; Sande Loureiro, M.; Freire de Melo, F. Immunotherapy in glioblastoma treatment: Current state and future prospects. World J. Clin. Oncol. 2023, 14, 138–159. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, L.; Zhang, H.; Zhang, Y.; Ju, H.; Wang, X.; Ren, H.; Zhu, X.; Dong, Y. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. Front. Immunol. 2022, 13, 1003651. [Google Scholar] [CrossRef]
- Mangani, D.; Weller, M.; Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharmacol. 2017, 130, 1–9. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Zheng, H.; Li, C.; Xiong, J.; Wang, W.; Bao, H.; Jin, H.; Liang, P. Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2019, 38, 380. [Google Scholar] [CrossRef]
- Mittal, S.K.; Cho, K.J.; Ishido, S.; Roche, P.A. Interleukin 10 (IL-10)-mediated Immunosuppression: March-I Induction Regulates Antigen Presentation by Macrophages but not Dendritic Cells. J. Biol. Chem. 2015, 290, 27158–27167. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Jiang, J.; Qiu, J.; Li, Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends Cancer 2017, 3, 75–78. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Linz, U.; Schleithoff, L.; Janssen, J.W.; Bartram, C.R.; Muller, C.R. A PvuII-polymorphism within the AXL gene on chromosome 19q13.1. Hum. Mol. Genet 1993, 2, 492. [Google Scholar] [CrossRef]
- Liu, E.; Hjelle, B.; Bishop, J.M. Transforming genes in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 1988, 85, 1952–1956. [Google Scholar] [CrossRef]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef]
- Kimani, S.G.; Kumar, S.; Bansal, N.; Singh, K.; Kholodovych, V.; Comollo, T.; Peng, Y.; Kotenko, S.V.; Sarafianos, S.G.; Bertino, J.R.; et al. Small molecule inhibitors block Gas6-inducible TAM activation and tumorigenicity. Sci. Rep. 2017, 7, 43908. [Google Scholar] [CrossRef]
- Ireland, L.; Luckett, T.; Schmid, M.C.; Mielgo, A. Blockade of Stromal Gas6 Alters Cancer Cell Plasticity, Activates NK Cells, and Inhibits Pancreatic Cancer Metastasis. Front. Immunol. 2020, 11, 297. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef]
- Brien, F.S. Desirable standards for clinical tests of drugs. Chemotherapia 1964, 9, 220–222. [Google Scholar] [CrossRef]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; MacDonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal. Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef]
- Colavito, S.A. AXL as a Target in Breast Cancer Therapy. J. Oncol. 2020, 2020, 5291952. [Google Scholar] [CrossRef]
- Rigoni, T.S.; Vellozo, N.S.; Guimaraes-Pinto, K.; Cabral-Piccin, M.; Fabiano-Coelho, L.; Matos-Silva, T.C.; Filardy, A.A.; Takiya, C.M.; Lopes, M.F. Axl receptor induces efferocytosis, dampens M1 macrophage responses and promotes heart pathology in Trypanosoma cruzi infection. Commun. Biol. 2022, 5, 1421. [Google Scholar] [CrossRef]
- Du, W.; Phinney, N.Z.; Huang, H.; Wang, Z.; Westcott, J.; Toombs, J.E.; Zhang, Y.; Beg, M.S.; Wilkie, T.M.; Lorens, J.B.; et al. AXL Is a Key Factor for Cell Plasticity and Promotes Metastasis in Pancreatic Cancer. Mol. Cancer Res. 2021, 19, 1412–1421. [Google Scholar] [CrossRef]
- Bai, Y.; Li, J.; Fang, B.; Edwards, A.; Zhang, G.; Bui, M.; Eschrich, S.; Altiok, S.; Koomen, J.; Haura, E.B. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. 2012, 72, 2501–2511. [Google Scholar] [CrossRef]
- Bosurgi, L.; Bernink, J.H.; Delgado Cuevas, V.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef]
- Tai, K.Y.; Shieh, Y.S.; Lee, C.S.; Shiah, S.G.; Wu, C.W. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene 2008, 27, 4044–4055. [Google Scholar] [CrossRef]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Lemke, G. TAM receptor signaling and autoimmune disease. Curr. Opin. Immunol. 2010, 22, 740–746. [Google Scholar] [CrossRef]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Lin, J.Z.; Wang, Z.J.; De, W.; Zheng, M.; Xu, W.Z.; Wu, H.F.; Armstrong, A.; Zhu, J.G. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef]
- Sadahiro, H.; Kang, K.D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y.; et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef]
- Kang, C.H.; Kim, Y.; Lee, S.M.; Choi, S.U.; Park, C.H. Development of Antigen-specific Chimeric Antigen Receptor KHYG-1 Cells for Glioblastoma. Anticancer Res. 2021, 41, 1811–1819. [Google Scholar] [CrossRef]
- Kim, H.I.; Lee, S.J.; Choi, Y.J.; Kim, M.J.; Kim, T.Y.; Ko, S.G. Quercetin Induces Apoptosis in Glioblastoma Cells by Suppressing Axl/IL-6/STAT3 Signaling Pathway. Am. J. Chin. Med. 2021, 49, 767–784. [Google Scholar] [CrossRef]
- Sun, L.W.; Kao, S.H.; Yang, S.F.; Jhang, S.W.; Lin, Y.C.; Chen, C.M.; Hsieh, Y.H. Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis. Cells 2021, 10, 2919. [Google Scholar] [CrossRef]
- Zdzalik-Bielecka, D.; Poswiata, A.; Kozik, K.; Jastrzebski, K.; Schink, K.O.; Brewinska-Olchowik, M.; Piwocka, K.; Stenmark, H.; Miaczynska, M. The GAS6-AXL signaling pathway triggers actin remodeling that drives membrane ruffling, macropinocytosis, and cancer-cell invasion. Proc. Natl. Acad. Sci. USA 2021, 118, e2024596118. [Google Scholar] [CrossRef]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL receptor is required for Zika virus strain MR-766 infection in human glioblastoma cell lines. Mol. Ther. Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef]
- Chen, A.; Zhao, W.; Li, X.; Sun, G.; Ma, Z.; Peng, L.; Shi, Z.; Li, X.; Yan, J. Comprehensive Oncogenic Features of Coronavirus Receptors in Glioblastoma Multiforme. Front. Immunol. 2022, 13, 840785. [Google Scholar] [CrossRef]
- Liu, C.A.; Harn, H.J.; Chen, K.P.; Lee, J.H.; Lin, S.Z.; Chiu, T.L. Targeting the Axl and mTOR Pathway Synergizes Immunotherapy and Chemotherapy to Butylidenephthalide in a Recurrent GBM. J. Oncol. 2022, 2022, 3236058. [Google Scholar] [CrossRef]
- Scherschinski, L.; Prem, M.; Kremenetskaia, I.; Tinhofer, I.; Vajkoczy, P.; Karbe, A.G.; Onken, J.S. Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 982. [Google Scholar] [CrossRef]
- Zdzalik-Bielecka, D.; Kozik, K.; Poswiata, A.; Jastrzebski, K.; Jakubik, M.; Miaczynska, M. Bemcentinib and Gilteritinib Inhibit Cell Growth and Impair the Endo-Lysosomal and Autophagy Systems in an AXL-Independent Manner. Mol. Cancer Res. 2022, 20, 446–455. [Google Scholar] [CrossRef]
- Chen, D.; Varanasi, S.K.; Hara, T.; Traina, K.; Sun, M.; McDonald, B.; Farsakoglu, Y.; Clanton, J.; Xu, S.; Garcia-Rivera, L.; et al. CTLA-4 blockade induces a microglia-Th1 cell partnership that stimulates microglia phagocytosis and anti-tumor function in glioblastoma. Immunity 2023, 56, 2086–2104.e8. [Google Scholar] [CrossRef]
- Lecoultre, M.; Chliate, S.; Espinoza, F.I.; Tankov, S.; Dutoit, V.; Walker, P.R. Radio-chemotherapy of glioblastoma cells promotes phagocytosis by macrophages in vitro. Radiother. Oncol. 2023, 190, 110049. [Google Scholar] [CrossRef]
- Vo, T.T.; Tran, Q.; Hong, Y.; Lee, H.; Cho, H.; Kim, M.; Park, S.; Kim, C.; Bayarmunkh, C.; Boldbaatar, D.; et al. AXL is required for hypoxia-mediated hypoxia-inducible factor-1 alpha function in glioblastoma. Toxicol. Res. 2023, 39, 669–679. [Google Scholar] [CrossRef]
- Onken, J.; Vajkoczy, P.; Torka, R.; Hempt, C.; Patsouris, V.; Heppner, F.L.; Radke, J. Phospho-AXL is widely expressed in glioblastoma and associated with significant shorter overall survival. Oncotarget 2017, 8, 50403–50414. [Google Scholar] [CrossRef]
- Bergo, E.; Lombardi, G.; Guglieri, I.; Capovilla, E.; Pambuku, A.; Zagone, V. Neurocognitive functions and health-related quality of life in glioblastoma patients: A concise review of the literature. Eur. J. Cancer Care 2019, 28, e12410. [Google Scholar] [CrossRef]
- Aghababaei, F.; Hadidi, M. Recent Advances in Potential Health Benefits of Quercetin. Pharmaceuticals 2023, 16, 1020. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef]
- Pohlking, C.; Beier, S.; Formanski, J.P.; Friese, M.; Schreiber, M.; Schwalbe, B. Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1. Int. J. Mol. Sci. 2023, 24, 4467. [Google Scholar] [CrossRef]
- Ardizzone, A.; Basilotta, R.; Filippone, A.; Crupi, L.; Lanza, M.; Lombardo, S.P.; Colarossi, C.; Sciacca, D.; Cuzzocrea, S.; Esposito, E.; et al. Recent Emerging Immunological Treatments for Primary Brain Tumors: Focus on Chemokine-Targeting Immunotherapies. Cells 2023, 12, 841. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| First Author and Year of Publication | Title | Therapeutic Target | Conclusion | Reference |
|---|---|---|---|---|
| Kang et al., 2021 | Development of Antigen-specific Chimeric Antigen Receptor KHYG-1 Cells for Glioblastoma | CAR KHYG-1 cells can interact with c-Met, FOLR1, and AXL proteins | c-Met and AXL were over-expressed in several glioblastoma cell lines. CAR KHYG-1 cells can eradicate the positive cell of glioblastoma. | [90] |
| Kim et al., 2021 | Quercetin Induces Apoptosis in Glioblastoma Cells by Suppressing Axl/IL-6/STAT3 Signaling Pathway | The role of quercetin on AXL/IL-6/STAT3 pathway is investigated. | They propose quercetin as a possible anticancer drug that might enhance cancer treatment. | [91] |
| Sun et al., 2021 | Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis | Authors have experimented with the use of corosolic acid to treat glioblastoma. | The data show that CA reduces the invasiveness of glioblastoma cells by interacting with AXL, GAS6 and JAK2/MEK/ERK cascade. | [92] |
| Zdzalik-Bielecka et al., 2021 | The GAS6-AXL signaling pathway triggers actin remodeling that drives membrane ruffling, macropinocytosis, and cancer-cell invasion | The signaling pathway between AXL-GAS6 and how it affects the development of cancer cells. | Different actin-guided cytoskeletal rearrangements that the cell undergoes are caused by GAS6-AXL and contribute to the invasion of cancer cells. | [93] |
| Zwernik et al., 2021 | AXL receptor is required for Zika virus strain MR-766 infection in human glioblastoma cell lines | AXL and ZIKV are highly involved and can be exploited to treat glioblastoma. | ZIKV entry into glioblastoma cells through the AXL receptor produces cytotoxicity. | [94] |
| Chen et al., 2022 | Comprehensive Oncogenic Features of Coronavirus Receptors in Glioblastoma Multiforme | The authors study the connection between Coronavirus and glioblastoma, exploiting several receptors including AXL. | The work examines the connection between coronavirus receptors and glioblastoma for the first time and proposes the connection with ACE2, DPP4, ANPEP, AXL, TMPRSS2 and ENPEP | [95] |
| Liu et al., 2022 | Targeting the Axl and mTOR Pathway Synergizes Immunotherapy and Chemotherapy to Butylidenephthalide in a Recurrent glioblastoma | The authors studied the connection between (Z)-BP delivery through CWs and TMZ in glioblastoma, focusing on AXL and mTOR. | Simultaneous treatment allows blocking of the progression of several cancer pathways. | [96] |
| Scherschinski et al., 2022 | Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma | The role of AXL in the development of resistance-acquired therapy is explored. | RTK-AXL is required by the glioblastoma to develop drug resistance. | [97] |
| Zdzalik-Bielecka et al., 2022 | Bemcentinib and Gilteritinib Inhibit Cell Growth and Impair the Endo-Lysosomal and Autophagy Systems in an AXL-Independent Manner | Bemcentinib and Gilteritinib are highly involved in autophagy by AXL. | The endo-lysosomal and autophagy systems were compromised by bemcentinib and gilteritinib in a way that was independent of AXL. | [98] |
| Chen et al., 2023 | CTLA-4 blockade induces a microglia-Th1 cell partnership that stimulates microglia phagocytosis and anti-tumor function in glioblastoma | αCTLA-4 and αPD-1 are studied in a mouse model of glioblastoma. | In mesenchymal-like glioblastoma, αCTLA-4 inhibition activates CD4+ T cells and microglia via interferon-gamma. | [99] |
| Lecoultre et al., 2023 | Radio-chemotherapy of glioblastoma cells promotes phagocytosis by macrophages in vitro | The work aims to evaluate the interplay of the actual treatment in glioblastoma also evaluating the AXL and other receptors | The effects of radio-chemotherapy on phagocytic activity may enhance pro-tumoral and anti-inflammatory TAM activities. | [100] |
| Vo et al., 2023 | AXL is required for hypoxia-mediated hypoxia-inducible factor-1 alpha function in glioblastoma | AXL is involved in the release of (HIF-1α) in glioblastoma pattern. | HIF-1α and AXL co-expression was detected in human glioblastoma samples, but not in normal tissue. | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repici, A.; Ardizzone, A.; De Luca, F.; Colarossi, L.; Prestifilippo, A.; Pizzino, G.; Paterniti, I.; Esposito, E.; Capra, A.P. Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma. Cells 2024, 13, 361. https://doi.org/10.3390/cells13040361
Repici A, Ardizzone A, De Luca F, Colarossi L, Prestifilippo A, Pizzino G, Paterniti I, Esposito E, Capra AP. Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma. Cells. 2024; 13(4):361. https://doi.org/10.3390/cells13040361
Chicago/Turabian StyleRepici, Alberto, Alessio Ardizzone, Fabiola De Luca, Lorenzo Colarossi, Angela Prestifilippo, Gabriele Pizzino, Irene Paterniti, Emanuela Esposito, and Anna Paola Capra. 2024. "Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma" Cells 13, no. 4: 361. https://doi.org/10.3390/cells13040361
APA StyleRepici, A., Ardizzone, A., De Luca, F., Colarossi, L., Prestifilippo, A., Pizzino, G., Paterniti, I., Esposito, E., & Capra, A. P. (2024). Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma. Cells, 13(4), 361. https://doi.org/10.3390/cells13040361