
Persistent β-Hexachlorocyclohexane Exposure Impacts Cellular Metabolism with a Specific Signature in Normal Human Melanocytes
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Cultures and Treatments
2.3. Cell Count
2.4. MTT Assay
2.5. Melanin Content Determination
2.6. Western Blot Analysis
2.7. Gene Expression Analysis
2.8. Flow Cytometry Analysis
2.9. Annexin V/PI Staining
2.10. ATP Determination
2.11. MC1R Genomic Characterization
2.12. Extraction and GC-MS Analysis of β-HCH
2.13. Statistical Analysis
3. Results
3.1. β-HCH Impacts the Proliferation Capacity of Skin Cells
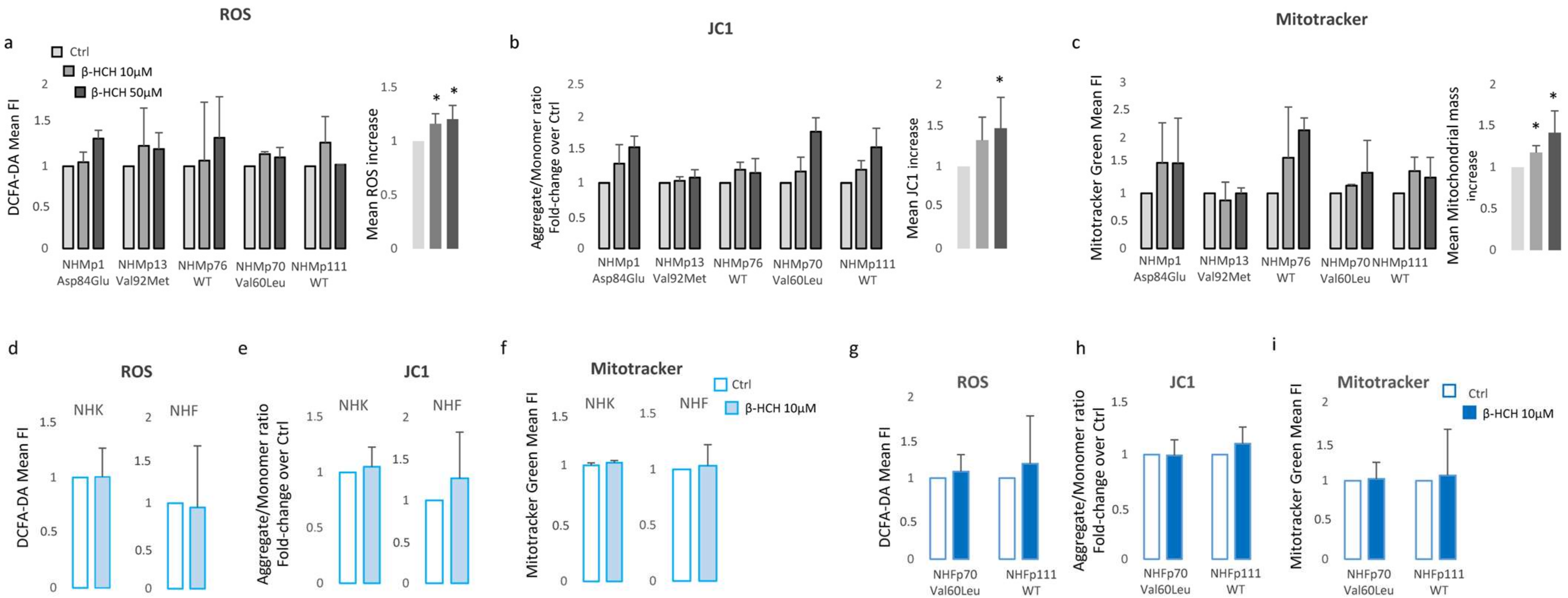
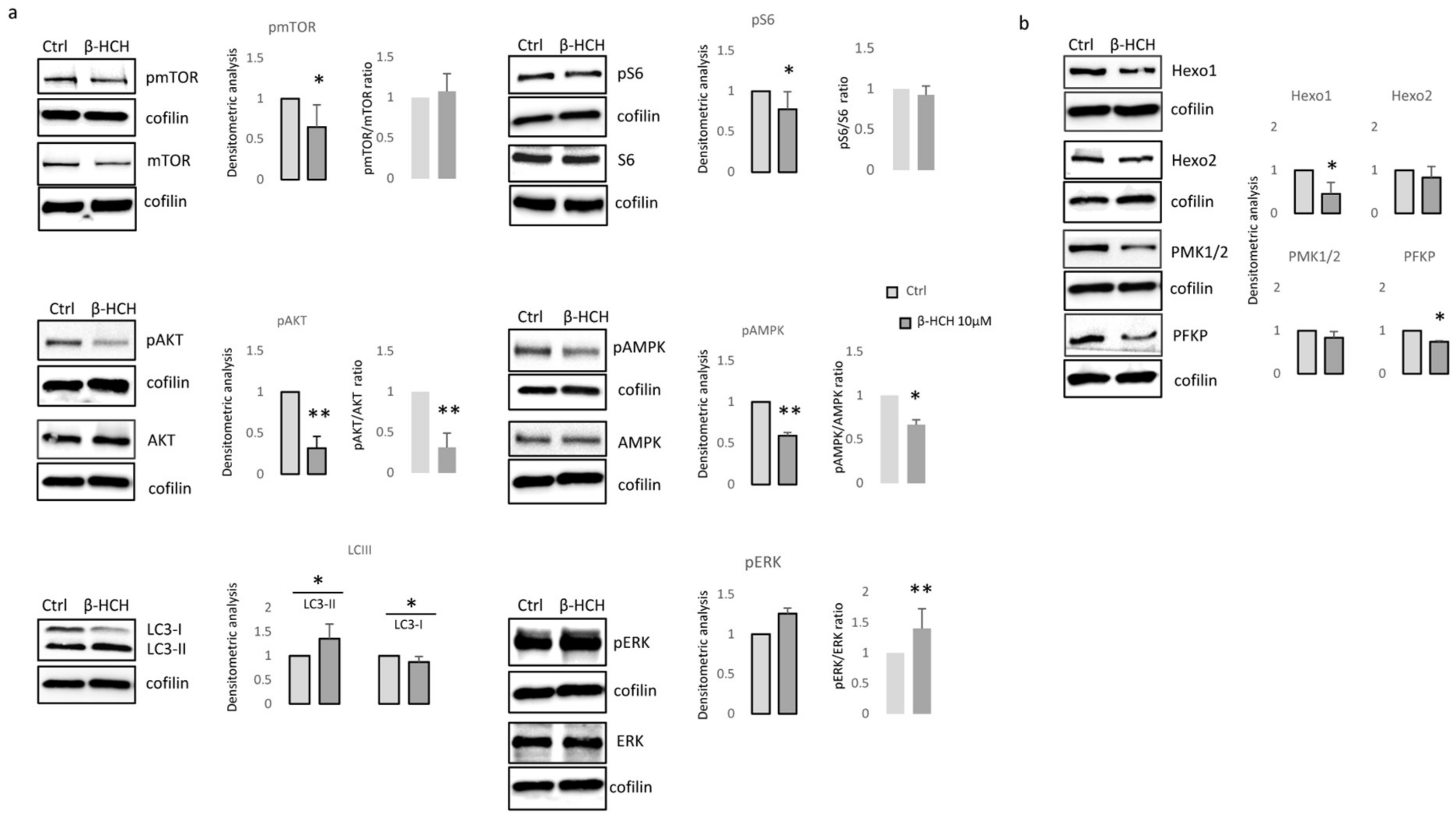
3.2. β-HCH Selectively Affects Melanocyte Intracellular Metabolism
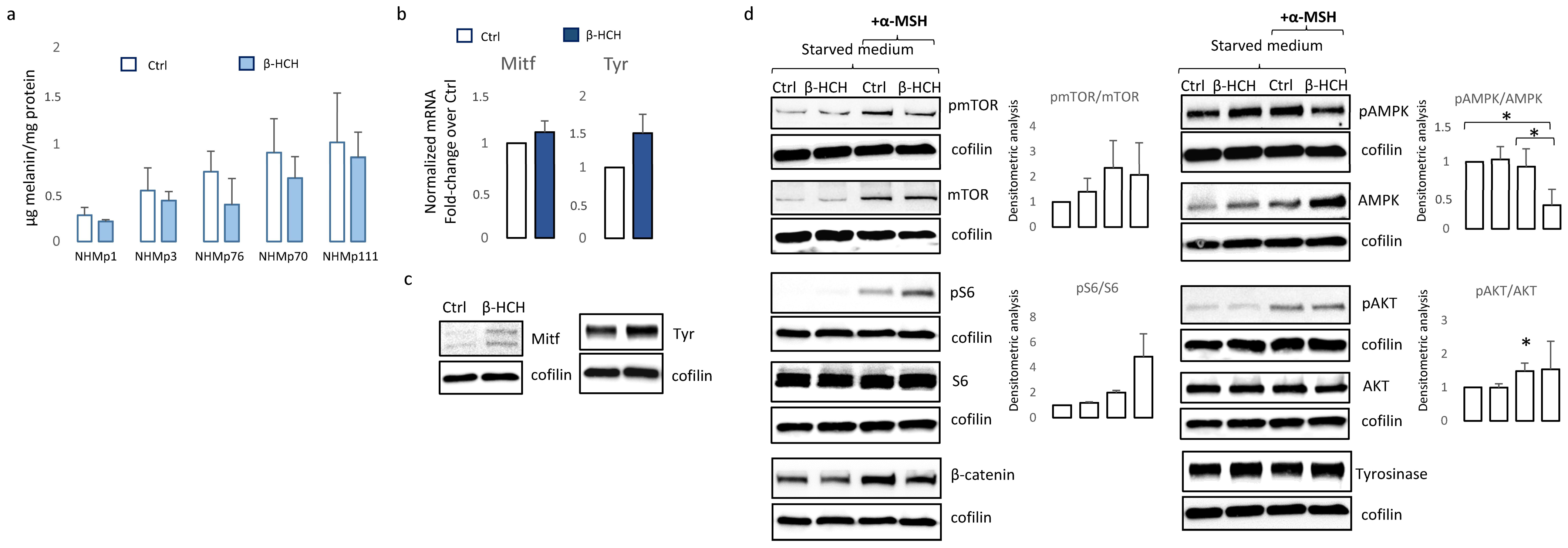
3.3. Chronic β-HCH Exposition Reduces Melanin Synthesis
3.4. Melanocyte-Specific Features Impact β-HCH Intracellular Accumulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthews, N.H.; Li, W.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Berwick, M.; Wiggins, C. The Current Epidemiology of Cutaneous Malignant Melanoma. Front. Biosci. 2006, 11, 1244–1254. [Google Scholar] [CrossRef]
- Houghton, A.N.; Polsky, D. Focus on Melanoma. Cancer. Cell. 2002, 2, 275–278. [Google Scholar] [CrossRef]
- Bressac-de-Paillerets, B.; Avril, M.; Chompret, A.; Demenais, F. Genetic and Environmental Factors in Cutaneous Malignant Melanoma. Biochimie 2002, 84, 67–74. [Google Scholar] [CrossRef]
- Serman, N.; Vranic, S.; Glibo, M.; Serman, L.; Bukvic Mokos, Z. Genetic Risk Factors in Melanoma Etiopathogenesis and the Role of Genetic Counseling: A Concise Review. Bosn J. Basic Med. Sci. 2022, 22, 673–682. [Google Scholar] [PubMed]
- Fargnoli, M.C.; Gandini, S.; Peris, K.; Maisonneuve, P.; Raimondi, S. MC1R Variants Increase Melanoma Risk in Families with CDKN2A Mutations: A Meta-Analysis. Eur. J. Cancer 2010, 46, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Longvert, C.; Saiag, P. Melanoma Update. Rev. Med. Interne 2019, 40, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.P.; Lee, T.K. Adverse Effects of Ultraviolet Radiation: A Brief Review. Prog. Biophys. Mol. Biol. 2006, 92, 119–131. [Google Scholar] [CrossRef]
- You, W.; Henneberg, R.; Coventry, B.J.; Henneberg, M. Cutaneous Malignant Melanoma Incidence is Strongly Associated with European Depigmented Skin Type Regardless of Ambient Ultraviolet Radiation Levels: Evidence from Worldwide Population-Based Data. AIMS Public Health 2022, 9, 378–402. [Google Scholar] [CrossRef]
- Kamiński, K.; Kazimierczak, U.; Kolenda, T. Oxidative Stress in Melanogenesis and Melanoma Development. Contemp. Oncol. 2022, 26, 1–7. [Google Scholar] [CrossRef]
- Gracia-Cazana, T.; Gonzalez, S.; Parrado, C.; Juarranz, A.; Gilaberte, Y. Influence of the Exposome on Skin Cancer. Actas Dermosifiliogr (Engl. Ed.) 2020, 111, 460–470. [Google Scholar] [CrossRef]
- Xu, H.; Jia, Y.; Sun, Z.; Su, J.; Liu, Q.S.; Zhou, Q.; Jiang, G. Corrigendum to “Environmental Pollution, a Hidden Culprit for Health Issues” [Eco-Environ. Health (2022) 31–45]. Eco Environ. Health 2022, 1, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.; Megha, P.; Sreedev, P. Organochlorine Pesticides, their Toxic Effects on Living Organisms and their Fate in the Environment. Interdiscip. Toxicol. 2016, 9, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Purdue, M.P.; Hoppin, J.A.; Blair, A.; Dosemeci, M.; Alavanja, M.C.R. Occupational Exposure to Organochlorine Insecticides and Cancer Incidence in the Agricultural Health Study. Int. J. Cancer 2007, 120, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.K.; Bhattacharya, B.D.; Bhattacharya, A.; Chatterjee, M.; Alam, A.; Satpathy, K.K.; Jonathan, M.P. Occurrence, Distribution and Possible Sources of Organochlorine Pesticide Residues in Tropical Coastal Environment of India: An Overview. Environ. Int. 2008, 34, 1062–1071. [Google Scholar] [CrossRef]
- Chang, C.; Chen, M.; Gao, J.; Luo, J.; Wu, K.; Dong, T.; Zhou, K.; He, X.; Hu, W.; Wu, W.; et al. Current Pesticide Profiles in Blood Serum of Adults in Jiangsu Province of China and a Comparison with Other Countries. Environ. Int. 2017, 102, 213–222. [Google Scholar] [CrossRef]
- Antignac, J.; Figiel, S.; Pinault, M.; Blanchet, P.; Bruyere, F.; Mathieu, R.; Lebdai, S.; Fournier, G.; Rigaud, J.; Maheo, K.; et al. Persistent Organochlorine Pesticides in Periprostatic Adipose Tissue from Men with Prostate Cancer: Ethno-Geographic Variations, Association with Disease Aggressiveness. Environ. Res. 2023, 216, 114809. [Google Scholar] [CrossRef]
- Cavalier, H.; Trasande, L.; Porta, M. Response to: Comments on “Exposures to Pesticides and Risk of Cancer: Evaluation of Recent Epidemiological Evidence in Humans and Paths Forward”. Int. J. Cancer 2023, 152, 2220–2221. [Google Scholar] [CrossRef]
- Moriceau, M.; Cano-Sancho, G.; Kim, M.; Coumoul, X.; Emond, C.; Arrebola, J.; Antignac, J.; Audouze, K.; Rousselle, C. Partitioning of Persistent Organic Pollutants between Adipose Tissue and Serum in Human Studies. Toxics 2022, 11, 41. [Google Scholar] [CrossRef]
- Arrebola, J.P.; Fernandez-Rodriguez, M.; Artacho-Cordon, F.; Garde, C.; Perez-Carrascosa, F.; Linares, I.; Tovar, I.; Gonzalez-Alzaga, B.; Exposito, J.; Torne, P.; et al. Associations of Persistent Organic Pollutants in Serum and Adipose Tissue with Breast Cancer Prognostic Markers. Sci. Total Environ. 2016, 566–567, 41–49. [Google Scholar] [CrossRef]
- Mustieles, V.; Perez-Carrascosa, F.M.; Leon, J.; Lange, T.; Bonde, J.; Gomez-Pena, C.; Artacho-Cordon, F.; Barrios-Rodriguez, R.; Olmedo-Requena, R.; Exposito, J.; et al. Adipose Tissue Redox Microenvironment as a Potential Link between Persistent Organic Pollutants and the 16-Year Incidence of Non-Hormone-Dependent Cancer. Environ. Sci. Technol. 2021, 55, 9926–9937. [Google Scholar] [CrossRef]
- Lal, R.; Pandey, G.; Sharma, P.; Kumari, K.; Malhotra, S.; Pandey, R.; Raina, V.; Kohler, H.E.; Holliger, C.; Jackson, C.; et al. Biochemistry of Microbial Degradation of Hexachlorocyclohexane and Prospects for Bioremediation. Microbiol. Mol. Biol. Rev. 2010, 74, 58–80. [Google Scholar] [CrossRef]
- Bonefeld-Jorgensen, E.C.; Ghisari, M.; Wielsoe, M.; Bjerregaard-Olesen, C.; Kjeldsen, L.S.; Long, M. Biomonitoring and Hormone-Disrupting Effect Biomarkers of Persistent Organic Pollutants in Vitro and Ex Vivo. Basic Clin. Pharmacol. Toxicol. 2014, 115, 118–128. [Google Scholar] [CrossRef]
- Annamalai, J.; Namasivayam, V. Endocrine Disrupting Chemicals in the Atmosphere: Their Effects on Humans and Wildlife. Environ. Int. 2015, 76, 78–97. [Google Scholar] [CrossRef]
- Darvishian, M.; Bhatti, P.; Gaudreau, E.; Abanto, Z.; Choi, C.; Gallagher, R.P.; Spinelli, J.J.; Lee, T.K. Persistent Organic Pollutants and Risk of Cutaneous Malignant Melanoma among Women. Cancer Rep. 2022, 5, e1536. [Google Scholar] [CrossRef]
- Fantini, F.; Porta, D.; Fano, V.; De Felip, E.; Senofonte, O.; Abballe, A.; D’Ilio, S.; Ingelido, A.M.; Mataloni, F.; Narduzzi, S.; et al. Epidemiologic Studies on the Health Status of the Population Living in the Sacco River Valley. Epidemiol. Prev. 2012, 36, 44–52. [Google Scholar]
- Narduzzi, S.; Fantini, F.; Blasetti, F.; Rantakokko, P.; Kiviranta, H.; Forastiere, F.; Michelozzi, P.; Porta, D. Predictors of Beta-Hexachlorocyclohexane Blood Levels among People Living Close to a Chemical Plant and an Illegal Dumping Site. Environ. Health 2020, 19, 7–9. [Google Scholar] [CrossRef]
- Rubini, E.; Altieri, F.; Chichiarelli, S.; Giamogante, F.; Carissimi, S.; Paglia, G.; Macone, A.; Eufemi, M. STAT3, a Hub Protein of Cellular Signaling Pathways, is Triggered by Beta-Hexaclorocyclohexane. Int. J. Mol. Sci. 2018, 19, 2108. [Google Scholar] [CrossRef] [PubMed]
- Rubini, E.; Minacori, M.; Paglia, G.; Altieri, F.; Chichiarelli, S.; Romaniello, D.; Eufemi, M. Beta-Hexachlorocyclohexane Drives Carcinogenesis in the Human Normal Bronchial Epithelium Cell Line BEAS-2B. Int. J. Mol. Sci. 2021, 22, 5834. [Google Scholar] [CrossRef] [PubMed]
- Rubini, E.; Paglia, G.; Cannella, D.; Macone, A.; Di Sotto, A.; Gulli, M.; Altieri, F.; Eufemi, M. Beta-Hexachlorocyclohexane: A Small Molecule with a Big Impact on Human Cellular Biochemistry. Biomedicines 2020, 8, 505. [Google Scholar] [CrossRef] [PubMed]
- Rubini, E.; Minacori, M.; Paglia, G.; Macone, A.; Chichiarelli, S.; Altieri, F.; Eufemi, M. Tomato and Olive Bioactive Compounds: A Natural Shield Against the Cellular Effects Induced by Beta-Hexachlorocyclohexane-Activated Signaling Pathways. Molecules 2021, 26, 7135. [Google Scholar] [CrossRef] [PubMed]
- van Tonder, A.; Joubert, A.M.; Cromarty, A.D. Limitations of the 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyl-2H-Tetrazolium Bromide (MTT) Assay when Compared to Three Commonly used Cell Enumeration Assays. BMC Res. Notes 2015, 8, 47–48. [Google Scholar] [CrossRef]
- Herraiz, C.; Garcia-Borron, J.C.; Jiménez-Cervantes, C.; Olivares, C. MC1R Signaling. Intracellular Partners and Pathophysiological Implications. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2448–2461. [Google Scholar] [CrossRef]
- Gergely, P.J.; Niland, B.; Gonchoroff, N.; Pullmann, R.J.; Phillips, P.E.; Perl, A. Persistent Mitochondrial Hyperpolarization, Increased Reactive Oxygen Intermediate Production, and Cytoplasmic Alkalinization Characterize Altered IL-10 Signaling in Patients with Systemic Lupus Erythematosus. J. Immunol. 2002, 169, 1092–1101. [Google Scholar] [CrossRef]
- Lebiedzinska, M.; Karkucinska-Wieckowska, A.; Wojtala, A.; Suski, J.M.; Szabadkai, G.; Wilczynski, G.; Wlodarczyk, J.; Diogo, C.V.; Oliveira, P.J.; Tauber, J.; et al. Disrupted ATP Synthase Activity and Mitochondrial Hyperpolarisation-Dependent Oxidative Stress is Associated with p66Shc Phosphorylation in Fibroblasts of NARP Patients. Int. J. Biochem. Cell Biol. 2013, 45, 141–150. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Tao, Z.; Aslam, H.; Parke, J.; Sanchez, M.; Cheng, Z. Mechanisms of Autophagic Responses to Altered Nutritional Status. J. Nutr. Biochem. 2022, 103, 108955. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Ali, M.M.; Holail, J.; Houssein, M. Growth Or Death? Control of Cell Destiny by mTOR and Autophagy Pathways. Prog. Biophys. Mol. Biol. 2023, 185, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Fang, W.; Lin, J.; Li, J.; Wu, W.; Xu, J. Vitamin D Protects Human Melanocytes Against Oxidative Damage by Activation of Wnt/Β-Catenin Signaling. Lab. Investig. 2018, 98, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Cheng, L.; Bi, H.; Zhang, Z.; Yao, J.; Zhou, X.; Jiang, Q. Alpha-Melanocyte Stimulating Hormone Protects Retinal Pigment Epithelium Cells from Oxidative Stress through Activation of Melanocortin 1 Receptor-Akt-mTOR Signaling. Biochem. Biophys. Res. Commun. 2014, 443, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Dua, V.K.; Pant, C.S.; Sharma, V.P.; Pathak, G.K. HCH and DDT in Surface Extractable Skin Lipid as a Measure of Human Exposure in India. Bull. Environ. Contam. Toxicol. 1998, 60, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.M.; Lo, J.; Fisher, D.E. How does Pheomelanin Synthesis Contribute to Melanomagenesis?: Two Distinct Mechanisms could Explain the Carcinogenicity of Pheomelanin Synthesis. Bioessays 2013, 35, 672–676. [Google Scholar] [CrossRef]
- Ranadive, N.S.; Shirwadkar, S.; Persad, S.; Menon, I.A. Effects of Melanin-Induced Free Radicals on the Isolated Rat Peritoneal Mast Cells. J. Investig. Dermatol. 1986, 86, 303–307. [Google Scholar] [CrossRef]
- Yang, L.; Mei, G.; Yang, Y.; Cui, J.; Peng, S.; Peng, Z.; Cheng, Y. Hexachlorocyclohexane Impairs Human Sperm Motility by Affecting Lysine Glutarylation and Mitochondrial Functions. Food Chem. Toxicol. 2023, 179, 113991. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Kriete, A.; Bosl, W.J.; Booker, G. Rule-Based Cell Systems Model of Aging using Feedback Loop Motifs Mediated by Stress Responses. PLoS Comput. Biol. 2010, 6, e1000820. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, J.; Ahn, Y.; Lee, E.J.; Hwang, S.; Almurayshid, A.; Park, K.; Chung, H.; Kim, H.J.; Lee, S.; et al. Autophagy Induction can Regulate Skin Pigmentation by Causing Melanosome Degradation in Keratinocytes and Melanocytes. Pigment Cell. Melanoma Res. 2020, 33, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Karnik, A.B.; Majumder, S.K.; Visweswariah, K.; Raju, G.S.; Bai, K.M.; Lakkad, B.C.; Thakore, K.N.; Chatterjee, B.B. Serum Hexachlorocyclohexane Residues in Workers Engaged at a HCH Manufacturing Plant. Int. Arch. Occup. Environ. Health 1986, 57, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Quintana, P.J.E.; Delfino, R.J.; Korrick, S.; Ziogas, A.; Kutz, F.W.; Jones, E.L.; Laden, F.; Garshick, E. Adipose Tissue Levels of Organochlorine Pesticides and Polychlorinated Biphenyls and Risk of Non-Hodgkin’s Lymphoma. Environ. Health Perspect. 2004, 112, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Starr, H.G.J.; Clifford, N.J. Acute Lindane Intoxication: A Case Study. Arch. Environ. Health 1972, 25, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.; Angerer, J.; Heinrich, R.; Lehnert, G. Occupational Exposure to Hexachlorocyclohexane: I. Body Burden of HCH-Isomers. Int. Arch. Occup. Environ. Health 1980, 47, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Mahadevappa, K.L.; Radhakrishnamurty, R. Toxicity of Beta- and Gamma-Hexachlorocyclohexane in Rats of Different Ages. Bull. Environ. Contam. Toxicol. 1991, 47, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Ishizaka, T.; Suzuki, T.; Takeda, M.; Uchiyama, M. Organochlorine Chemicals in Skin Lipids as an Index of their Accumulation in the Human Body. Arch. Environ. Contam. Toxicol. 1991, 21, 190–194. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-HCH | 10 μM | 50 μM | 100 μM |
| NHF | 0.65 ± 0.02 | 0.73 ± 0.26 | 1.11 ± 0.19 |
| NHK | 0.46 ± 0.11 | 0.38 ± 0.14 | 0.27 ± 0.15 |
| NHM | 1.05 ± 0.17 | 1.11 ± 0.38 | 1.21 ± 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papaccio, F.; Caputo, S.; Iorio, A.; De Simone, P.; Ottaviani, M.; Del Brocco, A.; Frascione, P.; Bellei, B. Persistent β-Hexachlorocyclohexane Exposure Impacts Cellular Metabolism with a Specific Signature in Normal Human Melanocytes. Cells 2024, 13, 374. https://doi.org/10.3390/cells13050374
Papaccio F, Caputo S, Iorio A, De Simone P, Ottaviani M, Del Brocco A, Frascione P, Bellei B. Persistent β-Hexachlorocyclohexane Exposure Impacts Cellular Metabolism with a Specific Signature in Normal Human Melanocytes. Cells. 2024; 13(5):374. https://doi.org/10.3390/cells13050374
Chicago/Turabian StylePapaccio, Federica, Silvia Caputo, Alessandra Iorio, Paola De Simone, Monica Ottaviani, Antonella Del Brocco, Pasquale Frascione, and Barbara Bellei. 2024. "Persistent β-Hexachlorocyclohexane Exposure Impacts Cellular Metabolism with a Specific Signature in Normal Human Melanocytes" Cells 13, no. 5: 374. https://doi.org/10.3390/cells13050374