

Potential of Plant-Derived Compounds in Preventing and Reversing Organ Fibrosis and the Underlying Mechanisms



Abstract
:
1. Introduction
2. Major Plant-Derived Compounds that Modulate Fibrosis
2.1. Curcumin
2.2. Capsaicin
2.3. Ellagic Acid
2.4. Epigallocatechin-3-Gallate
2.5. Resveratrol
2.6. Genistein
2.7. Quercetin
2.8. Naringin/Naringenin
2.9. Sulforaphane
2.10. α-Lipoic Acid
2.11. Emodin
3. Conclusions

{kind=link}
{kind=link}
| Compound | Mechanisms of Anti-Fibrotic Effects | References |
|---|---|---|
| Curcumin |
| [29,30,31,32,33,34,35,36,37,38,39,40,45,46,47,48,49,50] |
| Capsaicin |
| [51,52,53,54,55,56,57,58] |
| Ellagic acid |
| [59,60,61,62,63,64,66,67,68,69,70,71,72,73] |
| Epigallocatechin-3-gallate (egcg) |
| [74,75,76,77,78,79,80,81,82,83,84,85,86] |
| Resveratrol |
| [87,88,89,90,91,92,93,94,95,96,97] |
| Genistein |
| [98,99,100,101,102,103,104,105,106,107,108,109,110] |
| Quercetin |
| [111,112,113,114,115,116,117,118,119,120,121,122,123,124,127] |
| Naringenin |
| [131,132,133,134,135,136,137,138,139,140,141,142,143,144,146,147,148,149] |
| Sulforaphane |
| [150,151,152,153,154,155,156,157,158,159,160,163,164,165,166] |
| α-Lipoic acid |
| [167,168,169,170,171,172,173] |
| Emodin |
| [174,175,176,177,178,179,180,181,182,183,184,185,186,187,188] |
| Plant-Derived Compound | Structure | Origin | Reference |
|---|---|---|---|
| Curcumin |  | Asia | [190,191] |
| Capsaicin | 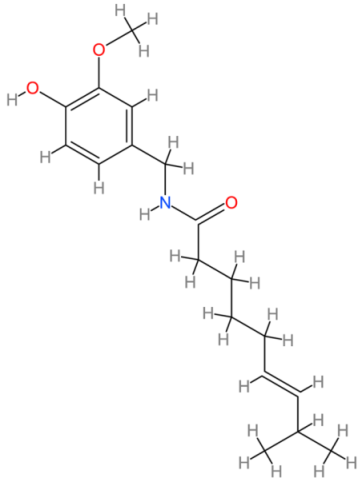 | Americas | [192,193] |
| Ellagic acid | 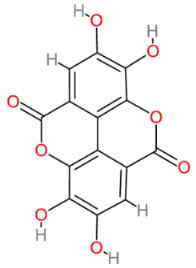 | Multinational | [194,195] |
| Epigallocatechin gallate | 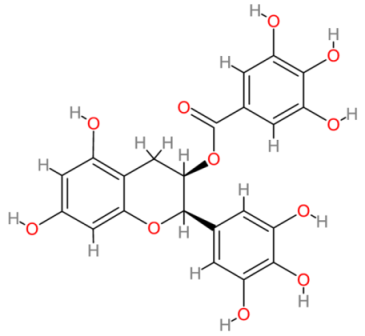 | Multinational | [196,197] |
| Resveratrol |  | China and Japan | [198,199] |
| Genistein |  | Multinational | [200,201] |
| Quercetin |  | Multinational | [202,203] |
| Naringenin | 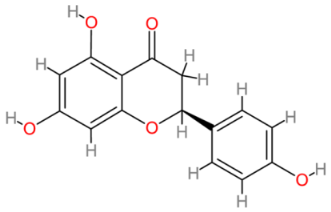 | Multinational | [204,205] |
| Sulforaphane | 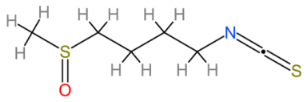 | Multinational | [206,207] |
| α-Lipoic acid | 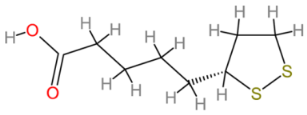 | Multinational | [208,209] |
| Emodin | 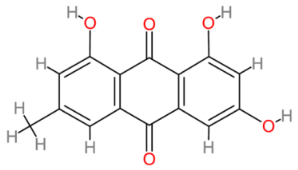 | Multinational | [175,210] |
Author Contributions
Funding
Conflicts of Interest
References
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef]
- Löhler, J.; Timpl, R.; Jaenisch, R. Embryonic lethal mutation in mouse collagen I gene causes rupture of blood vessels and is associated with erythropoietic and mesenchymal cell death. Cell 1984, 38, 597–607. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Genetic disorders of the extracellular matrix. Anat. Rec. 2020, 303, 1527–1542. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; Merrild, C.; Norregaard, R.; Plana-Ripoll, O. The impact of fibrotic diseases on global mortality from 1990, to 2019. J. Transl. Med. 2023, 21, 818. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D.A. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2006, 21, S84–S87. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Can Myocardial fibrosis be reversed? J. Am. Coll. Cardiol. 2019, 73, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and molecular mechanisms underlying liver fibrosis regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef]
- Jia, H.; François, F.; Bourien, J.; Eybalin, M.; Lloyd, R.V.; Van De Water, T.R.; Puel, J.L.; Venail, F. Prevention of trauma-induced cochlear fibrosis using intracochlear application of anti-inflammatory and antiproliferative drugs. Neuroscience 2016, 316, 261–278. [Google Scholar] [CrossRef]
- Cholok, D.; Lee, E.; Lisiecki, J.; Agarwal, S.; Loder, S.; Ranganathan, K.; Qureshi, A.T.; Davis, T.A.; Levi, B. Traumatic muscle fibrosis: From pathway to prevention. J. Trauma Acute Care Surg. 2017, 82, 174–184. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S. Uremic toxins induce kidney fibrosis by activating intrarenal Renin–Angiotensin–Aldosterone System associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chane, X.; Cao, H.; Ziang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef]
- Pattabiraman, G.; Bell-Cohn, A.J.; Murphy, S.F.; Mazue, D.J.; Schaeffer, A.J.; Thumbikat, P. Mast cell function in prostate inflammation, fibrosis, and smooth muscle cell dysfunction. Am. J. Physiol. Renal. Physiol. 2021, 321, F466–F479. [Google Scholar] [CrossRef]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in chronic kidney disease: Pathogenesis and consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Rosenbloom, J.; Jimenez, S.A. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem. J. 1987, 247, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Zugmaier, G.; Paik, S.; Wilding, G.; Knabbe, C.; Bano, M.; Lupu, R.; Deschauer, B.; Simpson, S.; Dickson, R.B.; Lippman, M. Transforming growth factor beta 1 induces cachexia and systemic fibrosis without an antitumor effect in nude mice. Cancer Res. 1991, 51, 3590–3594. [Google Scholar]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef]
- Bharti, N.; Agrawal, V.; Kamthan, S.; Prasad, N.; Agarwal, V. Blood TGF-β1 and miRNA-21-5p levels predict renal fibrosis and outcome in IgA nephropathy. Int. Urol. Nephrol. 2023, 55, 1557–1564. [Google Scholar] [CrossRef]
- Hoffman, K.A.; Reynolds, C.; Bottazzi, M.E.; Hotez, P.; Jones, K. Improved biomarker and imaging analysis for characterizing progressive cardiac fibrosis in a mouse model of chronic chagasic cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e013365. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature 1992, 360, 361–364. [Google Scholar] [CrossRef]
- Arias, M.; Sauer-Lehnen, S.; Treptau, J.; Janoschek, N.; Theuerkauf, I.; Buettner, R.; Gressner, A.M.; Weiskirchen, R. Adenoviral expression of a transforming growth factor-beta1 antisense mRNA is effective in preventing liver fibrosis in bile-duct ligated rats. BMC Gastroenterol. 2003, 3, 29. [Google Scholar] [CrossRef]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, formation, and scope of molecular therapies for skin fibrosis. Biomolecules 2021, 11, 1095. [Google Scholar] [CrossRef]
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cells 2022, 11, 1386. [Google Scholar] [CrossRef]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The inflammatory speech of fibroblasts. Immunol. Rev. 2021, 302, 126–146. [Google Scholar] [CrossRef]
- Brenner, D.A.; Kisseleva, T.; Scholten, D.; Paik, Y.H.; Iwaisako, K.; Inokuchi, S.; Schnabl, B.; Seki, E.; De Minicis, S.; Oesterreicher, C.; et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair 2012, 5 (Suppl. S1), S17. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.C.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Hack, M.E.; Mohamed, T.-S.; Ayman, A.S.; Muhammad, A.; Mahmoud, M.A.G.; Mustafa, S.; Ahmed, N.; Ayman, E.T.; Khaled, A.E.-T. Curcumin, the active substance of turmeric: Its effects on health and ways to improve its bioavailability. J. Sci. Food Agric. 2021, 101, 5747–5762. [Google Scholar] [CrossRef] [PubMed]
- Urošević, M.; Nikolić, L.; Gajić, I.; Nikolić, V.; Dinić, A.; Miljković, V. Curcumin: Biological activities and modern pharmaceutical forms. Antibiotics 2022, 11, 135. [Google Scholar] [CrossRef]
- Kotha, R.R.; Luthria, D.L. Curcumin: Biological, pharmaceutical, nutraceutical, and analytical aspects. Molecules 2019, 24, 2930. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Hegde, M.; Parama, D.; Girisa, S.; Kumar, A.; Daimary, U.D.; Garodia, P.; Yenisetti, S.C.; Oommen, O.V.; Aggarwal, B.B. Role of tumeric and curcumin in prevention and treatment of chronic disease: Lessons learned from clinical trials. ACS Pharmacol. Transl. Sci. 2023, 6, 447–518. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Hajighasemi, S.; Kiaie, N.; Rosano, G.M.C.; Sathyapalan, T.; Al-Rasadi, K.; Sahebkar, A. Anti-fibrotic effects of curcumin and some of its analogues in the heart. Heart Fail. Rev. 2020, 25, 731–743. [Google Scholar] [CrossRef]
- Yu, W.K.; Hwang, W.L.; Wang, Y.C.; Tsai, C.C.; Wei, Y.H. Curcumin suppresses TGF-Β1-induced myofibroblast differentiation and attenuates angiogenic activity of orbital fibroblasts. Int. J. Mol. Sci. 2021, 22, 6829. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, A.; Shi, C.; Li, B. Curcumin suppresses transforming growth factor-β1-induced cardiac fibroblast differentiation via inhibition of Smad-2 and p38 MAPK signaling pathways. Exp. Ther. Med. 2016, 11, 998–1004. [Google Scholar] [CrossRef]
- Supriono, A.; Nugraheni, A.; Kalim, H.; Eko, M.H. The effect of curcumin on regression of liver fibrosis through decreased expression of transforming growth factor-Β1 (TGF-Β1). Indones. Biomed. J. 2019, 11, 52–58. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yang, W.H.; Wong, M.Y.; Chang, H.H.; Kuo, M.Y.-P. Curcumin inhibits thrombin-stimulated connective tissue growth factor (CTGF/CCN2) production through c-Jun NH2-terminal kinase suppression in human gingival fibroblasts. J. Periodontol. 2012, 83, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Wang, C.Y.; Chen, M.H. Curcumin inhibits TGF-Β1-induced connective tissue growth factor expression through the interruption of Smad2 signaling in human gingival fibroblasts. J. Formos. Med. Assoc. 2018, 117, 1115–1123. [Google Scholar] [CrossRef]
- Zhao, J.L.; Zhang, T.; Shao, X.; Zhu, J.J.; Guo, M.Z. Curcumin ameliorates peritoneal fibrosis via inhibition of transforming growth factor-activated kinase 1 (TAK1) pathway in a rat model of peritoneal dialysis. BMC Complement. Altern. Med. 2019, 19, 280. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Y.; Chen, Q.; Hong, T.; Zhong, Z.; He, J.; Ni, C. Curcumin ameliorates cardiac fibrosis by regulating macrophage-fibroblast crosstalk via IL18-P-SMAD2/3 signaling pathway inhibition. Front. Pharmacol. 2022, 12, 784041. [Google Scholar] [CrossRef]
- Morinelli, T.A.; Luttrell, L.M.; Strungs, E.G.; Ullian, M.E. Angiotensin II receptors and peritoneal dialysis-induced peritoneal fibrosis. Int. J. Biochem. Cell Biol. 2016, 77, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Sumners, C.; Peluso, A.A.; Haugaard, A.H.; Bertelsen, J.B.; Steckelings, U.M. Anti-fibrotic mechanisms of angiotensin AT2-receptor stimulation. Acta Physiol. 2019, 227, e13280. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, X.; Shen, Y.; Liu, C.; Kong, X.; Li, P. Alamandine attenuates angiotensin II-induced vascular fibrosis via inhibiting p38 MAPK pathway. Eur. J. Pharmacol. 2020, 883, 173384. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991, 83, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.F.; Zhang, L.H.; Bai, F.; Wang, N.P.; Garner, R.E.; McKallip, R.J.; Zhao, Z.Q. Attenuation of myocardial fibrosis with curcumin is mediated by modulating expression of angiotensin II AT1/AT2 receptors and ACE2 in rats. Drug Des. Devel. Ther. 2015, 9, 6043–6054. [Google Scholar] [CrossRef]
- Li, H.Y.; Yang, M.; Li, Z.; Meng, Z. Curcumin inhibits angiotensin II-induced inflammation and proliferation of rat vascular smooth muscle cells by elevating PPAR-γ activity and reducing oxidative stress. Int. J. Mol. Med. 2017, 39, 1307–1316. [Google Scholar] [CrossRef]
- Song, X.; Qian, X.; Shen, M.; Jiang, R.; Wagner, M.B.; Ding, G.; Chen, G.; Shen, B. Protein kinase C promotes cardiac fibrosis and heart failure by modulating galectin-3 expression. Biochim. Biophys. Acta 2015, 1853, 513–521. [Google Scholar] [CrossRef]
- Mahmmoud, Y.A. Modulation of Protein kinase C by curcumin; inhibition and activation switched by calcium ions. Br. J. Pharmacol. 2007, 150, 200–208. [Google Scholar] [CrossRef]
- Inoguchi, T.; Battan, R.; Handler, E.; Sportsman, J.R.; Heath, W.; King, G.L. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transplantation. Proc. Natl. Acad. Sci. USA 1992, 89, 11059–11063. [Google Scholar] [CrossRef]
- Soetikno, V.; Sari, F.R.; Sukumaran, V.; Lakshmanan, A.P.; Mito, S.; Harima, M.; Thandavarayan, R.A.; Suzuki, K.; Nagata, M.; Takagi, R.; et al. Curcumin prevents diabetic cardiomyopathy in strptozotocin-induced diabetic rats: Possible involvement of PKC-MAPK signaling pathway. Eur. J. Pharm. Sci. 2012, 47, 604–614. [Google Scholar] [CrossRef]
- Chapa-Oliver, A.M.; Mejía-Teniente, L. Capsaicin: From plants to a cancer-suppressing agent. Molecules 2016, 21, 931. [Google Scholar] [CrossRef]
- Rollyson, W.D.; Stover, C.A.; Brown, K.C.; Perry, H.E.; Stevenson, C.D.; McNees, C.A.; Ball, J.G.; Valentovic, M.A.; Dasgupta, P. Bioavailability of capsaicin and its implications for drug delivery. J. Control Release 2014, 196, 96–105. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, W.; Li, X.; Tang, S.; Meng, D.; Xia, W.; Wang, H.; Wu, Y.; Zhou, X.; Zhang, J. Capsaicin ameliorates renal fibrosis by inhibiting TGF-β1-Smad2/3 signaling. Phytomedicine 2022, 100, 154067. [Google Scholar] [CrossRef]
- Sheng, J.; Zhang, B.; Chen, Y.; Yu, F. Capsaicin attenuates liver fibrosis by targeting Notch signaling to inhibit TNF-α secretion from M1 macrophages. Immunopharmacol. Immunotoxicol. 2020, 42, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Bort, A.; Sánchez, B.G.; Mateos-Gómez, P.A.; Díaz-Laviada, I.; Rodríguez-Henche, N. Capsaicin targets lipogenesis in HepG2 cells through AMPK activation, AKT inhibition and PPARs regulation. Int. J. Mol. Sci. 2019, 20, 1660. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Wu, Y.H.; Li, X.H.; Li, D.; Du, J.; Hu, C.P.; Li, Y.J. Role of eukaryotic translation initiation factor 3a in bleomycin-induced pulmonary fibrosis. Eur. J. Pharmacol. 2015, 749, 89–97. [Google Scholar] [CrossRef]
- Lu, L.M.; Yu, T.T.; He, X.W.; Tang, J.; Li, X.W. Effect of small dose capsaicin for treatment of pulmonary fibrosis in mice and its mechanism. Chin. J. Appl. Physiol. 2020, 36, 216–222. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Li, S.; Zha, Z.; Chen, Y.; Wang, Q.; Zhou, S.; Huang, X.; Xu, M. Capsaicin alleviates doxorubicin-induced acute myocardial injury by regulating iron homeostasis and PI3K-Akt signaling pathway. Aging 2023, 15, 11845–11859. [Google Scholar] [CrossRef]
- Lim, S.C.; Hwang, H.; Han, S.L. Ellagic acid inhibits extracellular acidity-induced invasiveness and expression of COX1, COX2, Snail, Twist 1, and c-Myc in gastric carcinoma cells. Nutrients 2019, 11, 3023. [Google Scholar] [CrossRef]
- Karimi, M.Y.; Fatemi, I.; Kalantari, H.; Mombeini, M.A.; Mehrzadi, S.; Goudarzi, M. Ellagic acid prevents oxidative stress, inflammation, and histopathological alterations in acrylamide-induced hepatotoxicity in Wistar rats. J. Diet. Suppl. 2020, 17, 651–662. [Google Scholar] [CrossRef]
- Deepika, M.P.K. Ellagic acid: Insight into its protective effects in age-associated disorders. 3 Biotech 2022, 12, 340. [Google Scholar] [CrossRef]
- Nyamba, I.; Lechanteur, A.; Semdé, R.; Evrard, B. Physical formulation approaches for improving aqueous solubility and bioavailability of ellagic acid: A review. Eur. J. Pharm. Biopharm. 2021, 159, 198–210. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Wu, D.J.; Lv, D.L.; Zhang, B.D.; Chen, L.; Sun, Y.Q. Ellagic acid inhibits the formation of hypertrophic scars by suppressing TGF-β/Smad signaling pathway activity. Chem. Biol. Drug Des. 2023, 102, 773–781. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, M.; Ying, X.; Zhou, C. Ellagic acid protects rats from chronic renal failure via MiR-182/FOXO3a axis. Mol. Immunol. 2021, 138, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Brody, A.R. TNF-α induces TGF-Β1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J. Cell. Mol. Med. 2009, 13, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wei, D.; Xin, D.; Pan, J.; Huang, M. Ellagic acid inhibits proliferation and migration of cardiac fibroblasts by down-regulating expression of HDAC1. J. Toxicol. Sci. 2019, 44, 425–433. [Google Scholar] [CrossRef]
- Looi, D.; Goh, B.H.; Khan, S.U.; Ahemad, N.; Palanisamy, U.D. Metabolites of the ellagitannin, geraniin inhibit human ACE; in vitro and in silico evidence. Int. J. Food Sci. Nutr. 2020, 72, 470–477. [Google Scholar] [CrossRef]
- Baradaran, R.V.; Askari, V.R.; Mousavi, S.H. Ellagic acid dose and time-dependently abrogates d-galactose-induced animal model of aging: Investigating the role of PPAR-γ. Life Sci. 2019, 232, 116595. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Vinayak, M. Ellagic acid checks lymphoma promotion via regulation of PKC signaling pathway. Mol. Biol. Rep. 2013, 40, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Ou, H.C.; Lee, W.J.; Kuo, W.W.; Hwang, L.L.; Song, T.Y.; Huang, C.Y.; Chiu, T.H.; Tsai, K.L.; Tsai, C.S.; et al. Ellagic acid inhibits oxidized low-density lipoprotein (OxLDL)-induced metalloproteinase (MMP) expression by modulating the protein kinase C-α/extracellular signal-regulated kinase/peroxisome proliferator-activated receptor γ/nuclear factor-κB (PKC-α/ERK/PPAR-γ/NF-κB) signaling pathway in endothelial cells. J. Agric. Food Chem. 2011, 59, 5100–5108. [Google Scholar] [CrossRef]
- Zhu, S.; Chen, X.; Chen, Y.; Li, X.F.; Chen, S.Y.; Li, J.J.; Wang, A.; Huang, C.; Li, J. Role of microRNAs in hepatic stellate cells and hepatic fibrosis: An update. Curr. Pharm. Des. 2021, 27, 3000–3011. [Google Scholar] [CrossRef]
- Wei, D.Z.; Lin, C.; Huang, Y.Q.; Wu, L.P.; Huang, M.Y. Ellagic acid promotes ventricular remodeling after acute myocardial infarction by up-regulating miR-140-3p. Biomed. Pharmacother. 2017, 95, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jian, T.; Wu, Y.; Zuo, Y.; Li, J.; Lv, H.; Ma, L.; Ren, B.; Zhao, L.; Li, W.; et al. Ellagic acid ameliorates oxidative stress and insulin resistance in high glucose-treated HepG2 cells via miR-223/keap1-Nrf2 pathway. Biomed. Pharmacother. 2019, 110, 85–94. [Google Scholar] [CrossRef]
- Golam, J.A.; Yaxian, W.; Yameng, W.; Tianmeng, G.; Rubya, S.; Xin, L. Epigallocatechin-3-Gallate (EGCG): A unique secondary metabolite with diverse roles in plant-environment interaction. Environ. Exp. Bot. 2023, 209, 105299. [Google Scholar] [CrossRef]
- Alavi, M.; Hamblin, M.; Aghaie, E.; Mousavi, S.A.R.; Hajimolaali, M. Antibacterial and antioxidant activity of catechin, gallic acid, and epigallocatechin-3-gallate: Focus on nanoformulations. Cell. Mol. Biomed. Rep. 2023, 3, 62–72. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Barro, L.; Tsai, S.T.; Feng, T.W.; Wu, X.Y.; Chao, C.W.; Yu, R.S.; Chin, T.Y.; Hsieh, M.F. Epigallocatechin-3-Gallate-Loaded Liposomes Favor Anti-Inflammation of Microglia Cells and Promote Neuroprotection. Int. J. Mol. Sci. 2021, 22, 3037. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Tsuchishima, M.; Tsutsumi, M. Epigallocatechin-3-gallate inhibits osteopontin expression and prevents experimentally induced hepatic fibrosis. Biomed. Pharmacother. 2022, 151, 113111. [Google Scholar] [CrossRef] [PubMed]
- Kanlaya, R.; Thongboonkerd, V. Molecular mechanisms of epigallocatechin-3-gallate for prevention of chronic kidney disease and renal fibrosis: Preclinical evidence. Curr. Dev. Nutr. 2019, 3, nzz101. [Google Scholar] [CrossRef] [PubMed]
- Zhongyin, Z.; Wei, W.; Juan, X.; Guohua, F. Epigallocatechin gallate relieved PM2.5-induced lung fibrosis by inhibiting oxidative damage and epithelial-mesenchymal transition through AKT/mTOR pathway. Oxidative Med. Cell. Longev. 2022, 2022, 7291774. [Google Scholar] [CrossRef]
- Muhammed, I.; Sankar, S.; Govindaraj, S. Ameliorative effect of epigallocatechin gallate on cardiac hypertrophy and fibrosis in aged rats. J. Cardiovasc. Pharmacol. 2018, 71, 65–75. [Google Scholar] [CrossRef]
- Islam, M.S.; Parish, M.; Brennan, J.T.; Winer, B.L.; Segars, J.H. Targeting fibrotic signaling pathways by EGCG as a therapeutic strategy for uterine fibroids. Sci. Rep. 2023, 13, 8429. [Google Scholar] [CrossRef]
- Li, T.; Tong, Q.; Wang, Z.; Yang, Z.; Sun, Y.; Cai, J.; Xu, Q.; Lu, Y.; Liu, X.; Lin, K.; et al. Epigallocatechin-3-gallate inhibits atrial fibrosis and reduces the occurrence and maintenance of atrial fibrillation and its possible mechanisms. Cardiovasc. Drugs Ther. 2023, 1–22. [Google Scholar] [CrossRef]
- Murata, M.; Marugame, Y.; Morozumi, M.; Kyosuke, M.; Kumazoe, M.; Fujimura, Y.; Tachibana, H. (-)-Epigallocatechin-3-0-gallate upregulates the expression levels of miR-6757-3p, a suppressor of fibrosis-related gene expression, in extracellular vesicles derived from human umbilical vein endothelial cells. Biomed. Rep. 2023, 18, 19. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Moon, H.; Kim, R.; Kim, M.; Jeong, S.; Kim, H.; Kim, S.H.; Hwang, S.S.; Lee, M.Y.; et al. Epigallocatechin-3-gallate attenuates myocardial dysfunction via inhibition of endothelial-to-mesenchymal transition. Antioxidants 2023, 12, 1059. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Y.; Zeng, L.; An, X.; Su, D.; Qu, Y.; Ma, J.; Tang, X.; Wang, X.; Yang, J.; et al. Epigallocatechin-3-gallate allosterically activates Protein Kinase C-α and improves the cognition of estrogen deficiency mice. ACS Chem. Neurosci. 2021, 12, 3672–3682. [Google Scholar] [CrossRef]
- Jia, Q.; Yang, R.; Li, Y.; Mehmood, S. Epigallocatechin-3-gallate attenuates myocardial fibrosis in diabetic rats by activating autophagy. Exp. Biol. Med. 2022, 247, 1591–1600. [Google Scholar] [CrossRef]
- Meng, T.; Xiao, D.; Muhammed, A.; Deng, J.; Chen, L.; He, J. Anti-inflammatory action and mechanisms of resveratrol. Molecules 2021, 26, 229. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kang, S.; Yu, Y.; Jung, S.Y.; Park, K.; Kim, S.M.; Kim, H.J.; Kim, J.G.; Kim, S.E. In vitro anti-inflammatory and antioxidant activities of pH-responsive resveratrol-urocanic acid nano-assemblies. Int. J. Mol. Sci. 2023, 24, 3843. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Luo, D.; Chen, D.; Zhou, H.; Zhang, S.; Chen, X.; Lu, W.; Liu, W. Systematic study of resveratrol nanoliposomes transdermal delivery system for enhancing anti-aging and skin-brightening efficacy. Molecules 2023, 28, 2738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Liu, X.; Zhao, C.; Yin, J.; Li, X.; Zhang, X.; Wang, J.; Wang, S. Distinctive anti-inflammatory effects of resveratrol, dihydroresveratrol, and 3-(4-hydroxyphenyl)-propionic acid on DSS-induced colitis in pseudo-germ-free mice. Food Chem. 2023, 400, 133904. [Google Scholar] [CrossRef]
- Zhang, W.; Qian, S.; Tang, B.; Kang, P.; Zhang, H.; Shi, C. Resveratrol inhibits ferroptosis and decelerates heart failure progression via Sirt1/P53 pathway activation. J. Cell Mol. Med. 2023, 27, 3075–3089. [Google Scholar] [CrossRef]
- Fan, J.; Wei, S.; Zhang, X.; Chen, L.; Zhang, X.; Jiang, Y.; Sheng, M.; Chen, Y. Resveratrol inhibits TGF-β1-induced fibrotic effects in human pterygium fibroblasts. Environ. Health Prev. Med. 2023, 28, 59. [Google Scholar] [CrossRef]
- Liu, L.; Liu, B.; Li, L.; Zhou, X.; Li, Q. Effects of resveratrol on pulmonary fibrosis via TGF-β/Smad/ERK signaling pathway. Am. J. Chin. Med. 2023, 51, 651–676. [Google Scholar] [CrossRef]
- Wang, L.; Shao, M.; Jiang, W.; Huang, Y. Resveratrol alleviates bleomycin-induced pulmonary fibrosis by inhibiting epithelial-mesenchymal transition and down-regulating TLR4/NF-κB and TGF-β1/smad3 signalling pathways in rats. Tissue Cell 2022, 79, 101953. [Google Scholar] [CrossRef]
- Guo, S.; Zhou, Y.; Xie, X. Resveratrol inhibiting TGF/ERK signaling pathway can improve atherosclerosis: Backgrounds, mechanisms and effects. Biomed. Pharmacother. 2022, 155, 113775. [Google Scholar] [CrossRef]
- Qiao, Y.; Gao, K.; Wang, Y.; Wang, X.; Cui, B. Resveratrol ameliorates diabetic mephropathy in rats through negative regulation of the p38 MAPK/TGF-b1 pathway. Exp. Ther. Med. 2017, 13, 3223–3230. [Google Scholar] [CrossRef]
- Ma, E.; Wu, C.; Chen, J.; Wo, D.; Ren, D.N.; Yan, H.; Peng, L.; Zhu, W. Resveratrol prevents Ang II-induced cardiac hypertrophy by inhibition of NF-κB signaling. Biomed. Pharmacother. 2023, 165, 115275. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Borrás, C.; Viña, J. The multimodal action of genistein in Alzheimer’s and other age-related diseases. Free Radic. Biol. Med. 2022, 183, 127–137. [Google Scholar] [CrossRef]
- Křížová, L.; Dadáková, K.; Kašparovská, J.; Kašparovský, T. Isoflavones. Molecules 2019, 24, 1076. [Google Scholar] [CrossRef] [PubMed]
- Mukund, V.; Mukund, D.; Sharma, V.; Mannarapu, M.; Alam, A. Genistein: Its role in metabolic diseases and cancer. Crit. Rev. Oncol. Hematol. 2017, 119, 13–22. [Google Scholar] [CrossRef] [PubMed]
- You, L. Phytoestrogen genistein and its pharmacological interactions with synthetic endocrine-active compounds. Curr. Pharm. Des. 2004, 10, 2749–2757. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Q.; An, Q.; Gong, L.; Yang, S.; Zhang, B.; Su, B.; Yang, D.; Zhang, L.; Lu, Y.; et al. Optimized solubility and bioavailability of genistein based on cocrystal engineering. Nat. Prod. Bioprospect. 2023, 13, 30. [Google Scholar] [CrossRef]
- Steensma, A.; Faassen-Peters, M.A.; Noteborn, H.P.; Rietjens, I.M. Bioavailability of genistein and its glycoside genistin as measured in the portal vein of freely moving unanesthetized rats. J. Agric. Food Chem. 2006, 54, 8006–8012. [Google Scholar] [CrossRef]
- Kwon, S.H.; Kang, M.J.; Huh, J.S.; Ha, K.W.; Lee, J.R.; Lee, S.K.; Lee, B.S.; Han, I.H.; Lee, M.S.; Lee, M.W.; et al. Comparison of oral bioavailability of genistein and genistin in rats. Int. J. Pharm. 2007, 337, 148–154. [Google Scholar] [CrossRef]
- Yang, R.; Jia, Q.; Liu, X.F.; Ma, S.F. Effect of genistein on myocardial fibrosis in diabetic rats and its mechanism. Mol. Med. Rep. 2018, 17, 2929–2936. [Google Scholar] [CrossRef]
- Jia, Q.; Yang, R.; Liu, X.F.; Ma, S.F.; Wang, L. Genistein attenuates renal fibrosis in Streptozotocin-induced diabetic rats. Mol. Med. Rep. 2019, 19, 423–431. [Google Scholar] [CrossRef]
- Ning, Y.; Chen, J.; Shi, Y.; Song, N.; Yu, X.; Fang, Y.; Ding, X. Genistein ameliorates renal fibrosis through regulation Snail via M6A RNA demethylase ALKBH5. Front. Pharmacol. 2020, 11, 579265. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, D.; Yang, H.; Liu, Y.; Zhang, L.; Zhang, C.; Chen, G.; Hu, Y.; Chen, J.; Zhang, H.; et al. Hepatoprotective effect of genistein against dimethylnitrosamine-induced liver fibrosis in rats by regulating macrophage functional properties and inhibiting the JAK2/STAT3/SOCS3 signaling pathway. Front. Biosci. 2021, 26, 1572–1584. [Google Scholar] [CrossRef]
- Kwon, S.H.; Chung, H.; Seo, J.W.; Kim, H.S. Genistein alleviates pulmonary fibrosis by inactivating lung fibroblasts. BMB Rep. 2023, 11, 5967. [Google Scholar] [CrossRef]
- Jackson, I.L.; Zodda, A.; Gurung, G.; Pavlovic, R.; Kaytor, M.D.; Kuskowski, M.A.; Vujaskovic, Z. BIO 300, a nanosuspension of genistein, mitigates pneumonitis/fibrosis following high-dose radiation exposure in the C57L/J murine model. Br. J. Pharmacol. 2017, 174, 4738–4750. [Google Scholar] [CrossRef]
- Fideles, S.O.M.; de Cássia Ortiz, A.; Buchaim, D.V.; de Souza Bastos Mazuqueli Pereira, E.; Parreira, M.J.B.M.; de Oliveira Rossi, J.; da Cunha, M.R.; de Souza, A.T.; Soares, W.C.; Buchaim, R.L. Influence of the neuroprotective properties of quercetin on regeneration and functional recovery of the nervous system. Antioxidants 2023, 12, 149. [Google Scholar] [CrossRef]
- Nalini, T.; Basha, S.K.; Sadiq, A.M.; Kumari, V.S. Fabrication and evaluation of nanoencapsulated quercetin for wound healing application. Polym. Bull. 2023, 80, 515–540. [Google Scholar] [CrossRef]
- Xu, Z.; Xie, Y.; Song, J.; Huang, J.; Shi, W. Mechanism of action of a Chinese herbal compound containing quercetin, luteolin, and kaempferol in the treatment of vitiligo based on network pharmacology and experimental verification. Evid. Based Complement. Altern. Med. 2022, 2022, 7197533. [Google Scholar] [CrossRef]
- McKay, T.B.; Emmitte, K.A.; German, C.; Karamichos, D. Quercetin and related analogs as therapeutics to promote tissue repair. Bioengineering 2023, 10, 1127. [Google Scholar] [CrossRef]
- Xiao, Z.; He, L.; Hou, X.; Wei, J.; Ma, X.; Gao, Z.; Yuan, Y.; Xiao, J.; Li, P.; Yue, T. Relationships between structure and antioxidant capacity and activity of glycosylated flavonols. Foods 2021, 10, 849. [Google Scholar] [CrossRef]
- Alizadeh, S.R.; Savadkouhi, N.; Ebrahimzadeh, M.A. Drug design strategies that aim to improve the low solubility and poor bioavailability conundrum in quercetin derivatives. Expert Opin. Drug Discov. 2023, 18, 1117–1132. [Google Scholar] [CrossRef]
- Yan, Y.; Feng, Y.; Li, W.; Che, J.P.; Wang, G.C.; Liu, M.; Zheng, J.H. Protective effects of quercetin and hyperoside on renal fibrosis in rats with unilateral ureteral obstruction. Exp. Ther. Med. 2014, 8, 727–730. [Google Scholar] [CrossRef]
- Ren, J.; Li, J.; Liu, X.; Feng, Y.; Gui, Y.; Yang, J.; He, W.; Dai, C. Quercetin inhibits fibroblast activation and kidney fibrosis involving the suppression of mammalian target of rapamycin and β-catenin signaling. Sci. Rep. 2016, 6, 23968. [Google Scholar] [CrossRef]
- Lu, Q.; Ji, X.J.; Zhou, Y.X.; Yao, X.Q.; Liu, Y.Q.; Zhang, F.; Yin, X.X. Quercetin inhibits the mTORC1/p70S6K signaling-mediated renal tubular epithelial-mesenchymal transition and renal fibrosis in diabetic nephropathy. Pharmacol. Res. 2015, 99, 237–247. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhou, L.; Zhang, T.; Qin, C.; Wei, P.; Luo, L.; Luo, L.; Huang, G.; Chen, A.; Liu, G. Anti-fibrosis activity of quercetin attenuates rabbit tracheal stenosis via the TGF-β/AKT/mTOR signaling pathway. Life Sci. 2020, 250, 117552. [Google Scholar] [CrossRef]
- Geng, F.; Zhao, L.; Cai, Y.; Zhao, Y.; Jin, F.; Li, Y.; Li, T.; Yang, X.; Li, S.; Gao, X.; et al. Quercetin alleviates pulmonary fibrosis in silicotic mice by inhibiting macrophage transition and TGF-β-Smad2/3 pathway. Curr. Issues Mol. Biol. 2023, 45, 3087–3101. [Google Scholar] [CrossRef]
- Xiong, G.; Ji, W.; Wang, F.; Zhang, F.; Xue, P.; Cheng, M.; Sun, Y.; Wang, X.; Zhang, T. Quercetin inhibits inflammatory response induced by LPS from Porphyromonas gingivalis in human gingival fibroblasts via suppressing NF-κB signaling pathway. Biomed Res. Int. 2019, 2019, 6282635. [Google Scholar] [CrossRef]
- Turedi, S. Protective/preventive effects of quercetin against cyclophosphamide-induced hepatic inflammation, apoptosis and fibrosis in rats. Hepatol. Forum 2023, 4, 135–141. [Google Scholar] [CrossRef]
- Lu, H.; Wu, L.; Liu, L.; Ruan, Q.; Zhang, X.; Hong, W.; Wu, S.; Jin, G.; Bai, Y. Quercetin ameliorates kidney injury and fibrosis by modulating M1/M2 macrophage polarization. Biochem. Pharmacol. 2018, 154, 203–212. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging 2010, 2, 627–631. [Google Scholar] [CrossRef]
- Schafer, M.J.; Haak, A.J.; Tschumperlin, D.J.; LeBrasseur, N.K. Targeting senescent cells in fibrosis: Pathology, paradox, and practical considerations. Curr. Rheumatol. Rep. 2018, 20, 3. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A., Jr.; Hogaboam, C.M. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]
- Mourad, O.; Mastikhina, O.; Khan, S.; Sun, X.; Hatkar, R.; Williams, K.; Nunes, S.S. Antisenescence therapy improves function in a human model of cardiac fibrosis-on-a-chip. ACS Mater. Au 2023, 3, 360–370. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. eBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, A.; Monga, V.; Bhatia, R. Compendium of naringenin: Potential sources, analytical aspects, chemistry, nutraceutical potentials and pharmacological profile. Crit. Rev. Food Sci. Nutr. 2023, 63, 8868–8899. [Google Scholar] [CrossRef]
- Motallebi, M.; Bhia, M.; Rajani, H.F.; Bhia, I.; Tabarraei, H.; Mohammadkhani, N.; Pereira-Silva, M.; Kasaii, M.S.; Nouri-Majd, S.; Mueller, A.L.; et al. Naringenin: A potential flavonoid phytochemical for cancer therapy. Life Sci. 2022, 305, 120752. [Google Scholar] [CrossRef]
- Heidary, M.R.; Samimi, Z.; Moradi, S.Z.; Little, P.J.; Xu, S.; Farzaei, M.H. Naringenin and naringin in cardiovascular disease prevention: A preclinical review. Eur. J. Pharmacol. 2020, 887, 173535. [Google Scholar] [CrossRef]
- Goyal, A.; Verma, A.; Dubey, N.; Raghav, J.; Agrawal, A. Naringenin: A prospective therapeutic agent for Alzheimer’s and Parkinson’s disease. J. Food Biochem. 2022, 46, e14415. [Google Scholar] [CrossRef]
- Mehranfard, N.; Ghasemi, M.; Rajabian, A.; Ansari, L. Protective potential of naringenin and its nanoformulations in redox mechanisms of injury and disease. Heliyon 2023, 9, e22820. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, G.; Guan, H.; Li, M.; Liu, Y.; Tian, B.; He, Z.; Fu, Q. Naringenin nanocrystals for improving anti-rheumatoid arthritis activity. Asian J. Pharm. Sci. 2021, 16, 816–825. [Google Scholar] [CrossRef]
- Akamo, A.J.; Rotimi, S.O.; Akinloye, D.I.; Ugbaja, R.N.; Adeleye, O.O.; Dosumu, O.A.; Eteng, O.E.; Amah, G.; Obijeku, A.; Cole, O.E. Naringin prevents cyclophosphamide-induced hepatotoxicity in rats by attenuating oxidative stress, fibrosis, and inflammation. Food Chem. Toxicol. 2021, 153, 112266. [Google Scholar] [CrossRef]
- Meng, X.M.; Zhang, Y.; Huang, X.R.; Ren, G.L.; Li, J.; Lan, H.Y. Treatment of renal fibrosis by rebalancing TGF-β/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget 2015, 6, 36984–36997. [Google Scholar] [CrossRef]
- Wei, Y.; Sun, L.; Liu, C.; Li, L. Naringin regulates endoplasmic reticulum stress and mitophagy through the ATF3/PINK1 signaling axis to alleviate pulmonary fibrosis. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 1155–1169. [Google Scholar] [CrossRef]
- Adebiyi, O.A.; Adebiyi, O.O.; Owira, P.M. Naringin reduces hyperglycemia-induced cardiac fibrosis by relieving oxidative stress. PLoS ONE 2016, 11, e0149890. [Google Scholar] [CrossRef]
- Wang, R.; Wu, G.; Dai, T.; Lang, Y.; Chi, Z.; Yang, S.; Dong, D. Naringin attenuates renal interstitial fibrosis by regulating the TGF β/Smad signaling pathway and inflammation. Exp. Ther. Med. 2020, 21, 66. [Google Scholar] [CrossRef]
- Hernández-Aquino, E.; Quezada-Ramírez, M.A.; Silva-Olivares, A.; Casas-Grajales, S.; Ramos-Tovar, E.; Flores-Beltrán, R.E.; Segovia, J.; Shibayama, M.; Muriel, P. Naringenin attenuates the progression of liver fibrosis via inactivation of hepatic stellate cells and profibrogenic pathways. Eur. J. Pharmacol. 2019, 865, 172730. [Google Scholar] [CrossRef]
- Qin, M.C.; Li, J.J.; Zheng, Y.T.; Li, Y.J.; Zhang, Y.X.; Ou, R.X.; He, W.Y.; Zhao, J.M.; Liu, S.T.; Liu, M.H.; et al. Naringin ameliorates liver fibrosis in zebrafish by modulating IDO1-mediated lipid metabolism and inflammatory infiltration. Food Funct. 2023, 14, 10347–10361. [Google Scholar] [CrossRef]
- Amini, N.; Sarkaki, A.; Dianat, M.; Mard, S.A.; Ahangarpour, A.; Badavi, M. Protective effects of naringin and trimetazidine on remote effect of acute renal injury on oxidative stress and myocardial injury through Nrf-2 regulation. Pharmacol. Rep. 2019, 71, 1059–1066. [Google Scholar] [CrossRef]
- De Luca, A.; Carvalho, A.; Cunha, C.; Iannitti, R.G.; Pitzurra, L.; Giovannini, G.; Mencacci, A.; Bartolommei, L.; Moretti, S.; Massi-Benedetti, C.; et al. IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PLoS Pathog. 2013, 9, e1003486. [Google Scholar] [CrossRef]
- Li, Z.L.; Yang, B.C.; Gao, M.; Xiao, X.F.; Zhao, S.P.; Liu, Z.L. Naringin improves sepsis-induced intestinal injury by modulating macrophage polarization via PPARγ/MiR-21 axis. Mol. Ther. Nucleic Acids 2021, 25, 502–514. [Google Scholar] [CrossRef]
- Li, B.; Mei, X.F. Naringin may promote functional recovery following spinal cord injury by modulating microglial polarization through the PPAR-γ/NF-ΚB signaling pathway. Brain Res. 2023, 1821, 148563. [Google Scholar] [CrossRef]
- Zhang, J.; Qiu, H.; Huang, J.; Ding, S.; Huang, B.; Zhou, P.; Jiang, Q. EETs/PPARs activation together mediates the preventive effect of naringenin in high glucose-induced cardiomyocyte hypertrophy. Biomed. Pharmacother. 2019, 109, 1498–1505. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, X.; Xie, J.; Zhang, Y.; Li, H.; Zheng, D. Network pharmacology combined with molecular docking and experimental validation to reveal the pharmacological mechanism of naringin against renal fibrosis. Open Med. 2023, 18, 20230736. [Google Scholar] [CrossRef]
- Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Panjwani, A.A.; Liu, H.; Cornblatt, G.; Cornblatt, B.S.; Ownby, S.L.; Fuchs, E.; Holtzclaw, W.D.; et al. Bioavailability of sulforaphane following ingestion of glucoraphanin-rich broccoli sprout and seed extracts with active myrosinase: A pilot study of the effects of proton pump inhibitor administration. Nutrients 2019, 11, 1489. [Google Scholar] [CrossRef]
- Langston-Cox, A.G.; Anderson, D.; Creek, D.J.; Palmer, K.R.; Marshall, S.A.; Wallace, E.M. Sulforaphane bioavailability and effects on blood pressure in women with pregnancy hypertension. Reprod. Sci. 2021, 28, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Gamet-Payrastre, L.; Li, P.; Lumeau, S.; Cassar, G.; Dupont, M.A.; Chevolleau, S.; Gasc, N.; Tulliez, J.; Tercé, F. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000, 60, 1426–1433. [Google Scholar] [PubMed]
- Chaudhuri, D.; Orsulic, S.; Ashok, B.T. Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol. Cancer Ther. 2007, 6, 334–345. [Google Scholar] [CrossRef]
- Shan, Y.; Zhang, L.; Bao, Y.; Li, B.; He, C.; Gao, M.; Feng, X.; Xu, W.; Zhang, X.; Wang, S. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J. Nutr. Biochem. 2013, 24, 1062–1069. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, J.; Zheng, C.; Zhang, P.; Zhou, W.; Cui, W.; Mo, X.; Li, L.; Xu, L.; Gao, J. Nrf2 antioxidant pathway suppresses Numb-mediated epithelial-mesenchymal transition during pulmonary fibrosis. Cell Death Dis. 2018, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13. [Google Scholar] [CrossRef]
- Sun, C.; Li, S.; Li, D. Sulforaphane mitigates muscle fibrosis in mdx mice via Nrf2-mediated inhibition of TGF-beta/Smad signaling. J. Appl. Physiol. 2016, 120, 377–390. [Google Scholar] [CrossRef]
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Römer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox. Signal. 2013, 18, 66–79. [Google Scholar] [CrossRef]
- Fix, C.; Carver-Molina, A.; Chakrabarti, M.; Azhar, M.; Carver, W. Effects of the isothiocyanate sulforaphane on TGF-β1-induced rat cardiac fibroblast activation and extracellular matrix interactions. J. Cell. Physiol. 2019, 234, 13931–13941. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Wei, J.; Zheng, Z.; Su, J.; Bian, C.; Xin, Y.; Jiang, X. Sulforaphane regulates Nrf2-mediated antioxidant activity and downregulates TGF-Β1/Smad pathways to prevent radiation-induced muscle fibrosis. Life Sci. 2022, 311, 121197. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, J.; Xu, C.; Huang, X.; Ruan, Z.; Dai, Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3 signaling pathways. Mol. Med. 2020, 26, 49. [Google Scholar] [CrossRef] [PubMed]
- Umbarkar, P.; Tousif, S.; Singh, A.P.; Anderson, J.C.; Zhang, Q.; Tallquist, M.D.; Woodgett, J.; Lal, H. Fibroblast GSK-3α promotes fibrosis via RAF-MEK-ERK pathway in the injured heart. Circ. Res. 2022, 131, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Kaji, K.; Sato, S.; Ogawa, H.; Takagi, H.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane ameliorates ethanol plus carbon tetrachloride-induced liver fibrosis in mice through the Nrf2-mediated antioxidant response and acetaldehyde metabolization with inhibition of the LPS/TLR4 signaling pathway. J. Nutr. Biochem. 2021, 89, 108573. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wang, H.; Li, C.; Cheng, H.; Cui, Y.; Liu, L.; Zhao, Y. Sulforaphane ameliorates diabetes-induced renal fibrosis through epigenetic up-regulation of BMP-7. Diabetes Metab. J. 2021, 45, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wang, S.; Zhang, H.; Yang, G.; Bai, Y.; Liu, P.; Meng, L.; Jiang, X.; Xin, Y. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 through epigenetic modification. J. Cell. Mol. Med. 2021, 25, 4408–4419. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, Y.; Zhang, Q.; Liu, W.; Meng, L.; Jiang, X.; Xin, Y. Essential Role of Nrf2 in sulforaphane-induced protection against angiotensin II-induced aortic injury. Life Sci. 2022, 306, 120780. [Google Scholar] [CrossRef] [PubMed]
- Pop, A.L.; Crișan, S.; Bârcă, M.; Ciobanu, A.-M.; Varlas, V.N.; Pop, C.; Pali, M.-A.; Cauni, D.; Ozon, E.A.; Udeanu, D.; et al. Evaluation of dissolution profiles of a newly developed solid oral immediate-release formula containing alpha-lipoic acid. Processes 2021, 9, 176. [Google Scholar] [CrossRef]
- Salehi, B.; Yılmaz, Y.B.; Antika, G.; Tumer, T.B.; Mahomoodally, M.F.; Lobine, D.; Akram, M.; Riaz, M.; Capanoglu, E.; Sharopov, F.; et al. Insights on the use of α-lipoic acid for therapeutic purposes. Biomolecules 2019, 9, 356. [Google Scholar] [CrossRef]
- Banik, S.; Halder, S.; Sato, H.; Onoue, S. Self-emulsifying drug delivery system of (R)-α-lipoic acid to improve its stability and oral absorption. Biopharm. Drug Dispos. 2021, 42, 226–233. [Google Scholar] [CrossRef]
- Ibrahim, F.G.; Mousa, R.M. The protective potential of alpha lipoic acid on amiodarone-induced pulmonary fibrosis and hepatic injury in rats. Mol. Cell Biochem. 2021, 476, 3433–3448. [Google Scholar] [CrossRef] [PubMed]
- Kocak, A.; Ural, C.; Harmanci, D.; Oktan, M.A.; Afagh, A.; Sarioglu, S.; Yilmaz, O.; Birlik, M.; Akdogan, G.G.; Cavdar, Z. Protective effects of alpha-lipoic acid on bleomycin-induced skin fibrosis through the repression of NADPH Oxidase 4 and TGF-Β1/Smad3 signaling pathways. Hum. Exp. Toxicol. 2022, 41, 9603271211065975. [Google Scholar] [CrossRef]
- Baeeri, M.; Bahadar, H.; Rahimifard, M.; Navaei-Nigjeh, M.; Khorasani, R.; Rezvanfar, M.A.; Gholami, M.; Abdollahi, M. α-Lipoic acid prevents senescence, cell cycle arrest, and inflammatory cues in fibroblasts by inhibiting oxidative stress. Pharmacol. Res. 2019, 141, 214–223. [Google Scholar] [CrossRef]
- Yoon, J.; Lee, H.; Lim, J.W.; Kim, H. Inhibitory effect of alpha-lipoic acid on mitochondrial dysfunction and interleukin-8 expression in interleukin-1beta-stimulated ataxia teleangiectasia fibroblasts. J. Physiol. Pharmacol. 2020, 71, 155–165. [Google Scholar] [CrossRef]
- Cha, T.L.; Chuang, M.J.; Tang, S.H.; Wu, S.T.; Sun, K.H.; Chen, T.T.; Sun, G.H.; Chang, S.Y.; Yu, C.P.; Ho, J.Y.; et al. Emodin modulates epigenetic modifications and suppresses bladder carcinoma cell growth. Mol. Carcinog. 2015, 54, 167–177. [Google Scholar] [CrossRef]
- Stompor-Goracy, M. The heath benefits of emodin, a natural anthraquinone derived from rhubarb—A summary update. Int. J. Mol. Sci. 2021, 22, 9522. [Google Scholar] [CrossRef]
- Jia, X.; Yu, F.; Wang, J.; Iwanowycz, S.; Saaoud, F.; Wang, Y.; Hu, J.; Wang, Q.; Fan, D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res. Treat. 2014, 148, 291–302. [Google Scholar] [CrossRef]
- Iwanowycz, S.; Wang, J.; Hodge, J.; Wang, Y.; Yu, F.; Fan, D. Emodin bidirectionally modulates macrophage polarization and epigenetically regulates macrophage memory. J. Biol. Chem. 2016, 291, 11491–11503. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, Y.; Fu, S.; Qu, C.; Li, H.; Ni, J. Inhibition of mitochondrial complex function—The hepatotoxicity mechanism of emodin based on quantitative proteomic analyses. Cells 2019, 8, 264. [Google Scholar] [CrossRef]
- Sougiannis, A.T.; Enos, R.T.; VanderVeen, B.N.; Velazquez, K.T.; Kelly, B.; McDonald, S.; Cotham, W.; Chatzistamou, I.; Nagarkatti, M.; Fan, D.; et al. Safety of natural anthraquinone emodin: An assessment in mice. BMC Pharmacol. Toxicol. 2021, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Jia, X.; Liu, H.; Zhang, X.; Shao, Y. A comprehensive review of emodin in fibrosis treatment. Fitoterapia 2023, 165, 105358. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Niu, C.; Zhang, X.; Dong, M. Emodin suppresses activation of hepatic stellate cells through p38 mitogen-activated protein kinase and Smad signaling pathways in vitro. Phytother. Res. 2018, 32, 2436–2446. [Google Scholar] [CrossRef]
- Carver, W.; Fix, E.; Fix, C.; Fan, D.; Chakrabarti, M.; Azhar, M. Effects of emodin, a plant-derived anthraquinone, on TGF-β1-induced cardiac fibroblast activation and function. J. Cell. Physiol. 2021, 236, 7440–7449. [Google Scholar] [CrossRef]
- Yang, F.; Deng, L.; Li, J.; Chen, M.; Liu, Y.; Hu, Y.; Zhong, W. Emodin retarded renal fibrosis through regulating HGF and TGFbeta-Smad signaling pathway. Drug Des. Devel. Ther. 2020, 14, 3567–3575. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, J.; Qian, J.; Wu, G.; Ma, Z. Emodin alleviates CCl4-induced liver fibrosis by suppressing epithelial-mesenchymal transition and transforming growth factor-beta 1 in rats. Mol. Med. Rep. 2018, 18, 3262–3270. [Google Scholar] [CrossRef]
- Yang, T.; Wang, J.; Pang, Y.; Dang, X.; Ren, H.; Liu, Y.; Chen, M.; Shang, D. Emodin suppresses silica-induced lung fibrosis by promoting Sirt1 signaling via direct contact. Mol. Med. Rep. 2016, 14, 4643–4649. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, P.; Chen, Z.L.; Zhang, S.J.; Wang, Y.Q.; Cai, X.; Luo, L.; Zhou, X.; Zhao, L. Emodin attenuates lipopolysaccharide-induced acute liver injury via inhibiting the TLR4 signaling pathway in vitro and in vivo. Front. Pharmacol. 2018, 9, 962. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhou, T.; Huang, Y.; Chi, Q.; Shi, J.; Zhu, P.; Dong, N. Anthraquinone emodin inhibits tumor necrosis factor alpha-induced calcification of human aortic valve interstitial cells via the NF-κB pathway. Front. Pharmacol. 2018, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cui, X.; Zhao, R.; Hu, L.; Li, Y.; Liu, C. Emodin protects against lipopolysaccharide-induced inflammatory injury in HaCaT cells through upregulation of miR-21. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2654–2661. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, A.C.; Richardson, W.J.; Holmes, J.W.; Saucerman, J.J. Computational modeling of cardiac fibroblasts and fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 73–83. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 969516, Curcumin. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Curcumin (accessed on 12 February 2024).
- Pulido-Moran, M.; Moreno-Fernandez, J.; Ramirez-Tortosa, C.; Ramirez-Tortosa, M. Curcumin and health. Molecules 2016, 21, 264. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 1548943, Capsaicin. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Capsaicin (accessed on 12 February 2024).
- Chiou, K.L.; Hastorf, C.A. A systematic approach to species-level identification of Chile Pepper (Capsicum spp.) seeds: Establishing the groundwork for tracking the domestication and movement of Chile Peppers through the Americas and beyond. Econ. Bot. 2014, 68, 316–336. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5281855, Ellagic Acid. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ellagic-Acid (accessed on 12 February 2024).
- Landete, J.M. Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health. Food Res. Int. 2011, 44, 1150–1160. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 65064, Epigallocatechin Gallate. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Epigallocatechin-Gallate (accessed on 12 February 2024).
- Fareed, M.; Chaudhary, A.A. Herbal medicines for diabetes: Insights and recent advancement. In Herbal Medicines, 1st ed.; Sarwat, M., Siddique, H., Eds.; Academic Press: London, UK, 2022; Volume 1, pp. 207–222. ISBN 9780323905725. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 445154, Resveratrol. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Resveratrol (accessed on 12 February 2024).
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5280961, Genistein. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Genistein (accessed on 12 February 2024).
- Dixon, R.A.; Ferreira, D. Genistein. Phytochemistry 2002, 60, 205–211. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5280343, Quercetin. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Quercetin (accessed on 12 February 2024).
- Nishimuro, H.; Ohnishi, H.; Sato, M.; Ohnishi-Kameyama, M.; Matsunaga, I.; Naito, S.; Ippoushi, K.; Oike, H.; Nagata, T.; Akasaka, H.; et al. Estimated daily intake and seasonal food sources of quercetin in Japan. Nutrients 2015, 7, 2345–2358. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 439246, Naringenin. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Naringenin (accessed on 12 February 2024).
- Venkateswara, R.P.; Kiran, S.D.V.S.; Rohini, P.; Bhagyasree, P. Flavonoid: A review on Naringenin. J. Pharmacogn. Phytochem. 2017, 6, 2778–2783. [Google Scholar]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5350, Sulforaphane. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Sulforaphane (accessed on 12 February 2024).
- Men, X.; Han, X.; Oh, G.; Im, J.H.; Lim, J.S.; Cho, G.H.; Choi, S.I.; Lee, O.H. Plant sources, extraction techniques, analytical methods, bioactivity, and bioavailability of sulforaphane: A review. Food Sci. Biotechnol. 2023, 33, 539–556. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6112, Lipoic acid. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Lipoic-acid (accessed on 12 February 2024).
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 3220, Emodin. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Emodin (accessed on 12 February 2024).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azeredo, P.d.S.; Fan, D.; Murphy, E.A.; Carver, W.E. Potential of Plant-Derived Compounds in Preventing and Reversing Organ Fibrosis and the Underlying Mechanisms. Cells 2024, 13, 421. https://doi.org/10.3390/cells13050421
Azeredo PdS, Fan D, Murphy EA, Carver WE. Potential of Plant-Derived Compounds in Preventing and Reversing Organ Fibrosis and the Underlying Mechanisms. Cells. 2024; 13(5):421. https://doi.org/10.3390/cells13050421
Chicago/Turabian StyleAzeredo, Patrícia dos Santos, Daping Fan, E. Angela Murphy, and Wayne E. Carver. 2024. "Potential of Plant-Derived Compounds in Preventing and Reversing Organ Fibrosis and the Underlying Mechanisms" Cells 13, no. 5: 421. https://doi.org/10.3390/cells13050421
APA StyleAzeredo, P. d. S., Fan, D., Murphy, E. A., & Carver, W. E. (2024). Potential of Plant-Derived Compounds in Preventing and Reversing Organ Fibrosis and the Underlying Mechanisms. Cells, 13(5), 421. https://doi.org/10.3390/cells13050421