

Diverse Prophage Elements of Salmonella enterica Serovars Show Potential Roles in Bacterial Pathogenicity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Prevalent S. enterica Serovars, Collation of Genomes, and Extraction of Prophage Elements
2.2. Identification of Virulence Factors in Intact Prophages of Salmonella Strains
2.3. Genome Diversity and Relationships of Salmonella Intact Prophages Examined Here with Phages from Other Salmonella Serovars and Enterobacteria
3. Results
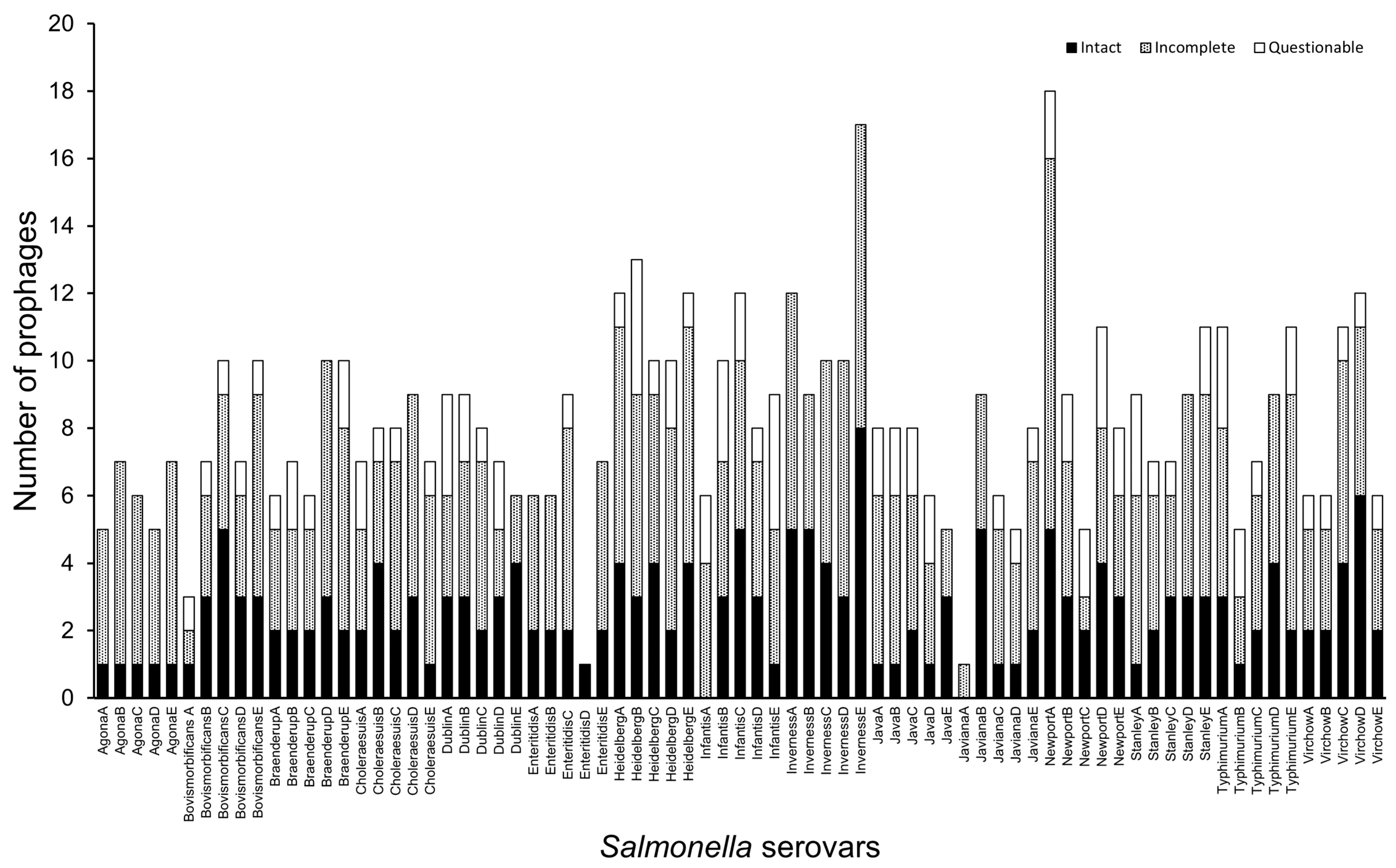
3.1. Diverse Prophage Elements Are Encoded in Salmonella Serovar Strains
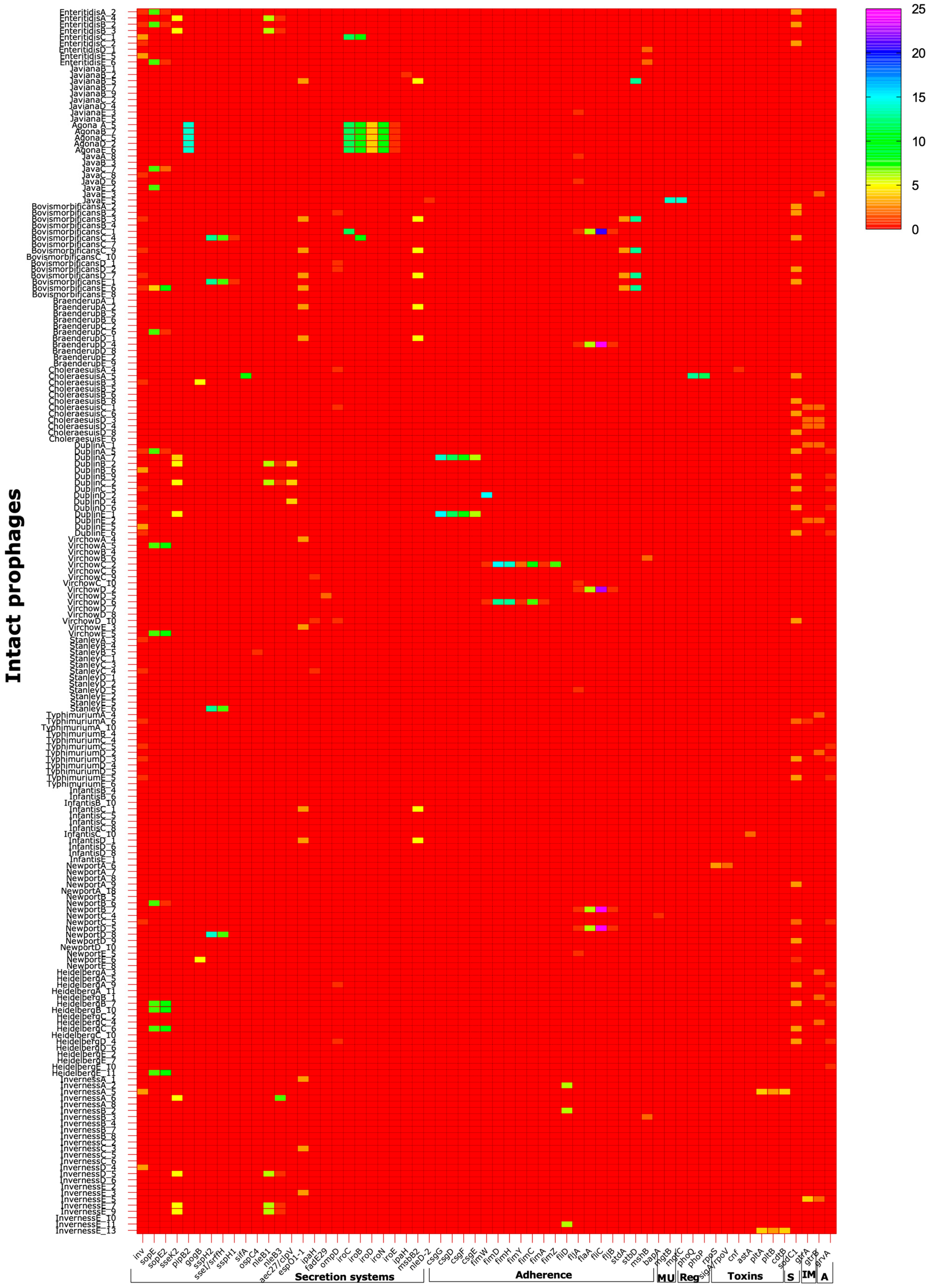
3.2. Salmonella Prophages Encode a Large Number of Virulence Factors
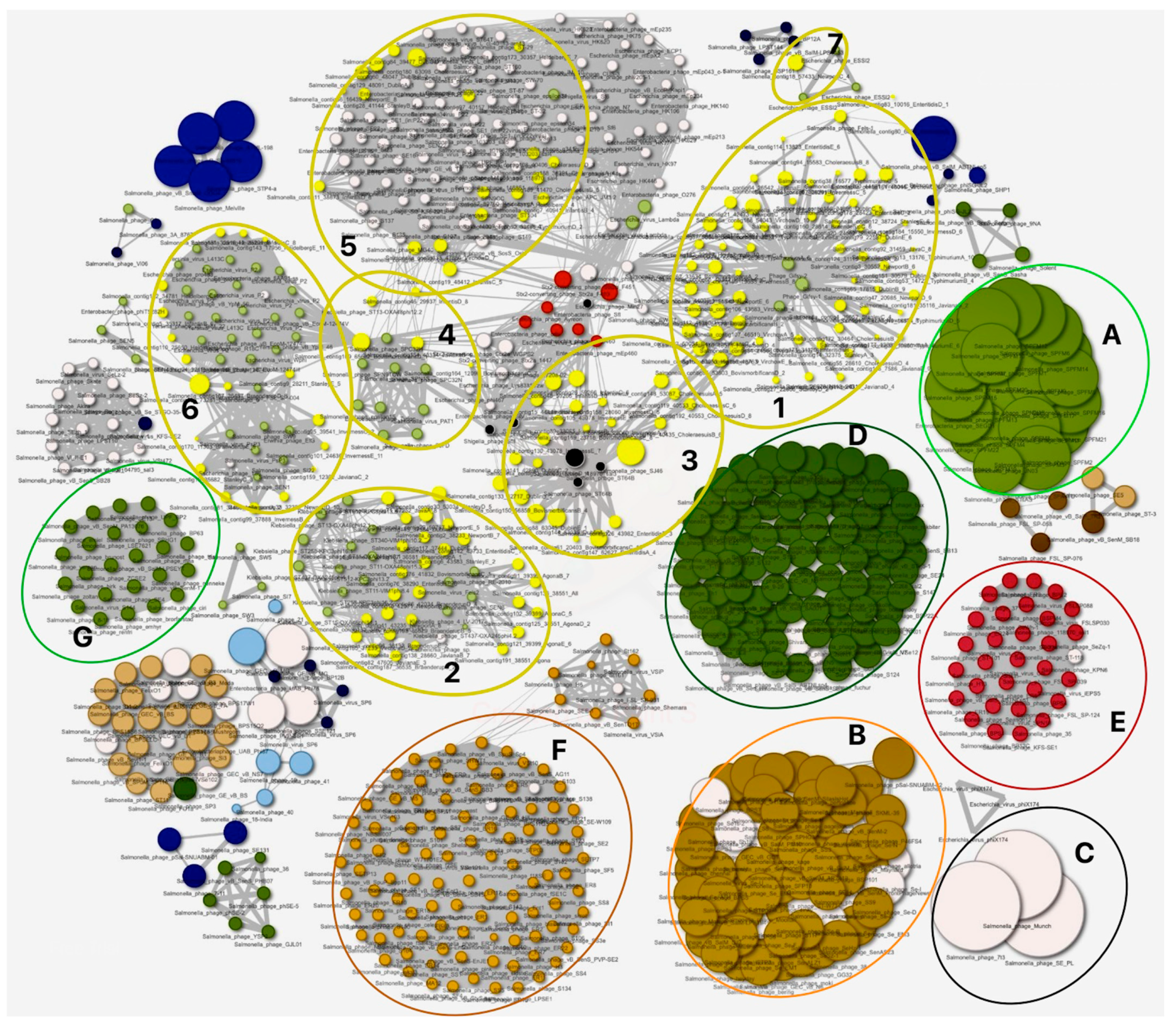
3.3. Genomes Diversity of Intact Prophages Identified in This Study and Their Relationships to Phages of Other Members of Enterobacteria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, H.-M.; Wang, Y.; Su, L.-H.; Chiu, C.-H. Nontyphoid Salmonella Infection: Microbiology, Clinical Features, and Antimicrobial Therapy. Pediatr. Neonatol. 2013, 54, 147–152. [Google Scholar] [CrossRef]
- Foley, S.L.; Lynne, A.M.; Nayak, R. Salmonella challenges: Prevalence in swine and poultry and potential pathogenicity of such isolates1,2. J. Anim. Sci. 2008, 86, E149–E162. [Google Scholar] [CrossRef]
- Collaborators, G.N.-T.S.I.D. The global burden of non-typhoidal Salmonella invasive disease: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect. Dis. 2019, 19, 1312–1324. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M. International Collaboration on Enteric Disease “Burden of Illness” Studies. The Global Burden of Nontyphoidal Salmonella Gastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar] [CrossRef]
- Eguale, T.; Gebreyes, W.A.; Asrat, D.; Alemayehu, H.; Gunn, J.S.; Engidawork, E. Non-typhoidal Salmonella serotypes, antimicrobial resistance and co-infection with parasites among patients with diarrhea and other gastrointestinal complaints in Addis Ababa, Ethiopia. BMC Infect. Dis. 2015, 15, 497. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Salmonella; EFSA: Parma, Italy, 2022.
- Darren, H.; Laura, T.; Nazmina, M.; Abdul, K. Estimating deaths from foodborne disease in the UK for 11 key pathogens. BMJ Open Gastroenterol. 2020, 7, e000377. [Google Scholar] [CrossRef]
- UKHSA. Non-Typhoidal Salmonella data 2010 to 2019; UKHSA: London, UK, 2021.
- HPS. Annual Summary of Salmonella Infections; HPS: London, UK, 2020.
- Branchu, P.; Bawn, M.; Kingsley, R.A. Genome Variation and Molecular Epidemiology of Salmonella enterica Serovar Typhimurium Pathovariants. Infect. Immun. 2018, 86, e00079-18. [Google Scholar] [CrossRef]
- Qamar, F.N.; Hussain, W.; Qureshi, S. Salmonellosis Including Enteric Fever. Pediatr. Clin. N. Am. 2022, 69, 65–77. [Google Scholar] [CrossRef]
- Anjum, M.F.; Duggett, N.A.; AbuOun, M.; Randall, L.; Nunez-Garcia, J.; Ellis, R.J.; Rogers, J.; Horton, R.; Brena, C.; Williamson, S.; et al. Colistin resistance in Salmonella and Escherichia coli isolates from a pig farm in Great Britain. J. Antimicrob. Chemother. 2016, 71, 2306–2313. [Google Scholar] [CrossRef] [PubMed]
- Petrovska, L.; Mather, A.E.; AbuOun, M.; Branchu, P.; Harris, S.R.; Connor, T.; Hopkins, K.L.; Underwood, A.; Lettini, A.A.; Page, A.; et al. Microevolution of Monophasic Salmonella Typhimurium during Epidemic, United Kingdom, 2005–2010. Emerg. Infect. Dis. 2016, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, J.A.; Steele-Mortimer, O. Salmonella—The ultimate insider. Salmonella virulence factors that modulate intracellular survival. Cell Microbiol. 2009, 11, 1579–1586. [Google Scholar] [CrossRef]
- Ruimin, G.; Linru, W.; Dele, O. Virulence Determinants of Non-typhoidal Salmonellae. In Microorganisms; Miroslav, B., Mona, S., Abdelaziz, E., Eds.; IntechOpen: Rijeka, Croatia, 2019; p. 9. [Google Scholar]
- van Asten, A.J.A.M.; van Dijk, J.E. Distribution of “classic” virulence factors among Salmonella spp. FEMS Immunol. Med. Microbiol. 2005, 44, 251–259. [Google Scholar] [CrossRef]
- Alenazy, R. Antibiotic resistance in Salmonella: Targeting multidrug resistance by understanding efflux pumps, regulators and the inhibitors. J. King Saud Univ. -Sci. 2022, 34, 102275. [Google Scholar] [CrossRef]
- Trofeit, L.; Sattler, E.; Künz, J.; Hilbert, F. Salmonella Prophages, Their Propagation, Host Specificity and Antimicrobial Resistance Gene Transduction. Antibiotics 2023, 12, 595. [Google Scholar] [CrossRef]
- Wahl, A.; Battesti, A.; Ansaldi, M. Prophages in Salmonella enterica: A driving force in reshaping the genome and physiology of their bacterial host? Mol. Microbiol. 2019, 111, 303–316. [Google Scholar] [CrossRef]
- Garcia-Russell, N.; Elrod, B.; Dominguez, K. Stress-induced prophage DNA replication in Salmonella enterica serovar Typhimurium. Infect. Genet. Evol. 2009, 9, 889–895. [Google Scholar] [CrossRef]
- Pattenden, T.; Eagles, C.; Wahl, L.M. Host life-history traits influence the distribution of prophages and the genes they carry. Philos. Trans. R. Soc. B Biol. Sci. 2022, 377, 20200465. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Davidson, A.R. When a virus is not a parasite: The beneficial effects of prophages on bacterial fitness. J. Microbiol. 2014, 52, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.; Lu, Y.T.; Brenner, T.; Falardeau, J.; Wang, S. Prophage Diversity Across Salmonella and Verotoxin-Producing Escherichia coli in Agricultural Niches of British Columbia, Canada. Front. Microbiol. 2022, 13, 853703. [Google Scholar] [CrossRef] [PubMed]
- Doub, J.B. Risk of Bacteriophage Therapeutics to Transfer Genetic Material and Contain Contaminants Beyond Endotoxins with Clinically Relevant Mitigation Strategies. Infect. Drug Resist. 2021, 14, 5629–5637. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.R.; Broadbent, S.E.; Harris, S.R.; Thomson, N.R.; van der Woude, M.W. Horizontally acquired glycosyltransferase operons drive Salmonellae lipopolysaccharide diversity. PLoS Genet. 2013, 9, e1003568. [Google Scholar] [CrossRef]
- Baños, R.C.; Aznar, S.; Madrid, C.; Juárez, A. Differential functional properties of chromosomal- and plasmid-encoded H-NS proteins. Res. Microbiol. 2011, 162, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Balbontín, R.; Rowley, G.; Pucciarelli, M.G.; López-Garrido, J.; Wormstone, Y.; Lucchini, S.; García-Del Portillo, F.; Hinton, J.C.; Casadesús, J. DNA adenine methylation regulates virulence gene expression in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2006, 188, 8160–8168. [Google Scholar] [CrossRef] [PubMed]
- Hershko-Shalev, T.; Odenheimer-Bergman, A.; Elgrably-Weiss, M.; Ben-Zvi, T.; Govindarajan, S.; Seri, H.; Papenfort, K.; Vogel, J.; Altuvia, S. Gifsy-1 Prophage IsrK with Dual Function as Small and Messenger RNA Modulates Vital Bacterial Machineries. PLoS Genet. 2016, 12, e1005975. [Google Scholar] [CrossRef] [PubMed]
- Cenens, W.; Makumi, A.; Mebrhatu, M.T.; Lavigne, R.; Aertsen, A. Phage-host interactions during pseudolysogeny: Lessons from the Pid/dgo interaction. Bacteriophage 2013, 3, e25029. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Bossi, N.; Bossi, L. Resuscitation of a Defective Prophage in Salmonella Cocultures. J. Bacteriol. 2004, 186, 4038–4041. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.; Baker, S.; Pickard, D.; Fookes, M.; Anjum, M.; Hamlin, N.; Wain, J.; House, D.; Bhutta, Z.; Chan, K.; et al. The Role of Prophage-like Elements in the Diversity of Salmonella enterica Serovars. J. Mol. Biol. 2004, 339, 279–300. [Google Scholar] [CrossRef] [PubMed]
- Hiley, L.; Fang, N.X.; Micalizzi, G.R.; Bates, J. Distribution of Gifsy-3 and of variants of ST64B and Gifsy-1 prophages amongst Salmonella enterica Serovar Typhimurium isolates: Evidence that combinations of prophages promote clonality. PLoS ONE 2014, 9, e86203. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.D.; Slauch, J.M. Characterization of grvA, an antivirulence gene on the gifsy-2 phage in Salmonella enterica serovar typhimurium. J. Bacteriol. 2001, 183, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.D.; Slauch, J.M. OmpC is the receptor for Gifsy-1 and Gifsy-2 bacteriophages of Salmonella. J. Bacteriol. 2001, 183, 1495–1498. [Google Scholar] [CrossRef]
- ECDC. European Centre for Disease Prevention and Control. Salmonellosis. In Annual Epidemiological Report for 2019; ECDC: Stockholm, Sweden, 2023. [Google Scholar]
- PHS. Public Health Scotland: Gastrointestinal & Zoonoses. Biennial Report 2020/2021. 2023. Available online: https://publichealthscotland.scot/publications/gastrointestinal-and-zoonoses/gastrointestinal-and-zoonoses-biennial-report-2020-to-2021/ (accessed on 7 March 2024).
- FSA. Food Safety Agency—Advisory Committee on the Microbiological Safety of Food: Epidemiology of Foodborne Infections Group (EFIG); FSA: Tokyo, Japan, 2020.
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Hahsler, M.; Piekenbrock, M.; Doran, D. dbscan: Fast Density-Based Clustering with R. J. Stat. Softw. 2019, 91, 1–30. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2018, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.R.; Clayton, D.J.; Windhorst, D.; Vernikos, G.; Davidson, S.; Churcher, C.; Quail, M.A.; Stevens, M.; Jones, M.A.; Watson, M.; et al. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 2008, 18, 1624–1637. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Pineros, G.; Millard, A.; Michniewski, S.; Scanlan, D.; Sirén, K.; Reyes, A.; Petersen, B.; Clokie, M.R.J.; Sicheritz-Pontén, T. From Trees to Clouds: PhageClouds for Fast Comparison of ~640,000 Phage Genomic Sequences and Host-Centric Visualization Using Genomic Network Graphs. PHAGE 2021, 2, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, H.G.; Nilsson, A.; Melo, L.D.R.; Oliveira, A. Analysis of intact prophages in genomes of Paenibacillus larvae: An important pathogen for bees. Front. Microbiol. 2022, 13, 903861. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.L.; Falkow, S. Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect. Immun. 1988, 56, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Pepe, J.C.; Miller, V.L. Yersinia enterocolitica invasin: A primary role in the initiation of infection. Proc. Natl. Acad. Sci. USA 1993, 90, 6473–6477. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Wang, S.; Zhao, J.-H.; Liu, S.-L. Salmonella secretion systems: Differential roles in pathogen-host interactions. Microbiol. Res. 2020, 241, 126591. [Google Scholar] [CrossRef]
- Kane, C.D.; Schuch, R.; Day, W.A., Jr.; Maurelli, A.T. MxiE regulates intracellular expression of factors secreted by the Shigella flexneri 2a type III secretion system. J. Bacteriol. 2002, 184, 4409–4419. [Google Scholar] [CrossRef]
- Mirold, S.; Rabsch, W.; Tschäpe, H.; Hardt, W.-D. Transfer of the Salmonella type III effector sopE between unrelated phage families11Edited by M. Gottesman. J. Mol. Biol. 2001, 312, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Sasakawa, C. Shigella IpaH Family Effectors as a Versatile Model for Studying Pathogenic Bacteria. Front. Cell. Infect. Microbiol. 2015, 5, 100. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.I.; Valdebenito, M.; Hantke, K. Salmochelin, the long-overlooked catecholate siderophore of Salmonella. Biometals 2009, 22, 691–695. [Google Scholar] [CrossRef]
- Wang, J.; Pritchard, J.R.; Kreitmann, L.; Montpetit, A.; Behr, M.A. Disruption of Mycobacterium avium subsp. paratuberculosis-specific genes impairs in vivo fitness. BMC Genomics 2014, 15, 415. [Google Scholar] [CrossRef]
- Karsten, V.; Murray, S.R.; Pike, J.; Troy, K.; Ittensohn, M.; Kondradzhyan, M.; Low, K.B.; Bermudes, D. msbB deletion confers acute sensitivity to CO2 in Salmonella enterica serovar Typhimurium that can be suppressed by a loss-of-function mutation in zwf. BMC Microbiol. 2009, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Moncrief, M.B.; Maguire, M.E. Magnesium and the role of MgtC in growth of Salmonella typhimurium. Infect Immun 1998, 66, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S.; Miller, S.I. PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J. Bacteriol. 1996, 178, 6857–6864. [Google Scholar] [CrossRef]
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. [Google Scholar] [CrossRef]
- Spanò, S.; Ugalde, J.E.; Galán, J.E. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38. [Google Scholar] [CrossRef]
- Nale, J.Y.; Thanki, A.M.; Rashid, S.J.; Shan, J.; Vinner, G.K.; Dowah, A.S.A.; Cheng, J.K.J.; Sicheritz-Pontén, T.; Clokie, M.R.J. Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages. Viruses 2022, 14, 2772. [Google Scholar] [CrossRef]
- Nale, J.Y.; Shan, J.; Hickenbotham, P.T.; Fawley, W.N.; Wilcox, M.H.; Clokie, M.R. Diverse temperate bacteriophage carriage in Clostridium difficile 027 strains. PLoS ONE 2012, 7, e37263. [Google Scholar] [CrossRef]
- Park, M.; Lee, J.H.; Shin, H.; Kim, M.; Choi, J.; Kang, D.H.; Heu, S.; Ryu, S. Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 2012, 78, 58–69. [Google Scholar] [CrossRef]
- Zeng, C.; Gilcrease, E.B.; Hendrix, R.W.; Xie, Y.; Jalfon, M.J.; Gill, J.J.; Casjens, S.R. DNA Packaging and Genomics of the Salmonella 9NA-Like Phages. J. Virol. 2019, 93, e00848-19. [Google Scholar] [CrossRef]
- Xie, Y.; Wahab, L.; Gill, J.J. Development and Validation of a Microtiter Plate-Based Assay for Determination of Bacteriophage Host Range and Virulence. Viruses 2018, 10, 189. [Google Scholar] [CrossRef]
- Rivera, D.; Moreno-Switt, A.I.; Denes, T.G.; Hudson, L.K.; Peters, T.L.; Samir, R.; Aziz, R.K.; Noben, J.-P.; Wagemans, J.; Dueñas, F. Novel Salmonella Phage, vB_Sen_STGO-35-1, Characterization and Evaluation in Chicken Meat. Microorganisms 2022, 10, 606. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Zhang, Y.; Cheng, M.; Le, S.; Gu, J.; Bao, J.; Qin, J.; Guo, X.; Zhu, T. Characterization of Klebsiella pneumoniae ST11 Isolates and Their Interactions with Lytic Phages. Viruses 2019, 11, 1080. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, I.; Trastoy, R.; Blasco, L.; Fernández-Cuenca, F.; Ambroa, A.; Fernández-García, L.; Pacios, O.; Perez-Nadales, E.; Torre-Cisneros, J.; Oteo-Iglesias, J.; et al. Genomic analysis of 40 prophages located in the genomes of 16 carbapenemase-producing clinical strains of Klebsiella pneumoniae. Microb. Genom. 2020, 6, e000369. [Google Scholar] [CrossRef] [PubMed]
- Mmolawa, P.T.; Schmieger, H.; Heuzenroeder, M.W. Bacteriophage ST64B, a genetic mosaic of genes from diverse sources isolated from Salmonella enterica serovar typhimurium DT 64. J. Bacteriol. 2003, 185, 6481–6485. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.Y.; Tran, E.N.H.; Morona, R. Bacteriophage Sf6 host range mutant that infects Shigella flexneri serotype 2a2 strains. FEMS Microbiol. Lett. 2022, 369, fnac020. [Google Scholar] [CrossRef] [PubMed]
- Christie, G.E.; Dokland, T. Pirates of the Caudovirales. Virology 2012, 434, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: Update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef]
- Islam, M.S.; Hu, Y.; Mizan, M.F.R.; Yan, T.; Nime, I.; Zhou, Y.; Li, J. Characterization of Salmonella Phage LPST153 That Effectively Targets Most Prevalent Salmonella Serovars. Microorganisms 2020, 8, 1089. [Google Scholar] [CrossRef]
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020, 18, e06007. [Google Scholar] [CrossRef]
- Ribeiro, J.M.; Pereira, G.N.; Durli Junior, I.; Teixeira, G.M.; Bertozzi, M.M.; Verri, W.A.; Kobayashi, R.K.T.; Nakazato, G. Comparative analysis of effectiveness for phage cocktail development against multiple Salmonella serovars and its biofilm control activity. Sci. Rep. 2023, 13, 13054. [Google Scholar] [CrossRef]
- Capparelli, R.; Nocerino, N.; Iannaccone, M.; Ercolini, D.; Parlato, M.; Chiara, M.; Iannelli, D. Bacteriophage Therapy of Salmonella enterica: A Fresh Appraisal of Bacteriophage Therapy. J. Infect. Dis. 2010, 201, 52–61. [Google Scholar] [CrossRef]
- Sanders, J.G.; Yan, W.; Mjungu, D.; Lonsdorf, E.V.; Hart, J.A.; Sanz, C.M.; Morgan, D.B.; Peeters, M.; Hahn, B.H.; Moeller, A.H. A low-cost genomics workflow enables isolate screening and strain-level analyses within microbiomes. Genome Biol. 2022, 23, 212. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.-R.; Nookaew, I.; Leuze, M.R.; Ahn, T.-H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Ashton, P.M.; Owen, S.V.; Kaindama, L.; Rowe, W.P.M.; Lane, C.R.; Larkin, L.; Nair, S.; Jenkins, C.; de Pinna, E.M.; Feasey, N.A.; et al. Public health surveillance in the UK revolutionises our understanding of the invasive Salmonella Typhimurium epidemic in Africa. Genome Med. 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- APHA. Salmonella in Animals and Feed in Great Britain 2022; APHA: Weybridge, Surrey, 2023. [Google Scholar]
- Directorate, V.M. Third UK One Health Report—Joint Report on Antibiotic Use, Antibiotic Sales and Antibiotic Resistance; Veterinary Medicines Directorate: New Haw, UK, 2023. [Google Scholar]
- Fu, Y.; Wu, Y.; Yuan, Y.; Gao, M. Prevalence and Diversity Analysis of Candidate Prophages to Provide An Understanding on Their Roles in Bacillus Thuringiensis. Viruses 2019, 11, 388. [Google Scholar] [CrossRef]
- Tucker, S.C.; Galán, J.E. Complex function for SicA, a Salmonella enterica serovar typhimurium type III secretion-associated chaperone. J. Bacteriol. 2000, 182, 2262–2268. [Google Scholar] [CrossRef]
- Vonaesch, P.; Sellin, M.E.; Cardini, S.; Singh, V.; Barthel, M.; Hardt, W.D. The Salmonella Typhimurium effector protein SopE transiently localizes to the early SCV and contributes to intracellular replication. Cell. Microbiol. 2014, 16, 1723–1735. [Google Scholar] [CrossRef]
- Kolenda, R.; Ugorski, M.; Grzymajlo, K. Everything You Always Wanted to Know About Salmonella Type 1 Fimbriae, but Were Afraid to Ask. Front. Microbiol. 2019, 10, 1017. [Google Scholar] [CrossRef]
- Guo, L.; Lim, K.B.; Gunn, J.S.; Bainbridge, B.; Darveau, R.P.; Hackett, M.; Miller, S.I. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science 1997, 276, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, B.; Tsai, C.N.; Coombes, B.K. Evolution of Salmonella-Host Cell Interactions through a Dynamic Bacterial Genome. Front. Cell. Infect. Microbiol. 2017, 7, 428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liao, Y.-T.; Salvador, A.; Sun, X.; Wu, V.C.H. Prediction, Diversity, and Genomic Analysis of Temperate Phages Induced From Shiga Toxin-Producing Escherichia coli Strains. Front. Microbiol. 2020, 10, 3093. [Google Scholar] [CrossRef] [PubMed]
- McClelland, M.; Sanderson, K.E.; Spieth, J.; Clifton, S.W.; Latreille, P.; Courtney, L.; Porwollik, S.; Ali, J.; Dante, M.; Du, F.; et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 2001, 413, 852–856. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Serovars | Total Prophage Elements | |||||
|---|---|---|---|---|---|---|
| Intact | Incomplete | Questionable | Total | Average per Strain | Carriage Category | |
| S. Enteritidis | 9 | 19 | 1 | 29 | 5.8 | Low |
| S. Javiana | 9 | 17 | 3 | 29 | 5.8 | Low |
| S. Agona | 5 | 25 | 0 | 30 | 6 | Moderate |
| S. Java | 8 | 19 | 8 | 35 | 7 | Moderate |
| S. Bovismorbificans | 15 | 17 | 5 | 37 | 7.4 | Moderate |
| S. Braenderup | 11 | 22 | 6 | 39 | 7.8 | Moderate |
| S. Choleraesuis | 12 | 22 | 5 | 39 | 7.8 | Moderate |
| S. Dublin | 15 | 16 | 8 | 39 | 7.8 | Moderate |
| S. Virchow | 16 | 20 | 5 | 41 | 8.2 | High |
| S. Stanley | 12 | 24 | 7 | 43 | 8.6 | High |
| S. Typhimurium | 12 | 23 | 8 | 43 | 8.6 | High |
| S. Infantis | 12 | 21 | 12 | 45 | 9 | High |
| S. Newport | 17 | 23 | 11 | 51 | 10.2 | Extreme |
| S. Heidelberg | 17 | 31 | 9 | 57 | 11.4 | Extreme |
| S. Inverness | 25 | 33 | 0 | 58 | 11.6 | Extreme |
| Totals: | 195 | 332 | 88 | 615 | 8.2 | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrews, K.; Landeryou, T.; Sicheritz-Pontén, T.; Nale, J.Y. Diverse Prophage Elements of Salmonella enterica Serovars Show Potential Roles in Bacterial Pathogenicity. Cells 2024, 13, 514. https://doi.org/10.3390/cells13060514
Andrews K, Landeryou T, Sicheritz-Pontén T, Nale JY. Diverse Prophage Elements of Salmonella enterica Serovars Show Potential Roles in Bacterial Pathogenicity. Cells. 2024; 13(6):514. https://doi.org/10.3390/cells13060514
Chicago/Turabian StyleAndrews, Kirstie, Toby Landeryou, Thomas Sicheritz-Pontén, and Janet Yakubu Nale. 2024. "Diverse Prophage Elements of Salmonella enterica Serovars Show Potential Roles in Bacterial Pathogenicity" Cells 13, no. 6: 514. https://doi.org/10.3390/cells13060514
APA StyleAndrews, K., Landeryou, T., Sicheritz-Pontén, T., & Nale, J. Y. (2024). Diverse Prophage Elements of Salmonella enterica Serovars Show Potential Roles in Bacterial Pathogenicity. Cells, 13(6), 514. https://doi.org/10.3390/cells13060514