
Characterization of Lysophospholipase D Activity in Mammalian Cell Membranes
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Cell Membranes
2.3. Phospholipase Assays
2.4. HPLC Separation of Lipids
2.5. Over-Expression of PLD2
2.6. Radioenzymatic Assay for LPA
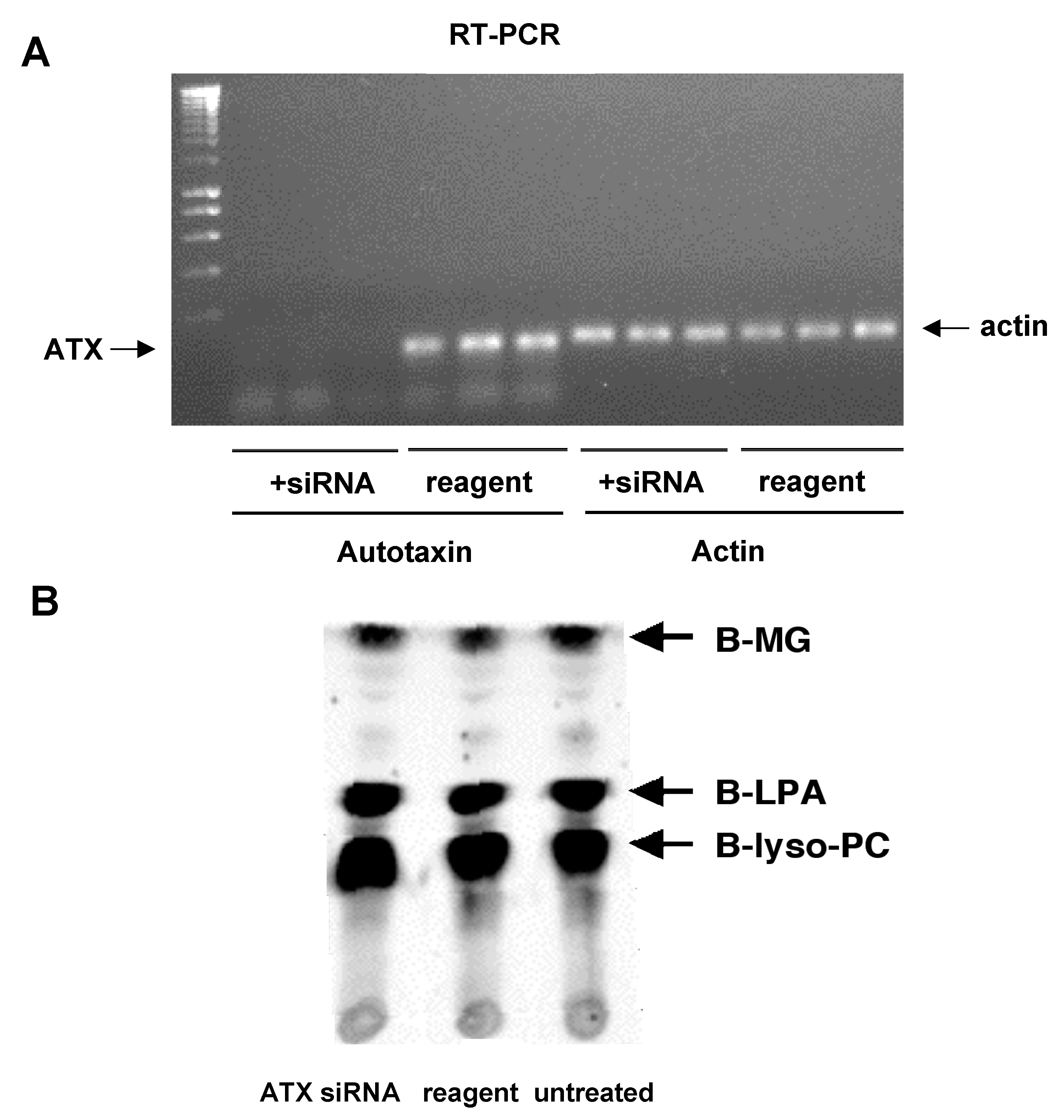
2.7. Autotaxin Knockdown
2.8. Proteomics Analysis
3. Results
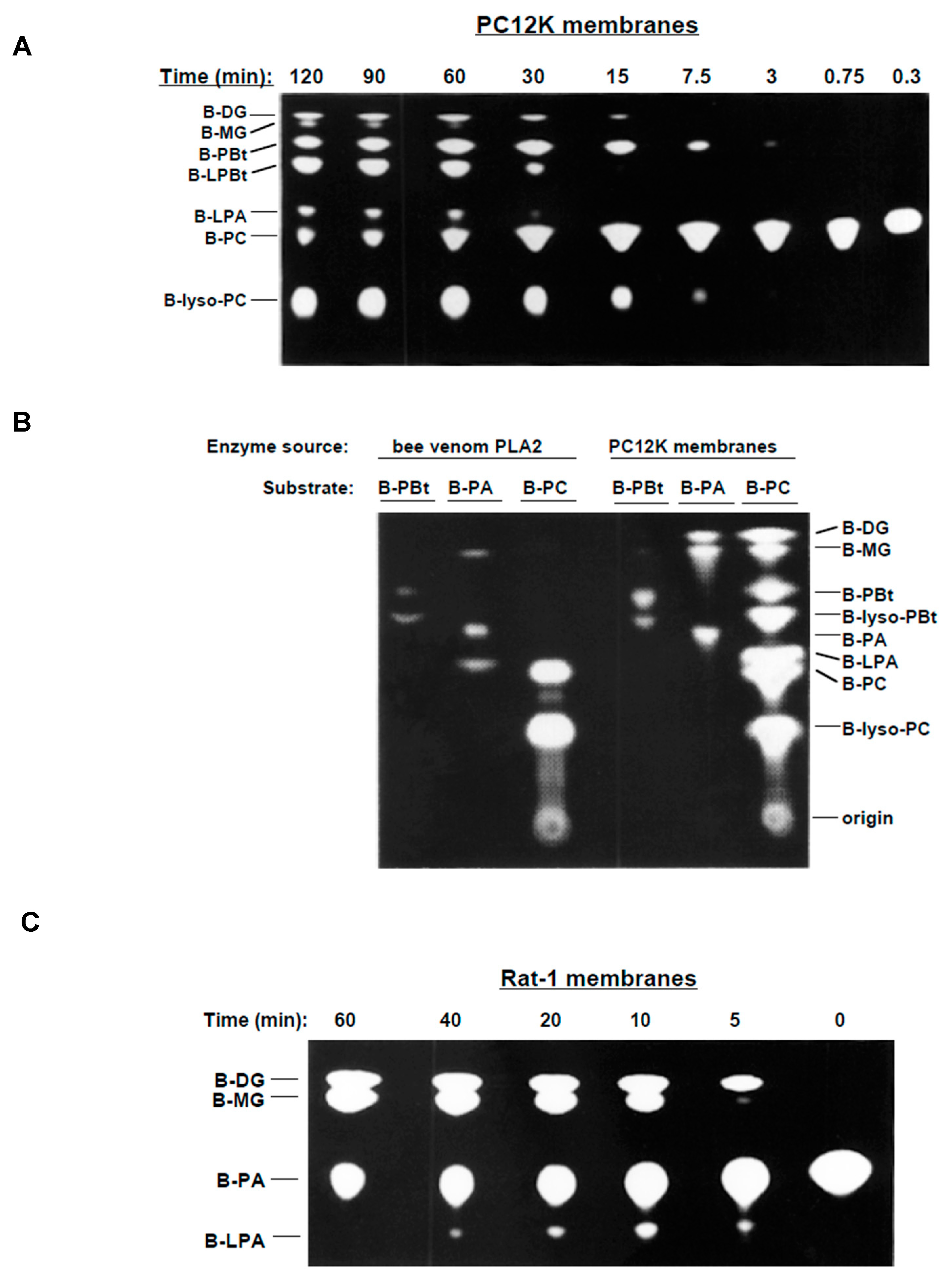
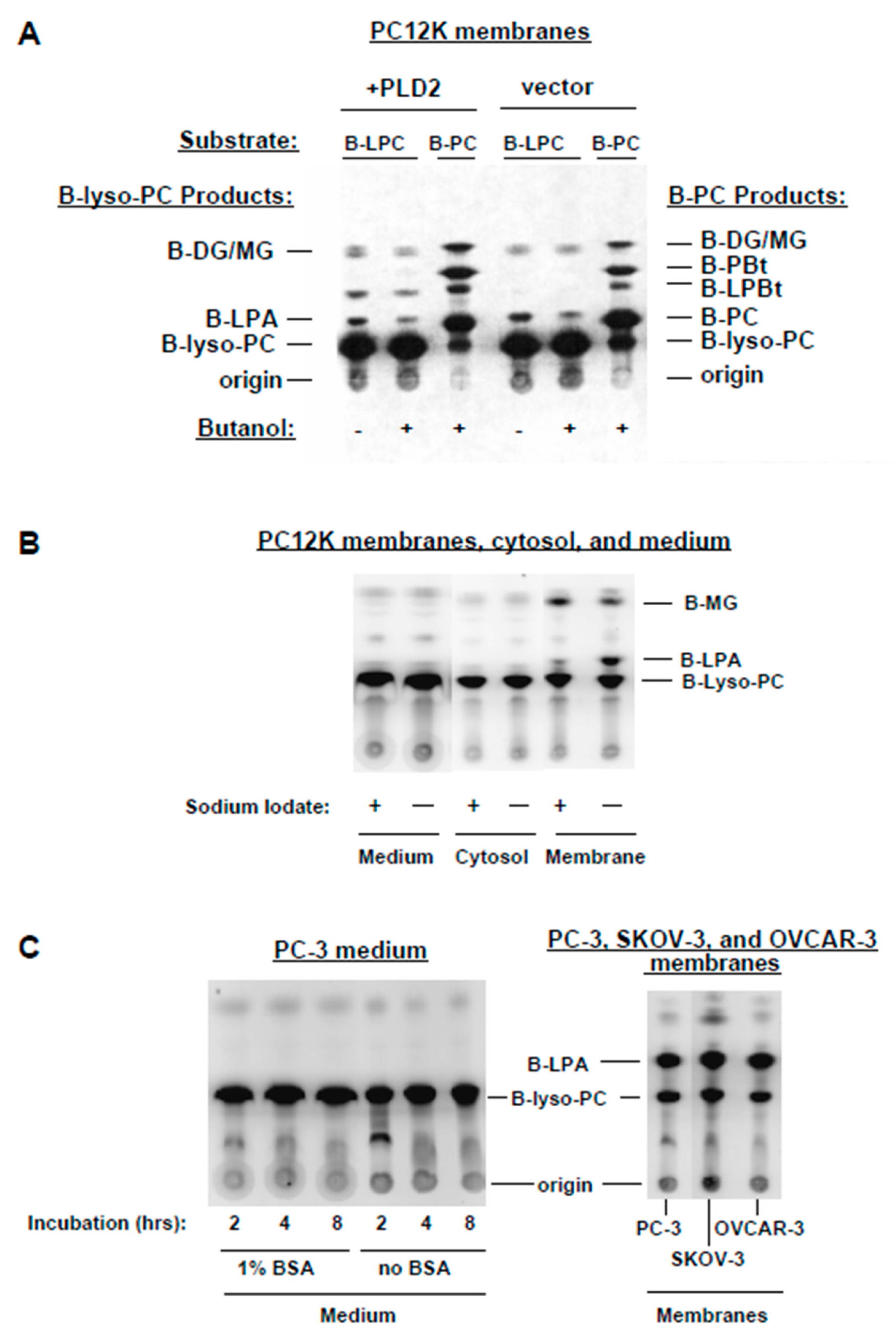
3.1. Metabolism of BODIPY-PC in Membrane Preparations
3.2. PA-PLA2 Activity
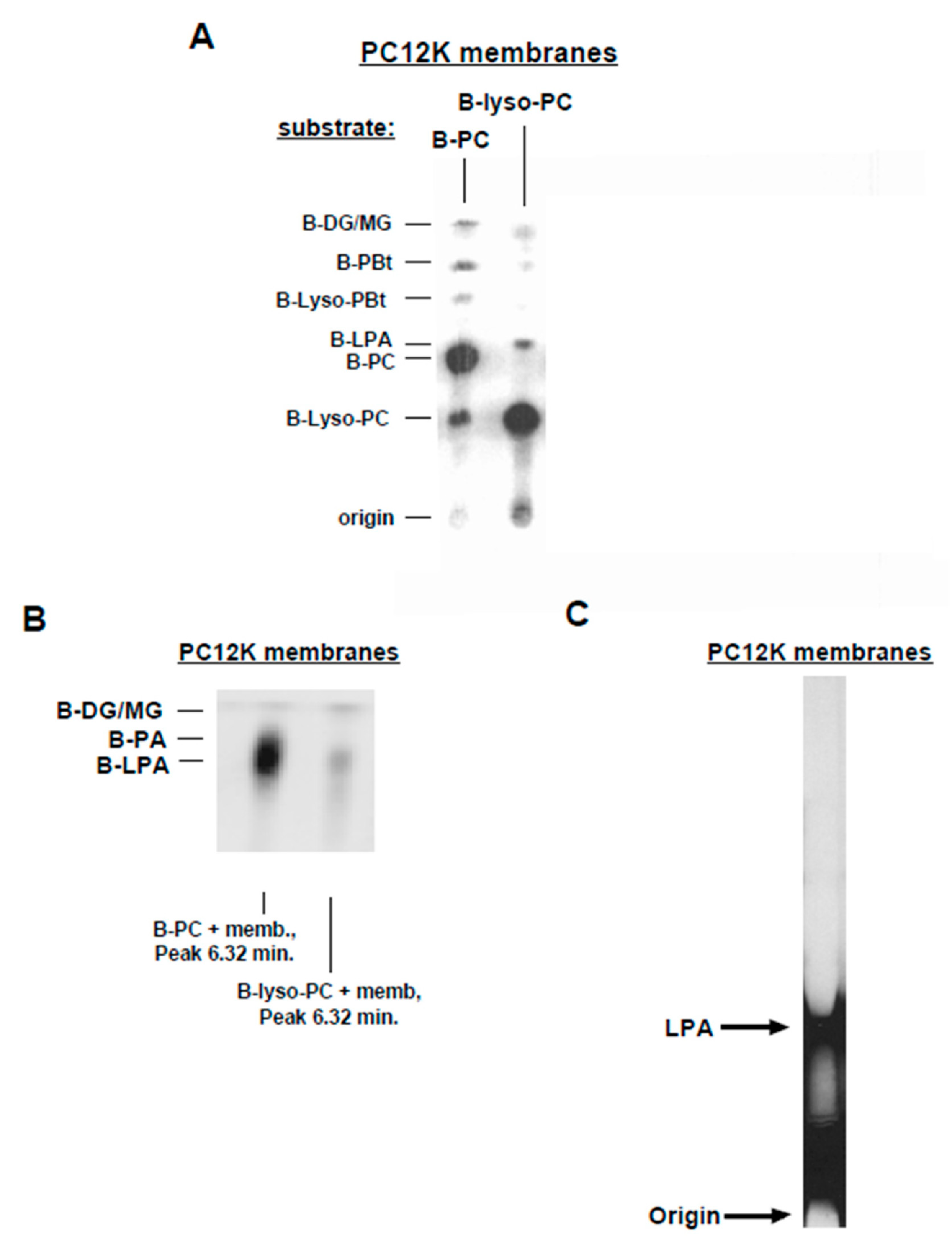
3.3. Lyso-PLD Activity
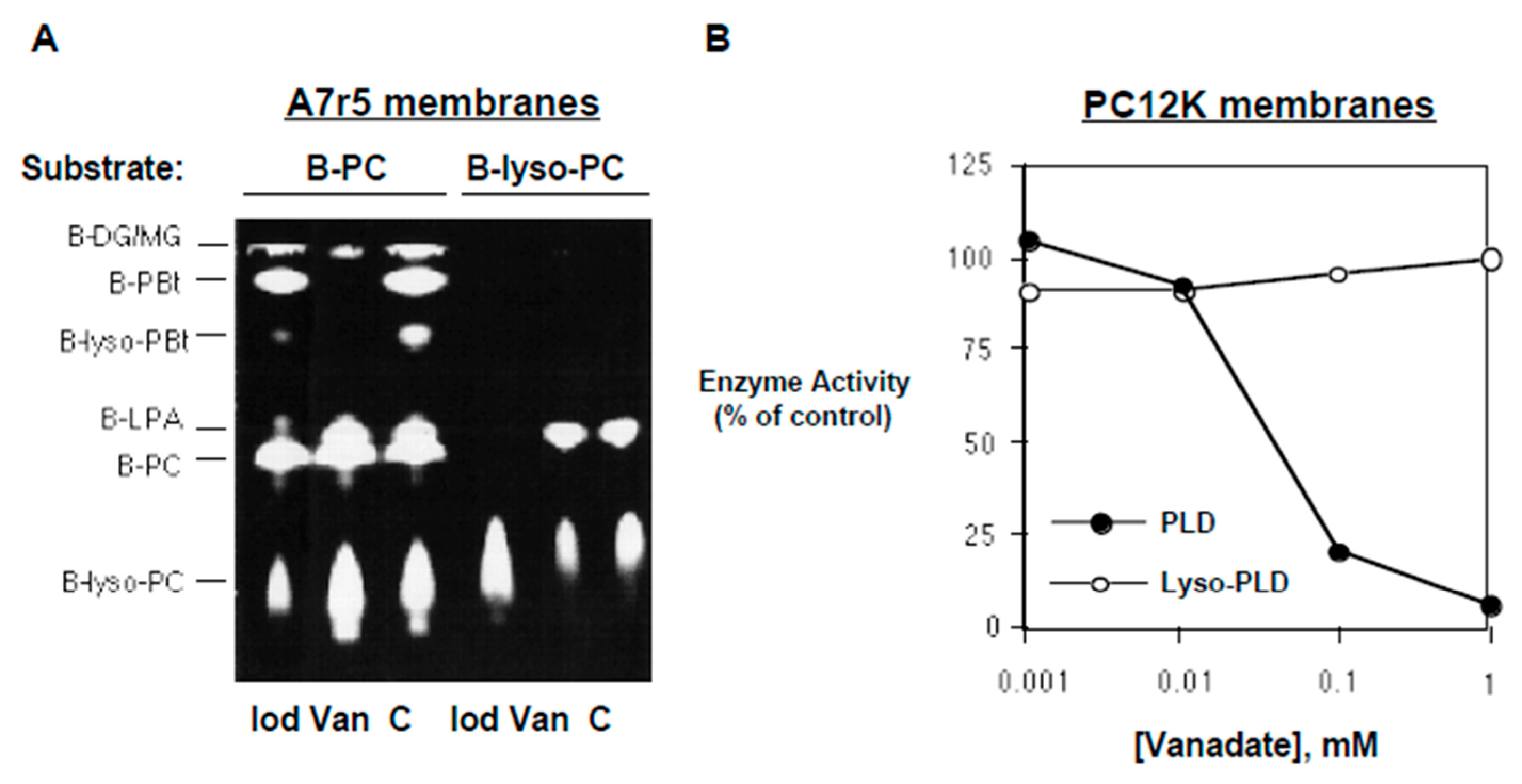
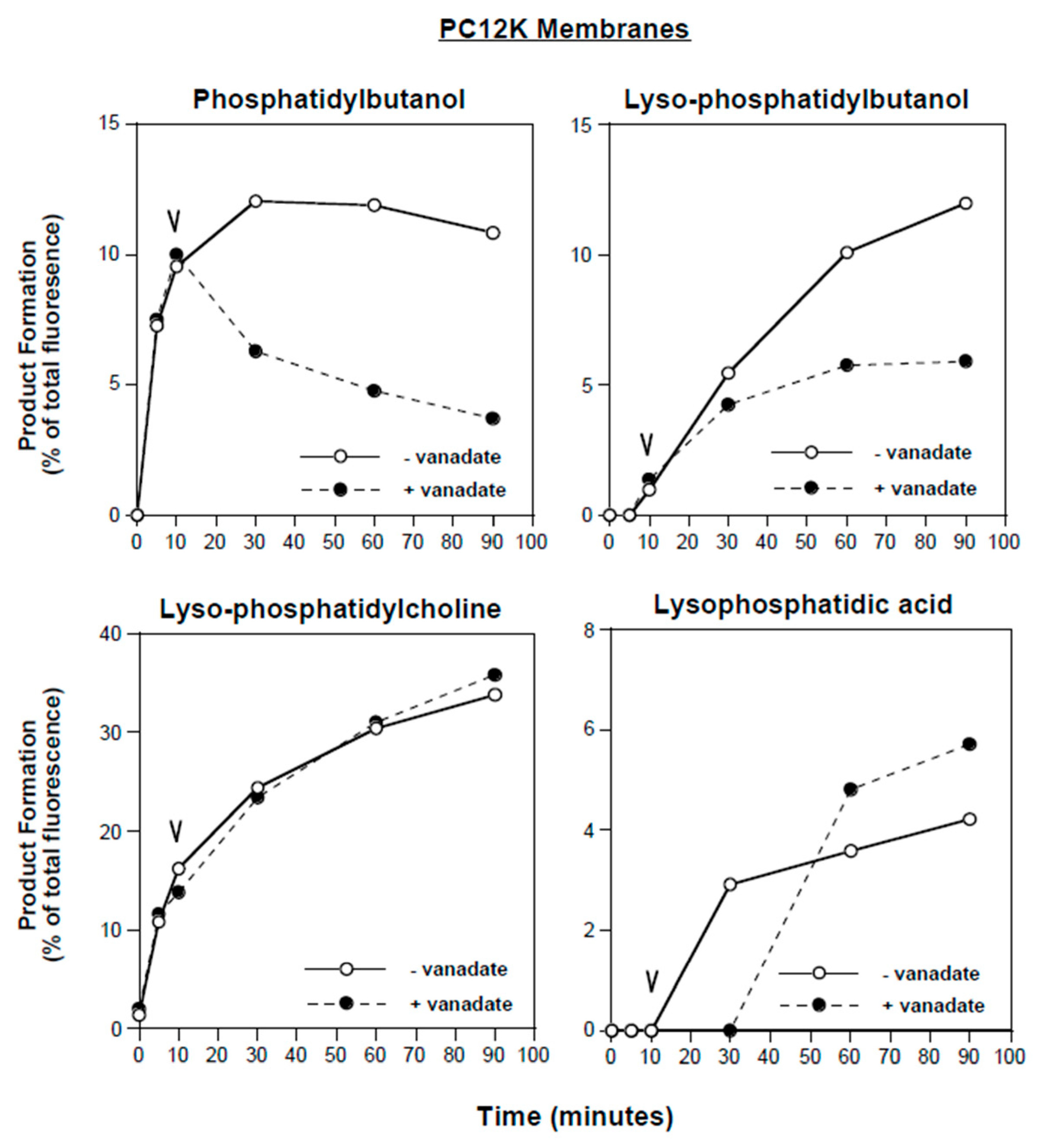
3.4. Inhibitors of PLD and PLA2
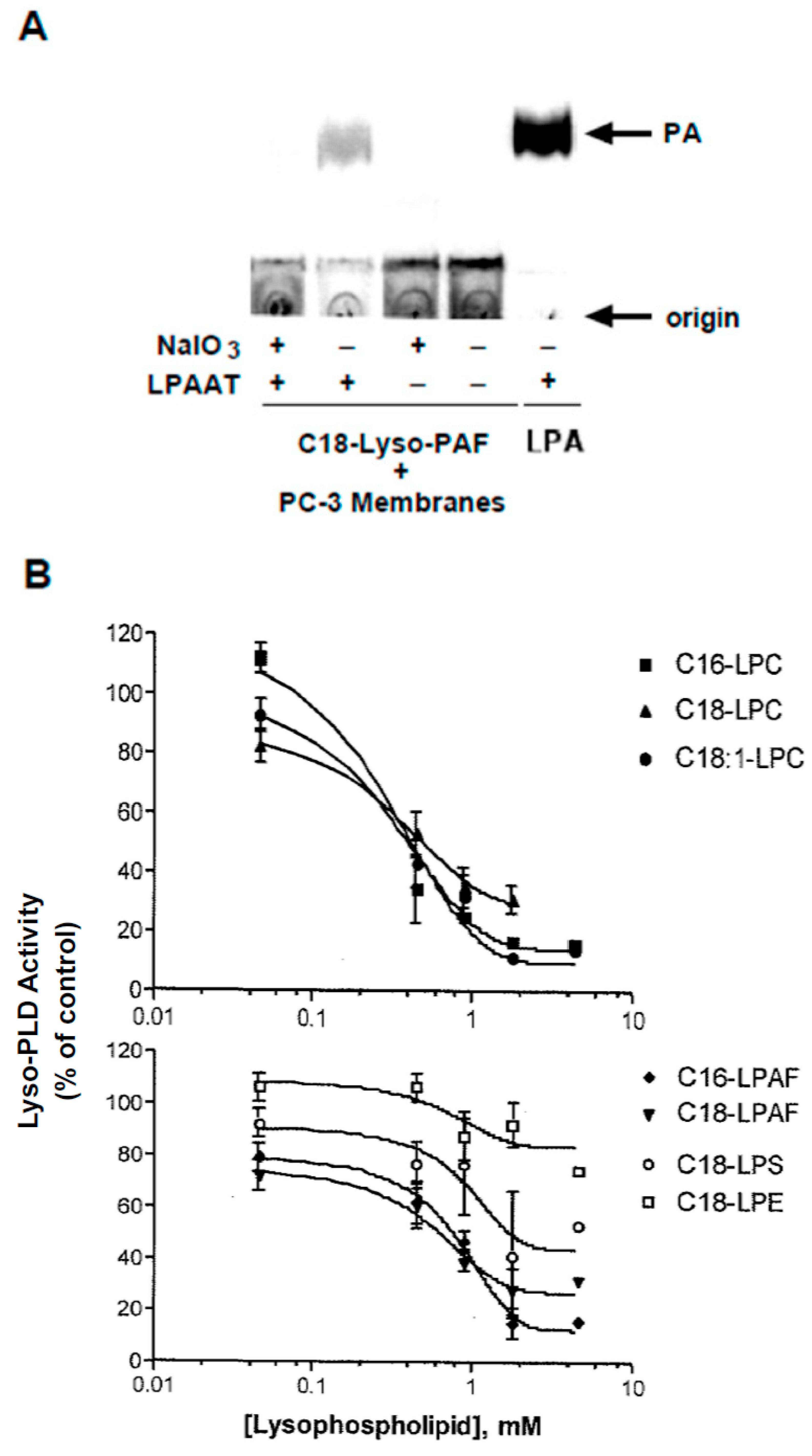
3.5. Further Characterization of Lyso-PLD Activity
3.6. Autotaxin Knockdown Experiments
3.7. Substrate Utilization by Lyso-PLD
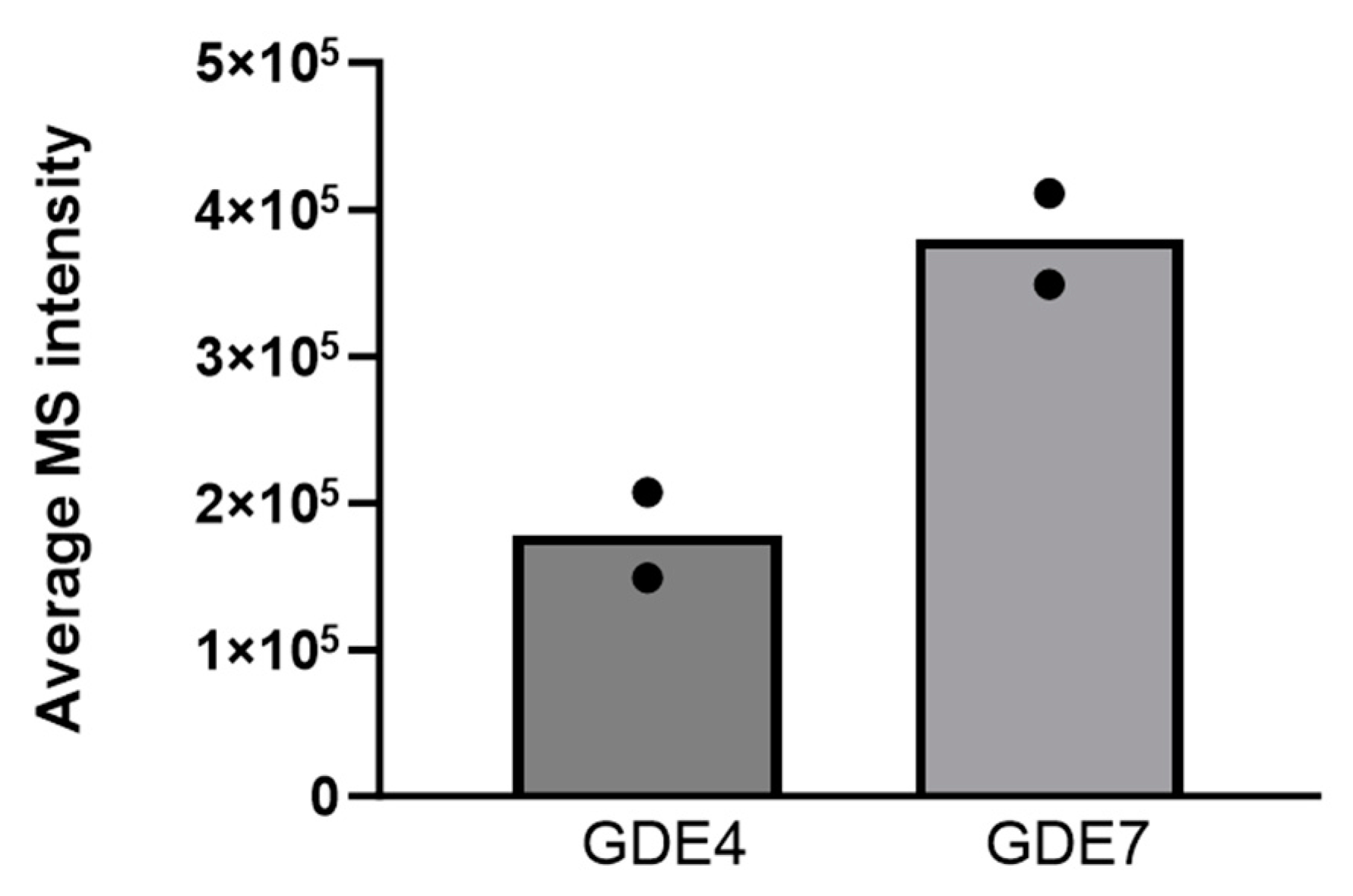
3.8. Analysis of GDE4 and GDE7 Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukushima, N.; Ishii, I.; Contos, J.J.; Weiner, J.A.; Chun, J. Lysophospholipid receptors. Ann. Rev. Pharm. Toxicol. 2001, 41, 507–534. [Google Scholar] [CrossRef]
- Geraldo, L.H.M.; de Sampaio Spohr, T.C.L.; do Amaral, R.F.; da Fonseca, A.C.C.; Garcia, C.; de Almeida Mendes, R.; Freitas, C.; dos Santos, M.F.; Lima, F.R.S. Role of lysophophatidic acid and its receptors in health and disease; novel therapeutic strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef]
- Goetzl, D.J. Pleiotypic mechanisms of cellular responses to biologically active lysophospholipids. Prost. Lipid Med. 2001, 64, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, P.; Sitton, C.C.; Meier, K.E. Lysophosphatidic Acid Signaling in Cancer Cells: What Makes LPA So Special? Cells 2021, 10, 2059. [Google Scholar] [CrossRef] [PubMed]
- Eichholtz, T.; Jalink, K.; Fahrenfort, I.; Moolenaar, W.H. The bioactive phospholipid lysophophatidic acid is released from activated platelets. Biochem. J. 1993, 291, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Pages, C.; Simon, M.F.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid synthesis and release. Prost. Lipid Med. 2001, 64, 1–10. [Google Scholar] [CrossRef]
- Xie, Y.; Gibbs, T.C.; Meier, K.E. Lysophosphatidic acid as an autocrine and paracrine mediator. Biochim. Biophys. Acta 2002, 1582, 270–281. [Google Scholar] [CrossRef]
- Xie, Y.; Gibbs, T.C.; Mukhin, Y.V.; Meier, K.E. Role for 18:1 lysophosphatidic acid as an autocrine mediator in prostate cancer cells. J. Biol. Chem. 2002, 277, 32516–32526. [Google Scholar] [CrossRef]
- Exton, J.H. Phospholipase D—Structure, regulation and function. Rev. Physiol. Biochem. Pharmacol. 2002, 144, 1–94. [Google Scholar]
- Munnik, T. Phosphatidic acid: An emerging plant lipid second messenger. Trends Plant Sci. 2001, 6, 227–233. [Google Scholar] [CrossRef]
- Martinson, E.A.; Trilivas, I.; Brown, J.H. Rapid protein kinase C-dependent activation of phospholipase D leads to delayed 1,2-diglyceride accumulation. J. Biol. Chem. 1990, 265, 22282–22287. [Google Scholar] [CrossRef] [PubMed]
- Billah, M.M.; Lapetina, E.G.; Cuatrecasas, P. Phospholipase A2 activity specific for phosphatidic acid. J. Biol. Chem. 1981, 256, 5399–5403. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kriz, R.W.; Wolfman, N.; Shaffer, M.; Seehra, J.; Jones, S.S. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J. Biol. Chem. 1997, 272, 8567–8575. [Google Scholar] [CrossRef] [PubMed]
- Eder, A.M.; Sasagawa, T.; Mao, M.; Aoki, J.; Mills, G.B. Constitutive and lysophosphatidic acid (LPA)-induced LPA production: Role of phospholipase D and phospholipase A2. Clin. Canc. Res. 2000, 6, 2482–2491. [Google Scholar]
- Kitatani, K.; Akiba, S.; Sato, T. Role of phospholipase D-derived phosphatidic acid as a substrate for phospholipase A2 in RBL-2H3 cells. Biol. Pharm. Bull. 2000, 12, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Meier, K.E. Lysophospholipase D and its role in LPA production. Cell Signal. 2004, 16, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Wykle, R.L.; Schremmer, J.M. A lysophospholipase D pathway in the metabolism of ether-linked lipids in brain microsomes. J. Biol. Chem. 1974, 249, 1742–1746. [Google Scholar] [CrossRef]
- Wykle, R.L.; Kraemer, W.F.; Schremmer, J.M. Studies of lysophospholipase D of rat liver and other tissues. Arch. Biochem. Biophys. 1997, 184, 149–155. [Google Scholar] [CrossRef]
- Wykle, R.L.; Kraemer, W.F.; Schremmer, J.M. Specificity of lysophospholipase D. Biochim. Biophys. Acta 1980, 619, 58–67. [Google Scholar] [CrossRef]
- Wykle, R.L.; Strum, J.C. Lysophospholipase D. Meth. Enzymol. 1991, 197, 583–590. [Google Scholar]
- Kawasaki, T.; Snyder, F. The metabolism of lyso-platelet-activating factor (1-O-alkyl-2-lyso-sn-glycero-e-phosphocholine) by a calcium-dependent lysophospholipase D in rabbit kidney medulla. Biochim. Biophys. Acta 1987, 920, 85–93. [Google Scholar] [CrossRef]
- Furukawa, M.; Muguruma, K.; Frenkel, R.A.; Johnston, J.M. Metabolic fate of platelet-activating factor in the rat enterocyte: The role of a specific lysophospholipase D. Arch. Biochem. Biophys. 1995, 319, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Achterberg, V.; Gercken, G. Cytotoxicity of ester and ether lysophospholipids on Leishmania donovani promastigotes. Mol. Biochem. Parasitol. 1987, 26, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Homma, H.; Inoue, K.; Nojima, S. The metabolism of platelet activating factor in platelets and plasma of various animals. J. Toxicol. Sci. 1983, 8, 177–178. [Google Scholar] [CrossRef]
- Snyder, F. Metabolism of platelet activating factor and related ether lipids: Enzymatic pathways, subcellular sites, regulation, and membrane processing. Prog. Clin. Biol. Res. 1988, 282, 57–72. [Google Scholar] [PubMed]
- Fernandez-Gallardo, S.; Gijon, M.A.; Garcia, M.C.; Cano, E.; Sanchez Drespo, M. Biosynthesis of platelet-activating factor in glandular gastric mucose. Evidence for the involvement of the ‘de novo’ pathway and modulation by fatty acids. Biochem. J. 1988, 254, 707–714. [Google Scholar] [CrossRef]
- Qian, C.G.; Lee, T.C.; Snyder, F. Metabolism of platelet activating factor (PAF) and related ether lipids by neonatal rat myocytes. J. Lipid Med. 1989, 1, 113–123. [Google Scholar]
- Blank, M.L.; Fitzgerald, V.; Lee, T.C.; Snyder, F. Evidence for biosynthesis of plasmenylcholine from plasmenylethanolamine in HL-60 cells. Biochim. Biophys. Acta 1993, 1166, 309–312. [Google Scholar] [CrossRef]
- Muguruma, K.; Johnston, J.M. Metabolism of platelet-activating factor in rat epididymal spermatozoa. Biol. Reprod. 1997, 56, 529–536. [Google Scholar] [CrossRef]
- Tokumara, A.; Fuimoto, H.; Yoshimoto, O.; Nishioka, Y.; Miyake, M.; Fukuzawa, K. Production of lysophosphatidic acid by lysophospholipase D in incubated plasma of spontaneously hypertensive rats and Wistar Kyoto rats. Life Sci. 1999, 65, 245–253. [Google Scholar] [CrossRef]
- Tokumura, A.; Nishioka, Y.; Yoshimoto, O.; Shinomiya, J.; Fukuzawa, K. Substrate specificity of lysophospholipase D which produces bioactive lysophosphatidic acids in rat plasma. Biochim. Biophys. Acta 1999, 1437, 235–245. [Google Scholar] [CrossRef]
- Tokumura, A.; Kanaya, Y.; Kitahara, M.; Miyake, M.; Yoshioka, Y.; Fukuzawa, K. Increased formation of lysophophatidic acids by lysophospholipase D in serum of hypercholesterolemic rabbits. J. Lipid Res. 2002, 43, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Miyake, M.; Nishioka, Y.; Yamano, S.; Aono, T.; Fukuzawa, K. Production of lysophosphatidic acids by lysophospholipase D in human follicular fluids of in vitro fertilization patients. Biol. Reprod. 1999, 6, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Yamano, S.; Aono, T.; Fukuzawa, K. Lysophosphatidic acids produced by lysophospholipase D in mammalian serum and body fluid. Ann. N. Y. Acad. Sci. 2000, 905, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Gesta, S.; Simon, M.-F.; Rey, A.; Sibrac, D.; Girard, A.; Lafontan, M.; Valet, P.; Saulnier-Blache, J.S. Secretion of a lysophospholipase D activity by adipocytes: Involvement in lysophosphatidic acid synthesis. J. Lipid Res. 2002, 43, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 29436–39442. [Google Scholar] [CrossRef]
- Ohshima, N.; Kudo, T.; Yamashita, Y.; Mariggio, S.; Araki, M.; Honda, A.; Nagano, T.; Isaji, C.; Kato, N.; Corda, D.; et al. New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J. Biol. Chem. 2015, 290, 4260–4271. [Google Scholar] [CrossRef]
- Tsuboi, K.; Okamoto, Y.; Rahman, I.A.S.; Uyama, T.; Inoue, T.; Tokumura, A.; Ueda, N. Glycerophosphodiesterase GDE4 as a novel lysophospholipase D, a possible involvement in bioactive N-actylethanolamine biosynthesis. Biochim. Biophys. Acta 2015, 1851, 537–548. [Google Scholar] [CrossRef]
- Naka, K.; Ochiai, R.; Matsubara, E.; Kondo, C.; Yang, K.-M.; Hoshii, T.; Araki, M.; Araki, K.; Sotomaru, Y.; Sasaki, K.; et al. The lysophospholipase D enzyme Gdpd3 is required to maintain chronic myelogenous leukaemia stem cells. Nat. Commun. 2020, 11, 4681. [Google Scholar] [CrossRef]
- Tserendavga, B.; Ohshima, N.; Fujita, C.; Yuzawa, K.; Ohshima, M.; Yanaka, N.; Minamishima, Y.A.; Izumi, T. Characterization of recombinant murine GDE4 and GDE7, enzymes producing lysophosphatidic acid and/or cyclic phosphatidic acid. J. Biochem. 2021, 170, 713–727. [Google Scholar] [CrossRef]
- Hatoum, D.; Haddad, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphongosine kinase (SphK) isoenzymes and isoform expression: Challenges for an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef]
- Yamashita, A.; Kawagishi, N.; Miyashita, T.; Nagatsuka, T.; Sugiura, T.; Kume, K.; Shimizu, T.; Waku, K. ATP-independent fatty acyl-coenzyme A synthesis from phospholipid: Coenzyme A-dependent transacylation activity toward lysophosphatidic acid catalyzed by acyl-coenzyme A:lysophosphatidic acid acyltransferase. J. Biol. Chem. 2001, 276, 26745–26752. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Polo, F.; Borza, R.; Matsoukas, M.-T.; Marsais, F.; Jagerschmidt, C.; Waeckel, L.; Moolenaar, W.H.; Ford, P.; Heckmann, B.; Perrakis, A. Autotaxin facilitates selective LPA receptor signaling. Cell Chem. Biol. 2023, 30, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, R.; Takemoto, M.; Inoue, A.; Ishitani, R.; Nureki, O. Lateral access mechanism of LPA receptor probed by molecular dynamics simulation. PLoS ONE 2022, 17, e0263296. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Ella, K.M.; Qi, C.; McNair, A.F.; Park, J.-H.; Wisehart-Johnson, A.E.; Meier, K.E. Phospholipase D activity in PC12 cells. J. Biol. Chem. 1997, 272, 12909–12912. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, T.C.; Meier, K.E. Expression and regulation of phospholipase D isoforms in mammalian cell lines. J. Cell. Physiol. 2000, 182, 77–87. [Google Scholar] [CrossRef]
- Jones, L.G.; Ella, K.M.; Bradshaw, C.D.; Gause, K.C.; Dey, M.; Wisehart-Johnson, A.E.; Spivey, E.C.; Meier, K.E. Activations of mitogen-activated protein kinases and phospholipase D in A7r5 vascular smooth muscle cells. J. Biol. Chem. 1994, 269, 23790–23799. [Google Scholar] [CrossRef]
- Knoepp, S.M.; Wisehart-Johnson, A.E.; Buse, M.G.; Bradshaw, C.D.; Ella, K.M.; Meier, K.E. Synergistic effects of insulin and phorbol ester on mitogen-activated protein kinase in Rat-1 HIR cells. J. Biol. Chem. 1996, 371, 1678–1686. [Google Scholar] [CrossRef]
- Qi, C.; Park, J.-H.; Gibbs, T.C.; Shirley, D.W.; Bradshaw, C.D.; Ella, K.M.; Meier, K.E. Lysophosphatidic acid stimulates phospholipase D activity and cell proliferation in PC-3 human prostate cancer cells. J. Cell Physiol. 1997, 174, 261–272. [Google Scholar] [CrossRef]
- Balijepalli, P.; Guihua, Y.; Prasad, G.; Meier, K.E. Global proteomics analysis of lysophosphatidic acid signaling in PC-3 human prostate cancer cells: Role of CCN1. Int. J. Mol. Sci. 2024, in press. [Google Scholar] [CrossRef]
- Meier, K.E.; Gibbs, T.C. Phospholipase D and phosphatidylcholine metabolism. In Signal Transduction, A Practical Approach; Milligan, G., Ed.; Oxford University Press: Oxford, UK, 1999; pp. 301–320. [Google Scholar]
- Xie, Y.; Meier, K.E. Assays for phospholipase D metabolites in mammalian cells. Meth. Enzymol. 2001, 344, 294–305. [Google Scholar]
- Knoepp, S.M.; Chahal, M.S.; Xie, Y.; Zhang, Z.; Brauner, D.J.; Hallman, M.A.; Robinson, S.A.; Han, S.; Imai, M.; Tomlinson, S.; et al. Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 thymoma cells. Mol. Pharmacol. 2008, 74, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Saulnier-Blache, J.S.; Girard, A.; Simon, M.-F.; Lafontan, M.; Valet, P. A simple and highly sensitive radioenzymatic assay for lysophosphatidic acid quantification. J. Lipid Res. 2000, 41, 1947–1951. [Google Scholar] [CrossRef]
- Kume, K.; Shimizu, T. cDNA cloning and expression of murine 1-acyl-sn-glycerol-3-phosphate acyltransferase. Biochem. Biophys. Res. Commun. 1997, 237, 663–666. [Google Scholar] [CrossRef]
- Ella, K.M.; Meier, G.P.; Bradshaw, C.D.; Huffman, K.M.; Spivey, E.C.; Meier, K.E. A fluorescent assay for agonist-activated phospholipase D in mammalian cell extracts. Analyt. Biochem. 1994, 218, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Ella, K.M.; Meier, K.E.; Kumar, A.; Zhang, A.; Meier, G.P. Alcohol substrate effects in phospholipase D-mediated lipid metabolism. Biochem. Mol. Biol. Intl. 1997, 41, 715–724. [Google Scholar]
- Yamashita, A.; Sugiura, T.; Waku, K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J. Biochem. 1997, 122, 1–16. [Google Scholar] [CrossRef]
- Lio, Y.C.; Dennis, E.A. Interfacial activation, lysophospholipase and transacylase activity of Group VI Ca2+-independent phospholipase A2. Biochim. Biophys. Acta 1998, 1392, 320–332. [Google Scholar] [CrossRef]
- Ma, Z.; Turk, J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog. Nucl. Acid Res. Mol. Biol. 2001, 67, 1–33. [Google Scholar]
- Friedman, P.; Haimovitz, R.; Markman, O.; Roberts, M.F.; Shinitzky, M. Conversion of lysophospholipids to cyclic lysophosphatidic acid by phospholipase D. J. Biol. Chem. 1996, 271, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A.; Insel, P.A.; Dennis, E.A. Regulation and inhibition of phospholipase A2. Ann. Rev. Pharm. 1999, 39, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, I.D.; Nam, S.W.; Clair, T.; Jeong, E.M.; Hong, S.Y.; Han, J.W.; Lee, H.W.; Stracke, M.L.; Lee, H.Y. Enzymatic activation of autotaxin by divalent cations without EF-hand loop region involvement. Biochem. Pharm. 2001, 62, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Goding, J.W.; Terkeltaub, R.; Maurice, M.; Deterre, P.; Sali, A.; Belli, S.I. Ecto-phophodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: Structure and function of the PC-1 family. Immunol. Rev. 1998, 161, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, K.; Tsuboi, K.; Tsuda, M.; Takenouchi, Y.; Ishimaru, H.; Okamoto, Y. Development of a selective fluorescence-based enzyme assay for glycerophosphodiesterase family members GDE4 and GDE7. J. Lipid Res. 2021, 62, 100141. [Google Scholar] [CrossRef]
- Metz, S.A.; Dunlop, M. Production of phoshatidylethanol by phospholipase D phosphatidyl transferase in intact or dispersed pancreatic islets: Evidence for the in situ metabollsm of phosphatidylethanol. Arch. Biochem. Biophys. 1990, 283, 417–428. [Google Scholar] [CrossRef]
- Metz, S.A.; Dunlop, M. Inhibition of the metabolism of phosphatidylethanol and phosphatidic acid, and stimulation of insulin release, by propranolol in intact pancreatic islets. Biochem. Pharm. 1991, 41, R1–R4. [Google Scholar] [CrossRef]
- Ward, D.T.; Ohanian, J.; Heagerty, A.M.; Ohanian, V. Phospholipase D-induced phophatidate production in intact small arteries during noradrenaline stimulation: Involvement of both G-protein and tyrosine-phosphorylation-linked pathways. Biochem. J. 1995, 307, 451–456. [Google Scholar] [CrossRef]
- Moehren, G.; Gustavsson, L.; Hoek, J.B. Activation and desensitization of phospholipase D in intact rat hepatocytes. J. Biol. Chem. 1994, 269, 838–848. [Google Scholar] [CrossRef]
- Pai, J.K.; Liebl, E.C.; Tettenborn, C.S.; Ikegwuonu, F.I.; Mueller, G.C. 12-O-Tetradecanoylphorbol-13-acetate activates the synthesis of phosphatidylethanol in animal cells exposed to ethanol. Carcinogenesis 1987, 8, 173–178. [Google Scholar] [CrossRef]
- Fourcade, L.; Simon, M.-F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generate the novel lipid mediator lysophosphatidic acid in membrane vesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar] [CrossRef]
- Durieux, M.E. Lysophosphatidate Signaling: Cellular Effects and Molecular Mechanisms; R.G. Landes Co: Austin, TX, USA, 1995. [Google Scholar]
- Sonoda, H.; Aoiki, J.; Hiramatsu, T.; Ishida, M.; Bandoh, K.; Nagai, Y.; Taguchi, R.; Inoue, K.; Arai, H. A novel phosphatidic acid-selective phospholipase A1 that produces lysophosphatidic acid. J. Biol. Chem. 2002, 277, 34254–34263. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.; Vepa, S.; Shamlai, R.; Al-Hassani, M.; Ramasarma, T.; Ravishankar, H.N.; Scribner, W.M. Tyrosine kinases and calcium dependent activation of endothelial cell phospholipase D by peroxovanadate. Mol. Cell Biochem. 1998, 183, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.A.; Roberts, M.E. Vanadate is a potent competitive inhibitor of phospholipase C from Bacillus cereus. Biochem. Biophys. Acta 1996, 1298, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Hansen, L.K.; Hough, E. Crystal structures of phosphate, iodide and iodate-inhibited phospholipase C from Bacillus cereus and structural investigations of the binding of reaction products and a substrate analogue. J. Mol. Biol. 1992, 225, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fleming, N. Aluminum fluoride inhibition of cabbage phospholipase D by a phosphate-mimicking mechanism. FEBS Lett. 1999, 462, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Thomas, P.G.; Lindley, C.W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, C.; Horibata, Y.; Ando, H.; Mitsuhashi, S.; Arai, M.; Sugimoto, H. Characterization of glycerophosphodiesterase 4-interacting molecules Gαq/11 and Gβ, which mediate cellular lysophospholipase D activity. Biochem. J. 2019, 476, 3721–3736. [Google Scholar] [CrossRef]
- Kitakaze, K.; Ali, H.; Kimoto, R.; Takenouchi, Y.; Ishimaru, H.; Yamashita, A.; Ueda, N.; Tanaka, T.; Okamoto, Y.; Tsuboi, K. GDE7 produces cyclic phosphatidic acid in the ER lumen functioning as a lysophospholipid mediator. Commun. Biol. 2023, 6, 524. [Google Scholar] [CrossRef]
- Chang, P.A.; Shao, H.B.; Long, D.X.; Sun, Q.; Wu, Y.J. Isolation, characterization and molecular 3D model of human GDE4, a novel membrane protein containing glycerophosphodiester phosphodiesterase domain. Mol. Memb. Biol. 2008, 25, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Corda, D.; Mosca, M.G.; Ohshima, N.; Grauso, L.; Yanaka, N.; Mariggio, S. The emerging physiological roles of the glycerophosphodiesterase family. FEBS J. 2014, 281, 998–1016. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Ella, K.M.; Gibbs, T.C.; Yohannan, M.E.; Knoepp, S.M.; Balijepalli, P.; Meier, G.P.; Meier, K.E. Characterization of Lysophospholipase D Activity in Mammalian Cell Membranes. Cells 2024, 13, 520. https://doi.org/10.3390/cells13060520
Xie Y, Ella KM, Gibbs TC, Yohannan ME, Knoepp SM, Balijepalli P, Meier GP, Meier KE. Characterization of Lysophospholipase D Activity in Mammalian Cell Membranes. Cells. 2024; 13(6):520. https://doi.org/10.3390/cells13060520
Chicago/Turabian StyleXie, Yuhuan, Krishna M. Ella, Terra C. Gibbs, Marianne E. Yohannan, Stewart M. Knoepp, Pravita Balijepalli, G. Patrick Meier, and Kathryn E. Meier. 2024. "Characterization of Lysophospholipase D Activity in Mammalian Cell Membranes" Cells 13, no. 6: 520. https://doi.org/10.3390/cells13060520