
PROTAC-Mediated Dual Degradation of BCL-xL and BCL-2 Is a Highly Effective Therapeutic Strategy in Small-Cell Lung Cancer

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Chemical Compounds
2.3. Cell Viability Assays
2.4. Immunoblotting
2.5. Tumor Xenograft Studies
2.6. Platelet Counts
2.7. Statistical Analysis
3. Results
3.1. 753b Is a Dual BCL-xL/2 Degrader in SCLC Cells
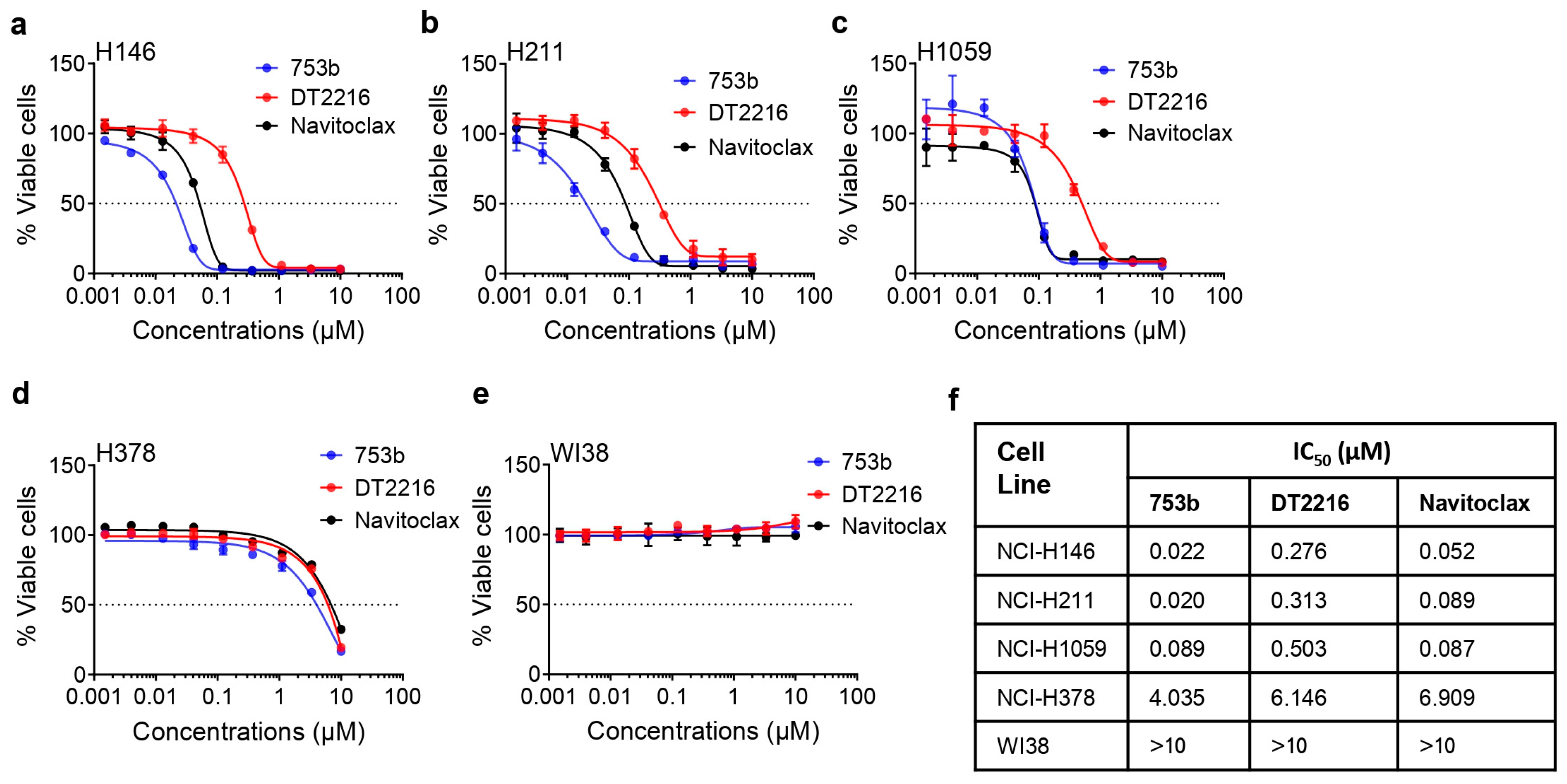
3.2. 753b Is Highly Potent to Kill BCL-xL/2-Dependent SCLC Cells
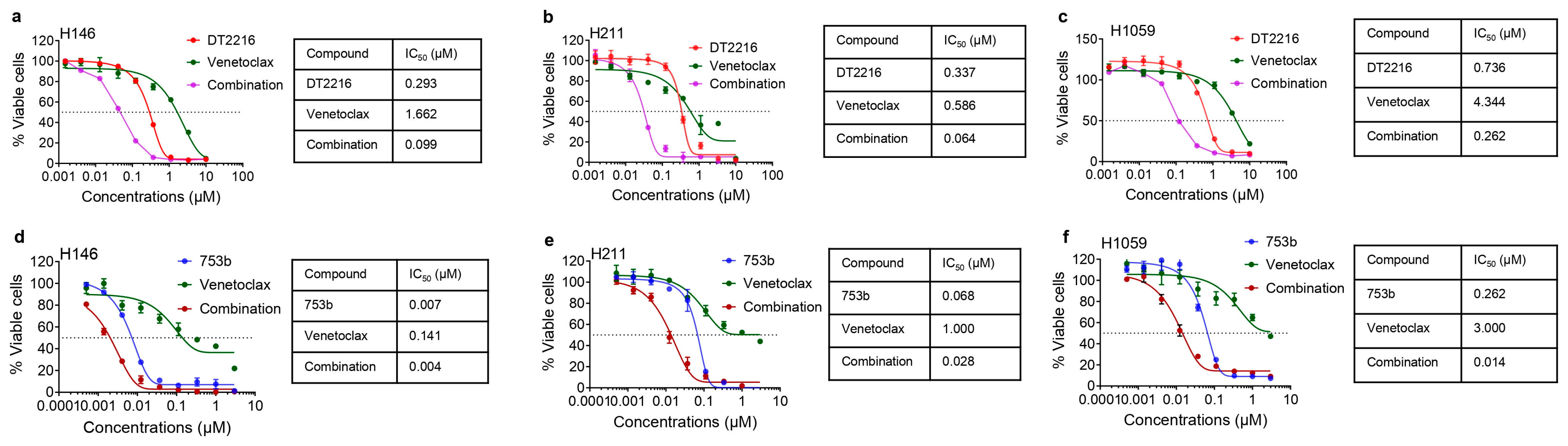
3.3. 753b Showed Similar Anti-Tumor Efficacy as DT2216 + Venetoclax by Dual BCL-xL/2 Degradation in H146 Xenograft Model
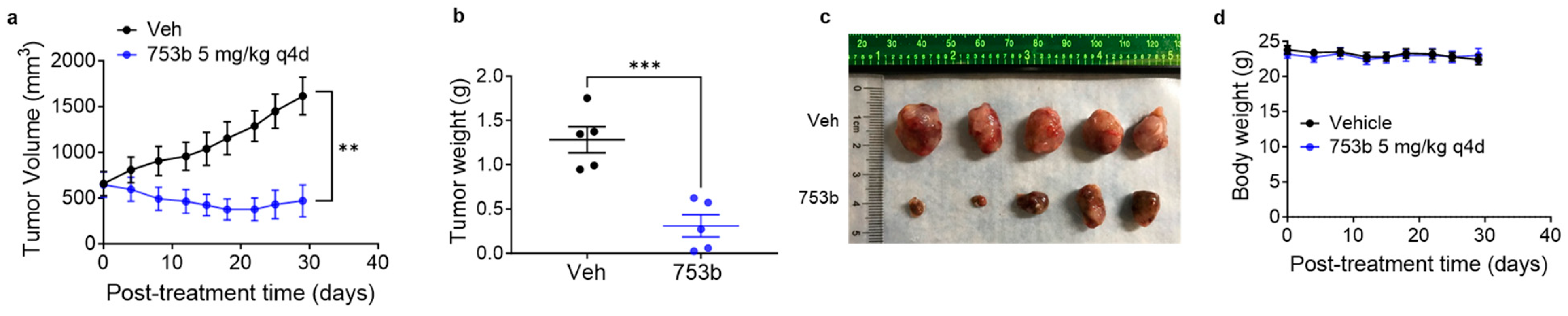
3.4. 753b Treatment Induced Tumor Regressions in H146 Xenograft Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, J.; Guan, X.; Bao, G.; Yao, Y.; Zhong, X. Molecular subtyping of small cell lung cancer. Semin. Cancer Biol. 2022, 86 Pt 2, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Megyesfalvi, Z.; Gay, C.M.; Popper, H.; Pirker, R.; Ostoros, G.; Heeke, S.; Lang, C.; Hoetzenecker, K.; Schwendenwein, A.; Boettiger, K.; et al. Clinical insights into small cell lung cancer: Tumor heterogeneity, diagnosis, therapy, and future directions. CA Cancer J. Clin. 2023, 73, 620–652. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Paz-Ares, L. Extensive-Stage Small-Cell Lung Cancer: First-Line and Second-Line Treatment Options. J. Clin. Oncol. 2022, 40, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, L.; Shah, S.; Pai-Scherf, L.; Larkins, E.; Vallejo, J.; Li, X.; Rodriguez, L.; Mishra-Kalyani, P.; Goldberg, K.B.; Kluetz, P.G.; et al. FDA Approval Summary: Atezolizumab and Durvalumab in Combination with Platinum-Based Chemotherapy in Extensive Stage Small Cell Lung Cancer. Oncologist 2021, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Lurbinectedin: First Approval. Drugs 2020, 80, 1345–1353. [Google Scholar] [CrossRef]
- Singh, S.; Jaigirdar, A.A.; Mulkey, F.; Cheng, J.; Hamed, S.S.; Li, Y.; Liu, J.; Zhao, H.; Goheer, A.; Helms, W.S.; et al. FDA Approval Summary: Lurbinectedin for the Treatment of Metastatic Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2378–2382. [Google Scholar] [CrossRef]
- Desai, A.; Smith, C.J.; Ashara, Y.; Orme, J.J.; Zanwar, S.; Potter, A.; Hocum, C.; Moffett, J.N.; Schwecke, A.J.; Manochakian, R.; et al. Real-World Outcomes with Lurbinectedin in Second-Line Setting and Beyond for Extensive Stage Small Cell Lung Cancer. Clin. Lung Cancer 2023, 24, 689–695.e1. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef]
- Shoemaker, A.R.; Mitten, M.J.; Adickes, J.; Ackler, S.; Refici, M.; Ferguson, D.; Oleksijew, A.; O’Connor, J.M.; Wang, B.; Frost, D.J.; et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin. Cancer Res. 2008, 14, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.K.; Wass, J.; Joseph, M.K.; Devanarayan, V.; Hessler, P.; Zhang, H.; Elmore, S.W.; Kroeger, P.E.; Tse, C.; Rosenberg, S.H.; et al. Identification of expression signatures predictive of sensitivity to the Bcl-2 family member inhibitor ABT-263 in small cell lung carcinoma and leukemia/lymphoma cell lines. Mol. Cancer Ther. 2010, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Faber, A.C.; Farago, A.F.; Costa, C.; Dastur, A.; Gomez-Caraballo, M.; Robbins, R.; Wagner, B.L.; Rideout, W.M.; Jakubik, C.T.; Ham, J.; et al. Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E1288–E1296. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Connis, N.; Poirier, J.T.; Cope, L.; Dobromilskaya, I.; Gallia, G.L.; Rudin, C.M.; Hann, C.L. Rapamycin rescues ABT-737 efficacy in small cell lung cancer. Cancer Res. 2014, 74, 2846–2856. [Google Scholar] [CrossRef]
- Inoue-Yamauchi, A.; Jeng, P.S.; Kim, K.; Chen, H.-C.; Han, S.; Ganesan, Y.T.; Ishizawa, K.; Jebiwott, S.; Dong, Y.; Pietanza, M.C.; et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat. Commun. 2017, 8, 16078. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Deng, K.; Han, W.; Hong, B.; Lin, W. The ratio of Bcl-2/Bim as a predictor of cisplatin response provides a rational combination of ABT-263 with cisplatin or radiation in small cell lung cancer. Cancer Biomark. 2019, 24, 51–59. [Google Scholar] [CrossRef]
- Potter, D.S.; Galvin, M.; Brown, S.; Lallo, A.; Hodgkinson, C.L.; Blackhall, F.; Morrow, C.J.; Dive, C. Inhibition of PI3K/BMX Cell Survival Pathway Sensitizes to BH3 Mimetics in SCLC. Mol. Cancer Ther. 2016, 15, 1248–1260. [Google Scholar] [CrossRef]
- Yasuda, Y.; Ozasa, H.; Kim, Y.H.; Yamazoe, M.; Ajimizu, H.; Yamamoto Funazo, T.; Nomizo, T.; Tsuji, T.; Yoshida, H.; Sakamori, Y.; et al. MCL1 inhibition is effective against a subset of small-cell lung cancer with high MCL1 and low BCL-X(L) expression. Cell Death Dis. 2020, 11, 177. [Google Scholar] [CrossRef]
- Mattoo, A.R.; FitzGerald, D.J. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int. J. Cancer 2013, 132, 978–987. [Google Scholar] [CrossRef]
- Nakajima, W.; Sharma, K.; Hicks, M.A.; Le, N.; Brown, R.; Krystal, G.W.; Harada, H. Combination with vorinostat overcomes ABT-263 (navitoclax) resistance of small cell lung cancer. Cancer Biol. Ther. 2016, 17, 27–35. [Google Scholar] [CrossRef]
- Wang, H.; Hong, B.; Li, X.; Deng, K.; Li, H.; Lui, V.W.Y.; Lin, W. JQ1 synergizes with the Bcl-2 inhibitor ABT-263 against MYCN-amplified small cell lung cancer. Oncotarget 2017, 8, 86312–86324. [Google Scholar] [CrossRef] [PubMed]
- Valko, Z.; Megyesfalvi, Z.; Schwendenwein, A.; Lang, C.; Paku, S.; Barany, N.; Ferencz, B.; Horvath-Rozsas, A.; Kovacs, I.; Schlegl, E.; et al. Dual targeting of BCL-2 and MCL-1 in the presence of BAX breaks venetoclax resistance in human small cell lung cancer. Br. J. Cancer 2023, 128, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kellish, P.; Connis, N.; Thummuri, D.; Wiegand, J.; Zhang, P.; Zhang, X.; Budamagunta, V.; Hua, N.; Yang, Y.; et al. Co-targeting BCL-X(L) and MCL-1 with DT2216 and AZD8055 synergistically inhibit small-cell lung cancer growth without causing on-target toxicities in mice. Cell Death Discov. 2023, 9, 1. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; De Oliveira, M.R.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A.; et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011, 118, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; A Iciek, L.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef]
- Negi, A.; Voisin-Chiret, A.S. Strategies to Reduce the On-Target Platelet Toxicity of Bcl-x(L) Inhibitors: PROTACs, SNIPERs and Prodrug-Based Approaches. ChemBioChem 2022, 23, e202100689. [Google Scholar] [CrossRef]
- Lakhani, N.J.; Rasco, D.; Wang, H.; Men, L.; Liang, E.; Fu, T.; Collins, M.C.; Min, P.; Yin, Y.; Davids, M.S.; et al. First-in-Human Study with Preclinical Data of BCL-2/BCL-xL Inhibitor Pelcitoclax in Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2024, 30, 506–521. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- He, Y.; Koch, R.; Budamagunta, V.; Zhang, P.; Zhang, X.; Khan, S.; Thummuri, D.; Ortiz, Y.T.; Zhang, X.; Lv, D.; et al. DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 2020, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, X.; Liu, X.; Khan, S.; Zhou, D.; Zheng, G. PROTACs are effective in addressing the platelet toxicity associated with BCL-X(L) inhibitors. Explor. Target. Antitumor. Ther. 2020, 1, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Thummuri, D.; Liu, X.; Hu, W.; Zhang, P.; Khan, S.; Yuan, Y.; Zhou, D.; Zheng, G. Discovery of PROTAC BCL-X(L) degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem. 2020, 192, 112186. [Google Scholar] [CrossRef]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Han, L.; Ramage, C.L.; Wang, Z.; Weng, C.C.; Yang, L.; Colla, S.; Ma, H.; Zhang, W.; Andreeff, M.; et al. Co-targeting BCL-XL and BCL-2 by PROTAC 753B eliminates leukemia cells and enhances efficacy of chemotherapy by targeting senescent cells. Haematologica 2023, 108, 2626–2638. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.; Hall, C.P.; Reynolds, C.P.; Kang, M.H. Caspase-dependent Mcl-1 cleavage and effect of Mcl-1 phosphorylation in ABT-737-induced apoptosis in human acute lymphoblastic leukemia cell lines. Exp. Biol. Med. 2014, 239, 1390–1402. [Google Scholar] [CrossRef]
- Wang, T.; Yang, Z.; Zhang, Y.; Zhang, X.; Wang, L.; Zhang, S.; Jia, L. Caspase cleavage of Mcl-1 impairs its anti-apoptotic activity and proteasomal degradation in non-small lung cancer cells. Apoptosis 2018, 23, 54–64. [Google Scholar] [CrossRef]
- Weng, C.; Li, Y.; Xu, D.; Shi, Y.; Tang, H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J. Biol. Chem. 2005, 280, 10491–10500. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Guerra, V.A.; DiNardo, C.; Konopleva, M. Venetoclax-based therapies for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Zengerle, M.; Testa, A.; Ciulli, A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J. Med. Chem. 2018, 61, 504–513. [Google Scholar] [CrossRef]
- Hughes, S.J.; Ciulli, A. Molecular recognition of ternary complexes: A new dimension in the structure-guided design of chemical degraders. Essays Biochem. 2017, 61, 505–516. [Google Scholar]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, L.; Cao, C.; Yu, J.; Luo, X.; Zhou, P.; Zhao, L.; Du, W.; Cheng, J.; Xie, Y.; et al. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur. J. Med. Chem. 2020, 187, 111952. [Google Scholar] [CrossRef]
- Augustyn, A.; Borromeo, M.; Wang, T.; Fujimoto, J.; Shao, C.; Dospoy, P.D.; Lee, V.; Tan, C.; Sullivan, J.P.; Larsen, J.E.; et al. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 14788–14793. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Cao, L.; Wiegand, J.; Zhang, P.; Zajac-Kaye, M.; Kaye, F.J.; Zheng, G.; Zhou, D. PROTAC-Mediated Dual Degradation of BCL-xL and BCL-2 Is a Highly Effective Therapeutic Strategy in Small-Cell Lung Cancer. Cells 2024, 13, 528. https://doi.org/10.3390/cells13060528
Khan S, Cao L, Wiegand J, Zhang P, Zajac-Kaye M, Kaye FJ, Zheng G, Zhou D. PROTAC-Mediated Dual Degradation of BCL-xL and BCL-2 Is a Highly Effective Therapeutic Strategy in Small-Cell Lung Cancer. Cells. 2024; 13(6):528. https://doi.org/10.3390/cells13060528
Chicago/Turabian StyleKhan, Sajid, Lin Cao, Janet Wiegand, Peiyi Zhang, Maria Zajac-Kaye, Frederic J. Kaye, Guangrong Zheng, and Daohong Zhou. 2024. "PROTAC-Mediated Dual Degradation of BCL-xL and BCL-2 Is a Highly Effective Therapeutic Strategy in Small-Cell Lung Cancer" Cells 13, no. 6: 528. https://doi.org/10.3390/cells13060528
APA StyleKhan, S., Cao, L., Wiegand, J., Zhang, P., Zajac-Kaye, M., Kaye, F. J., Zheng, G., & Zhou, D. (2024). PROTAC-Mediated Dual Degradation of BCL-xL and BCL-2 Is a Highly Effective Therapeutic Strategy in Small-Cell Lung Cancer. Cells, 13(6), 528. https://doi.org/10.3390/cells13060528