
Bitter Taste Receptor T2R14 and Autophagy Flux in Gingival Epithelial Cells

 , ,
, ,  and
and
Abstract
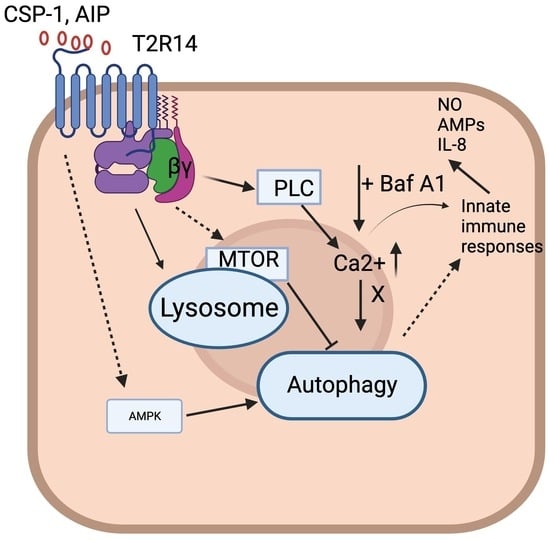
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents Used in the Study
2.2. Cell Line Used in the Study
2.3. Bacterial Strains Used in the Study
2.4. Co-Culture Assay of GECs
2.5. Transmission Electron Microscopy Imaging of GECs
2.6. Acridine Orange Acidic Vacuole Assay
2.7. Immunoblot Analysis
2.8. Intracellular Calcium Mobilization Assay
2.9. Immune Cell (dHL-60) Migration
2.10. Knockdown of ATG7
2.11. Statistical Analysis
3. Results
3.1. T2R14-Dependent Autophagy Flux in GEC
3.2. ATG7-Independent Autophagy Flux in GECs
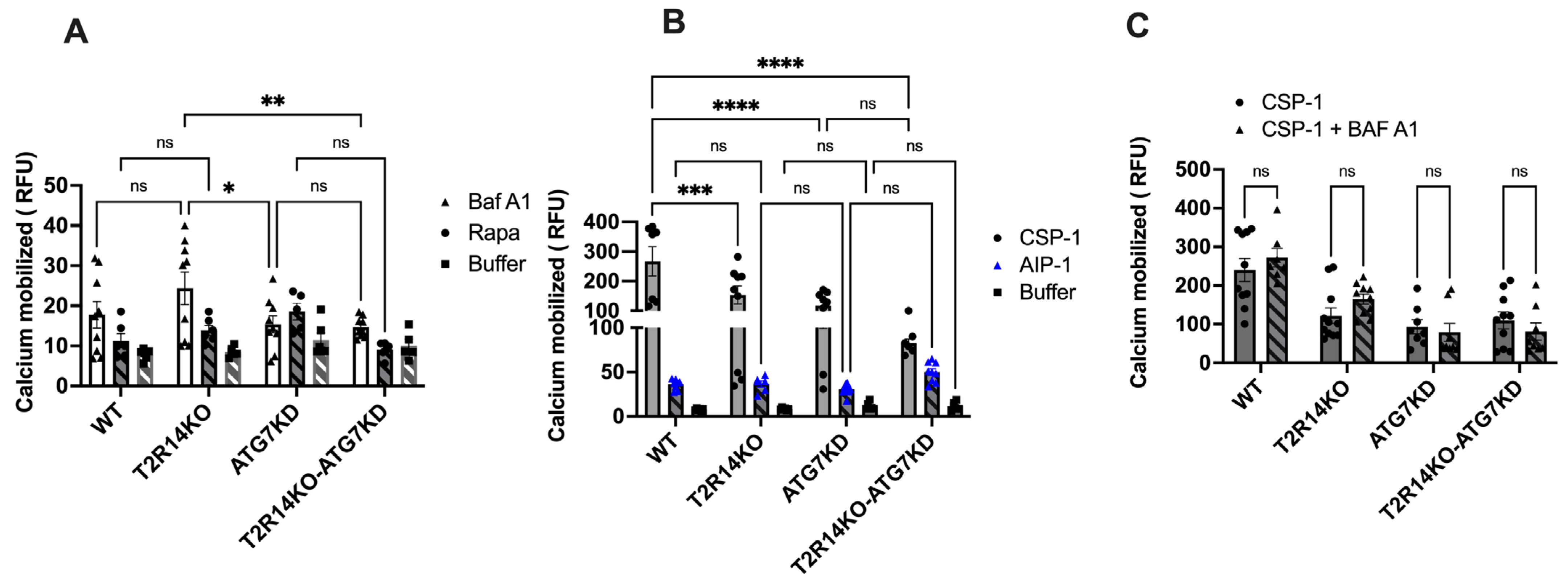
3.3. Intracellular Calcium Mobilization Assay of GECs
3.4. Immune Cell (dHL-60) Migration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wauson, E.M.; Dbouk, H.A.; Ghosh, A.B.; Cobb, M.H. G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 2014, 25, 274–282. [Google Scholar] [CrossRef]
- Behrooz, A.B.; Cordani, M.; Donadelli, M.; Ghavami, S. Metastatic outgrowth via the two-way interplay of autophagy and metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1870, 166824. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum, J.; Rollins, N.; Shah, P.; Bowman, D.; Lee, J.Y.; Tayber, O.; Bernard, H.; LeRoy, P.; Li, P.; Koenig, E.; et al. Identification of a lung cancer cell line deficient in atg7-dependent autophagy. Autophagy, 2015; Online ahead of print. [Google Scholar] [CrossRef]
- Chang, T.K.; Shravage, B.V.; Hayes, S.D.; Powers, C.M.; Simin, R.T.; Wade Harper, J.; Baehrecke, E.H. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat. Cell Biol. 2013, 15, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Corrigendum: Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2016, 533, 130. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Zhang, J.; Zhou, G. Autophagy and its implication in human oral diseases. Autophagy 2017, 13, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Morgan-Bathke, M.; Hill, G.A.; Harris, Z.I.; Lin, H.H.; Chibly, A.M.; Klein, R.R.; Burd, R.; Ann, D.K.; Limesand, K.H. Autophagy correlates with maintenance of salivary gland function following radiation. Sci. Rep. 2014, 4, 5206. [Google Scholar] [CrossRef]
- Wauson, E.M.; Zaganjor, E.; Lee, A.Y.; Guerra, M.L.; Ghosh, A.B.; Bookout, A.L.; Chambers, C.P.; Jivan, A.; McGlynn, K.; Hutchison, M.R.; et al. The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol. Cell 2012, 47, 851–862. [Google Scholar] [CrossRef]
- O’Rourke, E.J.; Kuballa, P.; Xavier, R.; Ruvkun, G. omega-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes. Dev. 2013, 27, 429–440. [Google Scholar] [CrossRef]
- Pan, S.; Sharma, P.; Shah, S.D.; Deshpande, D.A. Bitter taste receptor agonists alter mitochondrial function and induce autophagy in airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L154–L165. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Omrani, M.; Zarghooni, M.; Citi, V.; Brogi, S.; Calderone, V.; Sureda, A.; Lorzadeh, S.; da Silva Rosa, S.C.; Grabarek, B.O.; et al. New Visions on Natural Products and Cancer Therapy: Autophagy and Related Regulatory Pathways. Cancers 2022, 14, 5839. [Google Scholar] [CrossRef] [PubMed]
- Medapati, M.R.; Singh, N.; Bhagirath, A.Y.; Duan, K.; Triggs-Raine, B.; Batista, E.L., Jr.; Chelikani, P. Bitter taste receptor T2R14 detects quorum sensing molecules from cariogenic Streptococcus mutans and mediates innate immune responses in gingival epithelial cells. FASEB J. 2021, 35, e21375. [Google Scholar] [CrossRef]
- Medapati, M.R.; Bhagirath, A.Y.; Singh, N.; Schroth, R.J.; Bhullar, R.P.; Duan, K.; Chelikani, P. Bitter Taste Receptor T2R14 Modulates Gram-Positive Bacterial Internalization and Survival in Gingival Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 9920. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Dalvand, A.; da Silva Rosa, S.C.; Ghavami, S.; Marzban, H. Potential role of TGFBeta and autophagy in early crebellum development. Biochem. Biophys. Rep. 2022, 32, 101358. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef]
- McMahon, L.; Schwartz, K.; Yilmaz, O.; Brown, E.; Ryan, L.K.; Diamond, G. Vitamin D-mediated induction of innate immunity in gingival epithelial cells. Infect. Immun. 2011, 79, 2250–2256. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Singh, N.; De Jesus, V.C.; Gounni, M.S.; Dhanaraj, P.; Chelikani, P. Chemosensory bitter taste receptors (T2Rs) are activated by multiple antibiotics. FASEB J. 2019, 33, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Millius, A.; Weiner, O.D. Manipulation of neutrophil-like HL-60 cells for the study of directed cell migration. Methods Mol. Biol. 2010, 591, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Kochan, M.M.; Stewart, V.D.; Drewnik, D.A.; Hannila, S.S.; Ghavami, S. Inhibition of Autophagy Flux Promotes Secretion of Chondroitin Sulfate Proteoglycans in Primary Rat Astrocytes. Mol. Neurobiol. 2021, 58, 6077–6091. [Google Scholar] [CrossRef]
- Singh, N.; Ghavami, S.; Chelikani, P. Characterization Of Bitter Taste Receptor Dependent Autophagy in Oral Epithelial Cells. bioRxiv 2024. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed]
- Hajiahmadi, S.; Lorzadeh, S.; Iranpour, R.; Karima, S.; Rajabibazl, M.; Shahsavari, Z.; Ghavami, S. Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy. Biology 2023, 12, 302. [Google Scholar] [CrossRef]
- Pydi, S.P.; Sobotkiewicz, T.; Billakanti, R.; Bhullar, R.P.; Loewen, M.C.; Chelikani, P. Amino acid derivatives as bitter taste receptor (T2R) blockers. J. Biol. Chem. 2014, 289, 25054–25066. [Google Scholar] [CrossRef]
- Collins, S.J.; Gallo, R.C.; Gallagher, R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977, 270, 347–349. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Su, M.-W.; Cheng, Y.L.; Lin, Y.S.; Wu, J.J. Interplay between group A Streptococcus and host innate immune responses. Microbiol. Mol. Biol. Rev. 2024; e0005222, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xi, Y.; Dai, M.; Li, T.; Du, S.; Zhu, Y.; Li, M.; Li, Y.; Liu, S.; Ding, X.; et al. STING guides the STX17-SNAP29-VAMP8 complex assembly to control autophagy. Cell Insight 2024, 3, 100147. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Palmer, J.N.; Adappa, N.D.; Lee, R.J. Loss of CFTR function is associated with reduced bitter taste receptor-stimulated nitric oxide innate immune responses in nasal epithelial cells and macrophages. Front. Immunol. 2023, 14, 1096242. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef]
- Zhou, Y.W.; Sun, J.; Wang, Y.; Chen, C.P.; Tao, T.; Ma, M.; Chen, X.; Zhang, X.N.; Yang, L.Y.; Zhang, Z.L.; et al. Tas2R activation relaxes airway smooth muscle by release of Gα. Proc. Natl. Acad. Sci. USA 2022, 119, e2121513119. [Google Scholar] [CrossRef]
- Arakawa, S.; Honda, S.; Yamaguchi, H.; Shimizu, S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 378–385. [Google Scholar] [CrossRef]
- Ma, T.; Li, J.; Xu, Y.; Yu, C.; Xu, T.; Wang, H.; Liu, K.; Cao, N.; Nie, B.M.; Zhu, S.Y.; et al. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol. 2015, 17, 1379–1387. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Arakawa, S.; Kanaseki, T.; Miyatsuka, T.; Fujitani, Y.; Watada, H.; Tsujimoto, Y.; Shimizu, S. Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J. 2016, 35, 1991–2007. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Sample, A.; Shea, C.R.; Soltani, K.; Macleod, K.F.; He, Y.Y. Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis. Autophagy 2017, 13, 2086–2103. [Google Scholar] [CrossRef] [PubMed]
- Meyle, J.; Dommisch, H.; Groeger, S.; Giacaman, R.A.; Costalonga, M.; Herzberg, M. The innate host response in caries and periodontitis. J. Clin. Periodontol. 2017, 44, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Fu, V.; Hong, A.W.; Yu, F.X.; Meng, D.; Melick, C.H.; Wang, H.; Lam, W.M.; Yuan, H.X.; Taylor, S.S.; et al. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. Elife 2019, 8, e43038. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2011, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Wang, Y.C.; Zuchini, R.; Lan, K.Y.; Liu, H.S.; Lan, S.H. Secretory autophagy-promoted cargo exocytosis requires active RAB37. Autophagy, 2023; 1–2, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, M.; Dastghaib, S.; Aftabi, S.; Aghanoori, M.R.; Alizadeh, J.; Mokarram, P.; Mehrbod, P.; Ashrafizadeh, M.; Zarrabi, A.; McAlinden, K.D.; et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life 2020, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.D.; Resnik, R.; Vaccaro, M.I. Secretory Autophagy and Its Relevance in Metabolic and Degenerative Disease. Front. Endocrinol. 2020, 11, 266. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, K.K.; Patra, S.; Mishra, S.R.; Behera, B.P.; Patil, S.; Bhutia, S.K. Autophagy for secretory protein: Therapeutic targets in cancer. Adv. Protein Chem. Struct. Biol. 2023, 133, 159–180. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Ulmer, B.; Medapati, M.R.; Zhang, C.; Schroth, R.J.; Ghavami, S.; Chelikani, P. Bitter Taste Receptor T2R14 and Autophagy Flux in Gingival Epithelial Cells. Cells 2024, 13, 531. https://doi.org/10.3390/cells13060531
Singh N, Ulmer B, Medapati MR, Zhang C, Schroth RJ, Ghavami S, Chelikani P. Bitter Taste Receptor T2R14 and Autophagy Flux in Gingival Epithelial Cells. Cells. 2024; 13(6):531. https://doi.org/10.3390/cells13060531
Chicago/Turabian StyleSingh, Nisha, Ben Ulmer, Manoj Reddy Medapati, Christine Zhang, Robert J. Schroth, Saeid Ghavami, and Prashen Chelikani. 2024. "Bitter Taste Receptor T2R14 and Autophagy Flux in Gingival Epithelial Cells" Cells 13, no. 6: 531. https://doi.org/10.3390/cells13060531
APA StyleSingh, N., Ulmer, B., Medapati, M. R., Zhang, C., Schroth, R. J., Ghavami, S., & Chelikani, P. (2024). Bitter Taste Receptor T2R14 and Autophagy Flux in Gingival Epithelial Cells. Cells, 13(6), 531. https://doi.org/10.3390/cells13060531