


Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Ca2+ Imaging and IP3 Uncaging
2.3. Generation of Cell Lines with Inactivated IP3R Genes
2.3.1. Inactivation of IP3R1 Gene
2.3.2. Inactivation of IP3R2 and IP3R3 Genes
2.3.3. Generation of IP3R1-HEK, IP3R2-HEK, and IP3R3-HEK Lines
2.4. RT-PCR and RT-qPCR
2.5. Western Blot
3. Results
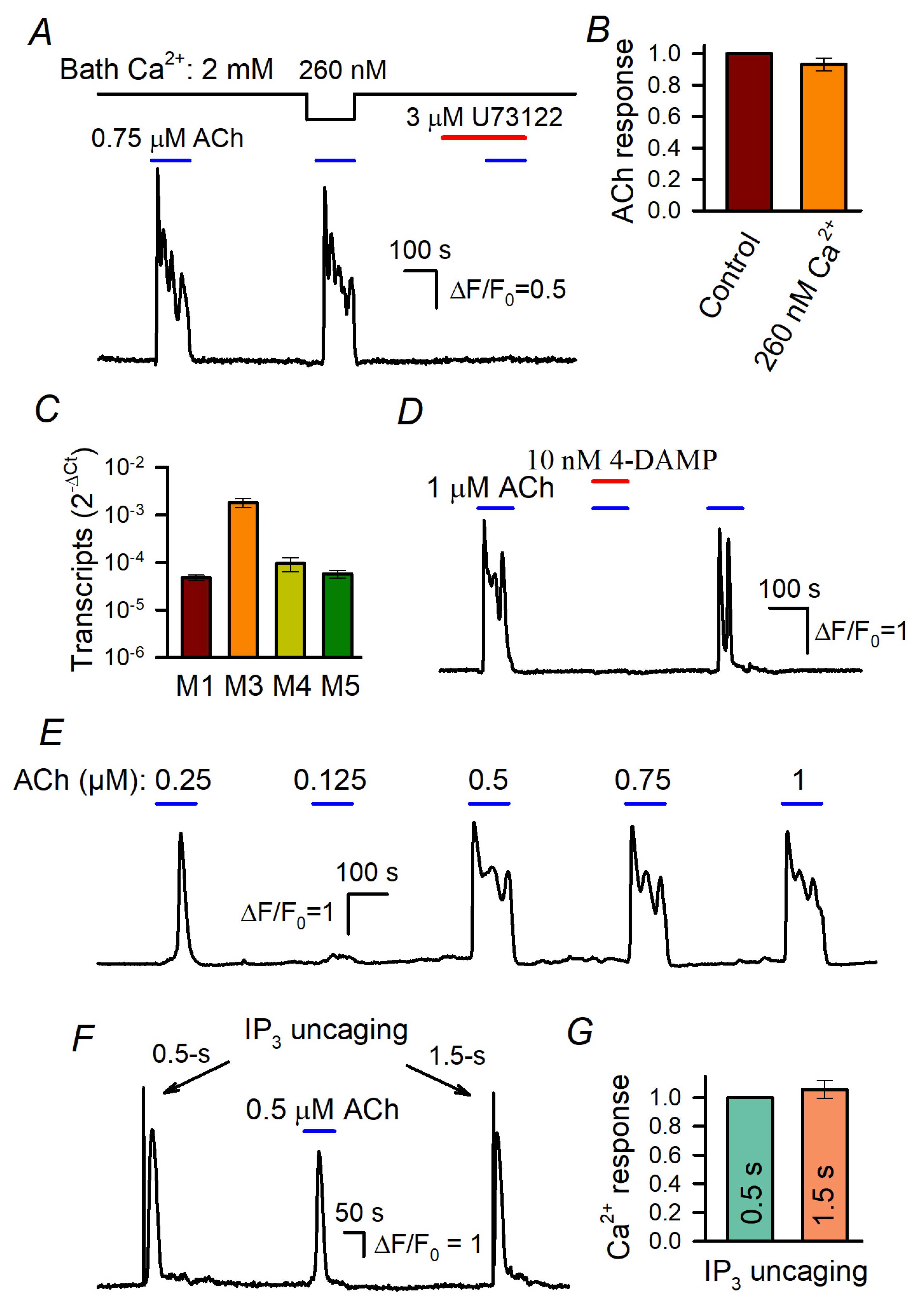
3.1. ACh Responses of WT-HEK Cells
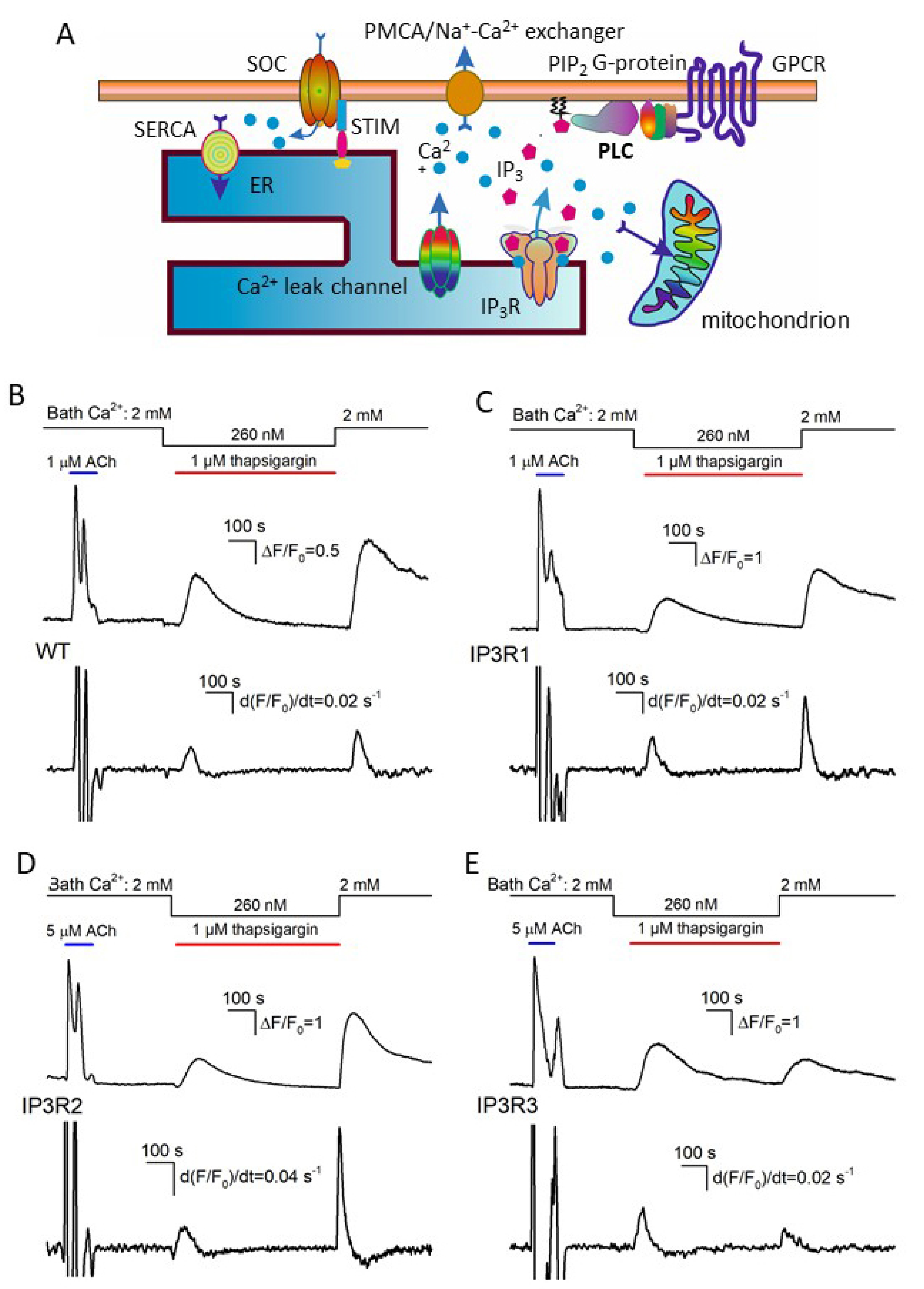
3.2. Responses of IP3R1-, IP3R2-, and IP3R3-HEK Cells to ACh
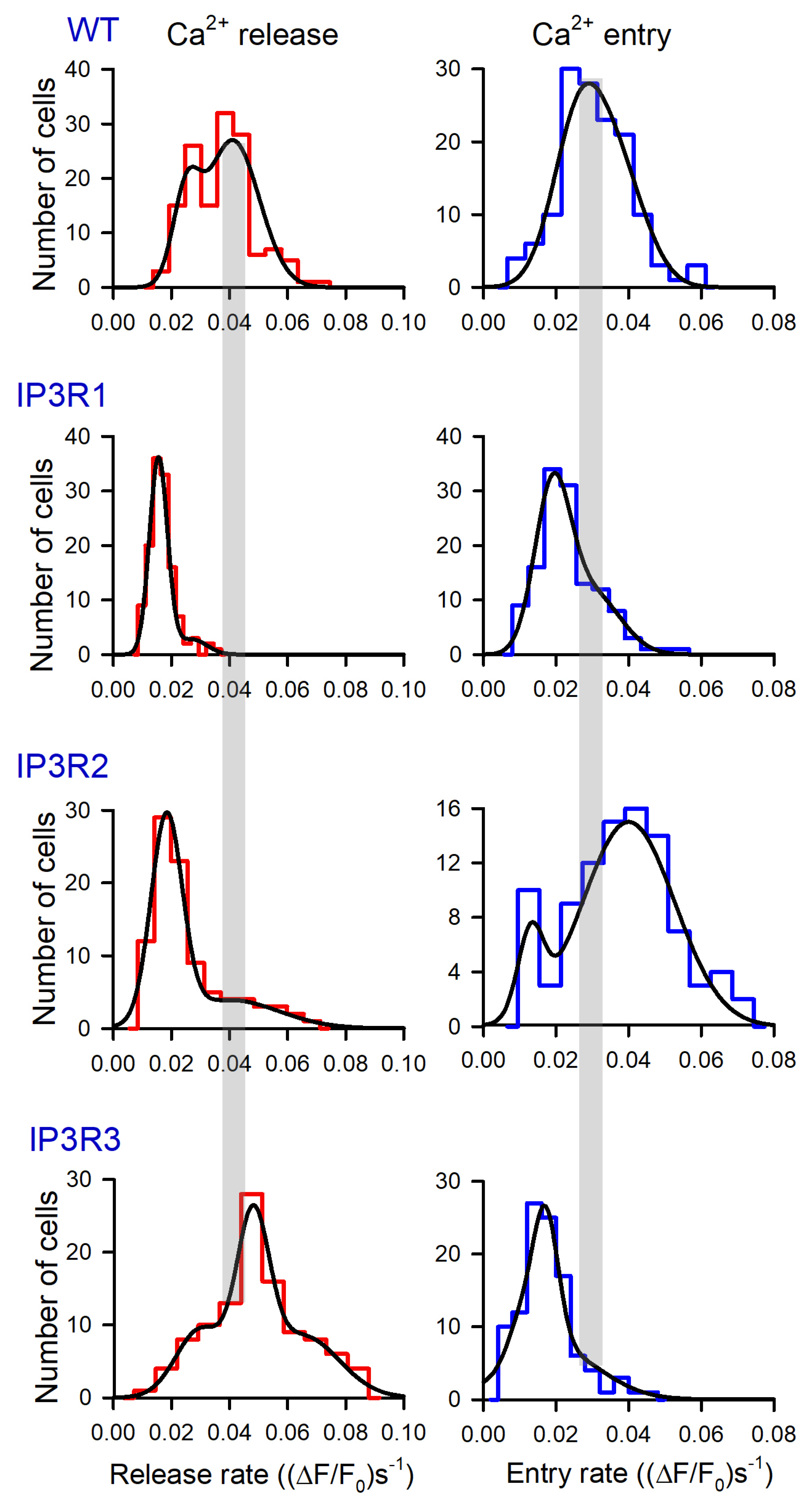
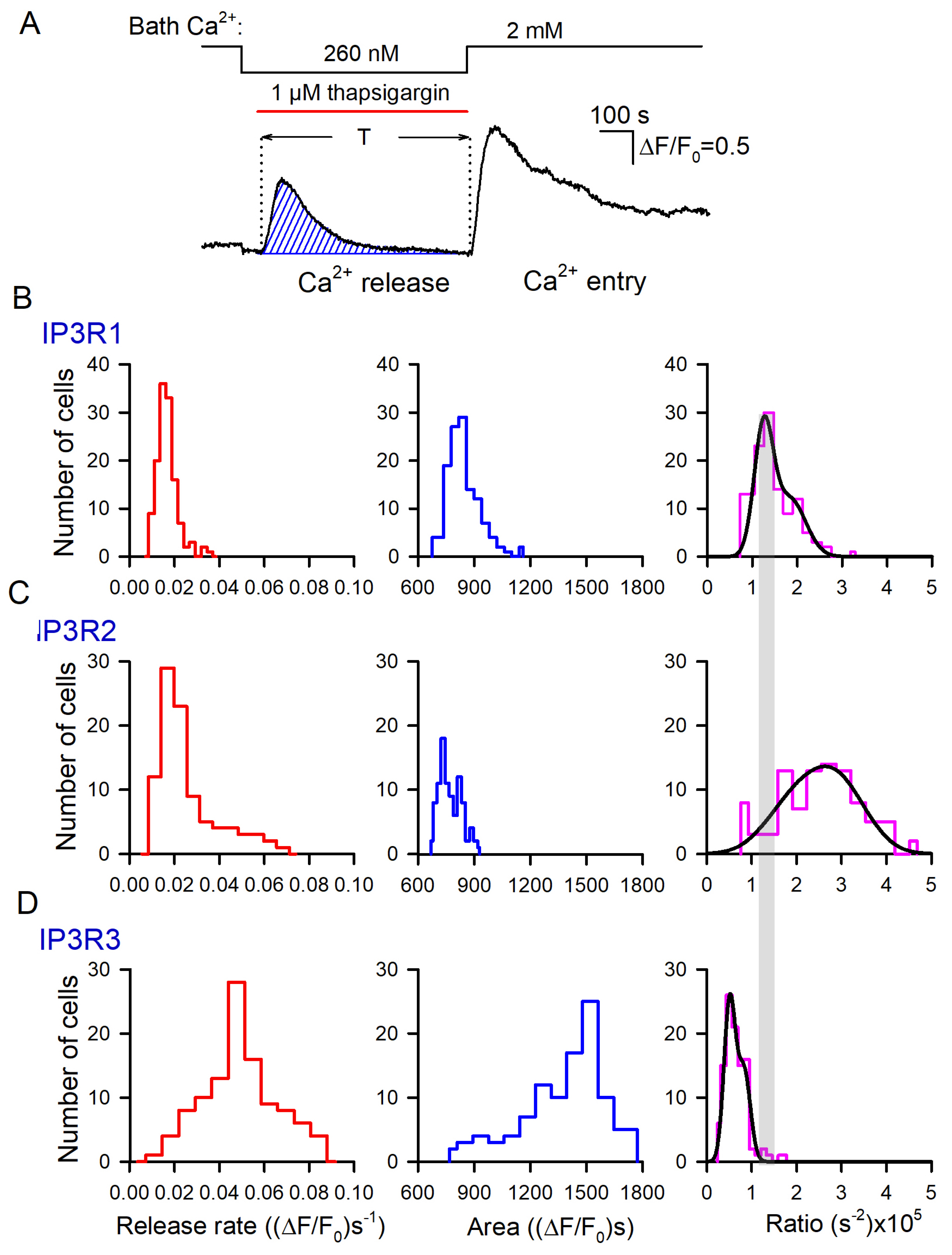
3.3. Thapsigargin Test of WT-, IP3R1-, IP3R2-, and IP3R3-HEK Cells
3.4. IP3R1-, IP3R2-, and IP3R3-HEK Cells with the Ca2+ Sensor R-CEPIA1er
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.A.; Thillaiappan, N.B.; Rossi, A.M. IP3 receptors: An “elementary” journey from structure to signals. Cell Calcium 2023, 113, 102761. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by G protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed]
- Thillaiappan, N.B.; Chakraborty, P.; Hasan, G.; Taylor, C.W. IP3 receptors and Ca2+ entry. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, R.; Alzayady, K.J.; Wagner, L.E., 2nd; Yule, D.I. Unique Regulatory Properties of Heterotetrameric Inositol 1,4,5-Trisphosphate Receptors Revealed by Studying Concatenated Receptor Constructs. J. Biol. Chem. 2016, 291, 4846–4860. [Google Scholar] [CrossRef] [PubMed]
- Wojcikiewicz, R.J.H. The making and breaking of inositol 1,4,5-trisphosphate receptor Tetramers. Messenger 2018, 6, 45–49. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Structure and function of IP3 receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef]
- Hamada, K.; Mikoshiba, K. IP3 receptor plasticity underlying diverse functions. Annu. Rev. Physiol. 2020, 82, 151–176. [Google Scholar] [CrossRef]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed]
- Lipp, P.; Laine, M.; Tovey, S.C.; Burrell, K.M.; Berridge, M.J.; Li, W.; Bootman, M.D. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr. Biol. 2000, 10, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Genazzani, A.A.; Morris, S.A. Expression of inositol trisphosphate receptors. Cell Calcium 1999, 26, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, M.A.; Abe, K.; Emori, Y. IP3 receptor type 3 and PLCbeta2 are co-expressed with taste receptors T1R and T2R in rat taste bud cells. Chem. Senses 2001, 26, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Clapp, T.R.; Stone, L.M.; Margolskee, R.F.; Kinnamon, S.C. Immunocytochemical evidence for co-expression of Type III IP3 receptor with signaling components of bitter taste transduction. BMC Neurosci. 2001, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Brann, J.H.; Dennis, J.C.; Morrison, E.E.; Fadool, D.A. Type-specific inositol 1,4,5-trisphosphate receptor localization in the vomeronasal organ and its interaction with a transient receptor potential channel, TRPC2. J. Neurochem. 2002, 83, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Parys, J.B.; Vervliet, T. New Insights in the IP3 Receptor and Its Regulation. Adv. Exp. Med. Biol. 2020, 1131, 243–270. [Google Scholar] [PubMed]
- Mak, D.O.; Foskett, J.K. Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: A single-channel point of view. Cell Calcium 2015, 58, 67–78. [Google Scholar] [CrossRef]
- Mikoshiba, K. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biol. Regul. 2015, 57, 217–227. [Google Scholar] [CrossRef]
- Wagner, L.E., 2nd; Yule, D.I. Differential regulation of the InsP₃ receptor type-1 and -2 single channel properties by InsP₃, Ca²⁺ and ATP. J. Physiol. 2012, 590, 3245–3259. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W. Regulation of IP3 receptors by cyclic AMP. Cell Calcium 2017, 63, 48–52. [Google Scholar] [CrossRef]
- Kaplin, A.I.; Snyder, S.H.; Linden, D.J. Reduced nicotinamide adenine dinucleotide-selective stimulation of inositol 1,4,5-trisphosphate receptors mediates hypoxic mobilization of calcium. J. Neurosci. 1996, 16, 2002–2011. [Google Scholar] [CrossRef]
- Worley, P.F.; Baraban, J.M.; Supattapone, S.; Wilson, V.S.; Snyder, S.H. Characterization of inositol trisphosphate receptor binding in brain. Regulation by pH and calcium. J. Biol. Chem. 1987, 262, 12132–12136. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Laude, A.J. IP3 receptors and their regulation by calmodulin and cytosolic Ca2+. Cell Calcium 2002, 32, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Mizutani, A.; Matsu-ura, T.; Mikoshiba, K. IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J. Biol. Chem. 2003, 278, 10602–10612. [Google Scholar] [CrossRef]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.X.; Allescher, H.D.; Korth, M.; Wilm, M.; et al. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 2000, 404, 197–201. [Google Scholar] [CrossRef]
- Vanderheyden, V.; Devogelaere, B.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim. Biophys. Acta 2009, 1793, 959–970. [Google Scholar] [CrossRef]
- Kotova, P.D.; Bystrova, M.F.; Rogachevskaja, O.A.; Khokhlov, A.A.; Sysoeva, V.Y.; Tkachuk, V.A.; Kolesnikov, S.S. Coupling of P2Y receptors to Ca2+ mobilization in mesenchymal stromal cells from the human adipose tissue. Cell Calcium 2018, 71, 1–14. [Google Scholar] [CrossRef]
- Kotova, P.D.; Sysoeva, V.Y.; Rogachevskaja, O.A.; Bystrova, M.F.; Kolesnikova, A.S.; Tyurin-Kuzmin, P.A.; Fadeeva, J.I.; Tkachuk, V.A.; Kolesnikov, S.S. Functional expression of adrenoreceptors in mesenchymal stromal cells derived from the human adipose tissue. Biochim. Biophys. Acta 2014, 1843, 1899–1908. [Google Scholar] [CrossRef]
- Kaimachnikov, N.P.; Kotova, P.D.; Kochkina, E.N.; Rogachevskaja, O.A.; Khokhlov, A.A.; Bystrova, M.F.; Kolesnikov, S.S. Modeling of Ca2+ transients initiated by GPCR agonists in mesenchymal stromal cells. BBA Adv. 2021, 1, 100012. [Google Scholar] [CrossRef]
- Cherkashin, A.P.; Rogachevskaja, O.A.; Kabanova, N.V.; Kotova, P.D.; Bystrova, M.F.; Kolesnikov, S.S. Taste Cells of the Type III Employ CASR to Maintain Steady Serotonin Exocytosis at Variable Ca2+ in the Extracellular Medium. Cells 2022, 11, 1369. [Google Scholar] [CrossRef]
- Sugawara, H.; Kurosaki, M.; Takata, M.; Kurosaki, T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 1997, 16, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Saleem, H.; Tovey, S.C.; Rahman, T.; Riley, A.M.; Potter, B.V.; Taylor, C.W. Stimulation of inositol 1,4,5-trisphosphate (IP3) receptor subtypes by analogues of IP3. PLoS ONE 2013, 8, e54877. [Google Scholar] [CrossRef] [PubMed]
- Alzayady, K.J.; Wang, L.; Chandrasekhar, R.; Wagner, L.E., 2nd; Van Petegem, F.; Yule, D.I. Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 2016, 9, ra35. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Hirose, M.; Mikoshiba, K. Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc. Natl. Acad. Sci. USA 2018, 115, 12259–12264. [Google Scholar] [CrossRef] [PubMed]
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef]
- Yue, L.; Wang, L.; Du, Y.; Zhang, W.; Hamada, K.; Matsumoto, Y.; Jin, X.; Zhou, Y.; Mikoshiba, K.; Gill, D.L.; et al. Type 3 Inositol 1,4,5-Trisphosphate receptor is a crucial regulator of calcium dynamics mediated by endoplasmic reticulum in HEK cells. Cells 2020, 9, 275. [Google Scholar] [CrossRef]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef]
- Chiang, T.W.; le Sage, C.; Larrieu, D.; Demir, M.; Jackson, S.P. CRISPR-Cas9(D10A) nickase-based genotypic and phenotypic screening to enhance genome editing. Sci. Rep. 2016, 6, 24356. [Google Scholar] [CrossRef] [PubMed]
- Atwood, B.K.; Lopez, J.; Wager-Miller, J.; Mackie, K.; Straiker, A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 2011, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Watson, N.; Daniels, D.V.; Ford, A.P.; Eglen, R.M.; Hegde, S.S. Comparative pharmacology of recombinant human M3 and M5 muscarinic receptors expressed in CHO-K1 cells. Br. J. Pharmacol. 1999, 127, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Mataragka, S.; Taylor, C.W. All three IP3 receptor subtypes generate Ca2+ puffs, the universal building blocks of IP3-evoked Ca2+ signals. J. Cell. Sci. 2018, 131, jcs220848. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Parker, I. IP3 mediated global Ca2+ signals arise through two temporally and spatially distinct modes of Ca2+ release. Elife 2020, 9, e55008. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihan, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef]
- Jones, L.; Ma, L.; Castro, J.; Litjens, T.; Barritt, G.J.; Rychkov, G.Y. The predominant role of IP₃ type 1 receptors in activation of store-operated Ca2+ entry in liver cells. Biochim. Biophys. Acta 2011, 1808, 745–751. [Google Scholar] [CrossRef]
- Gee, K.R.; Brown, K.A.; Chen, W.N.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Vais, H.; Wang, M.; Mallilankaraman, K.; Payne, R.; McKennan, C.; Lock, J.T.; Spruce, L.A.; Fiest, C.; Chan, M.Y.; Parker, I.; et al. ER-luminal [Ca2+] regulation of InsP3 receptor gating mediated by an ER-luminal peripheral Ca2+-binding protein. Elife 2020, 9, e53531. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Pizzo, P. Intracellular organelles in the saga of Ca2+ homeostasis: Different molecules for different purposes? Cell. Mol. Life Sci. 2012, 69, 1077–1104. [Google Scholar] [CrossRef]
- Takeshima, H.; Venturi, E.; Sitsapesan, R. New and notable ion-channels in the sarcoplasmic/endoplasmic reticulum: Do they support the process of intracellular Ca²⁺ release? J. Physiol. 2015, 593, 3241–3251. [Google Scholar] [CrossRef]
- Liu, Q. TMBIM-mediated Ca2+ homeostasis and cell death. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 850–857. [Google Scholar] [CrossRef]
- Camello, C.; Lomax, R.; Petersen, O.H.; Tepikin, A.V. Calcium leak from intracellular stores--the enigma of calcium signalling. Cell Calcium 2002, 32, 355–361. [Google Scholar] [CrossRef]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 Is an ER Ca2+ Load-Activated Ca2+ Channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef]
- Jardín, I.; López, J.J.; Salido, G.M.; Rosado, J.A. Functional relevance of the de novo coupling between hTRPC1 and type II IP3 receptor in store-operated Ca2+ entry in human platelets. Cell Signal. 2008, 20, 737–747. [Google Scholar] [CrossRef]
- Tai, K.; Hamaide, M.C.; Debaix, H.; Gailly, P.; Wibo, M.; Morel, N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. Eur. J. Pharmacol. 2008, 583, 135–147. [Google Scholar] [CrossRef]
- Ahmad, M.; Ong, H.L.; Saadi, H.; Son, G.Y.; Shokatian, Z.; Terry, L.E.; Trebak, M.; Yule, D.I.; Ambudkar, I. Functional communication between IP3R and STIM2 at subthreshold stimuli is a critical checkpoint for initiation of SOCE. Proc. Natl. Acad. Sci. USA 2022, 119, e2114928118. [Google Scholar] [CrossRef] [PubMed]
- Woodard, G.E.; López, J.J.; Jardín, I.; Salido, G.M.; Rosado, J.A. TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J. Biol. Chem. 2010, 285, 8045–8053. [Google Scholar] [CrossRef] [PubMed]
- Lur, G.; Sherwood, M.W.; Ebisui, E.; Haynes, L.; Feske, S.; Sutton, R.; Burgoyne, R.D.; Mikoshiba, K.; Petersen, O.H.; Tepikin, A.V. InsP₃ receptors and Orai channels in pancreatic acinar cells: Co-localization and its consequences. Biochem. J. 2011, 436, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Xu, X.; Mozhayeva, G.; Kuo, T.; Pessah, I.; Mignery, G.; Zhu, X.; Birnbaumer, L.; Muallem, S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 1998, 396, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Kiselyov, K.; Shin, D.M.; Chen, J.; Shcheynikov, N.; Kang, S.H.; Dehoff, M.H.; Schwarz, M.K.; Seeburg, P.H.; Muallem, S.; et al. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 2003, 114, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Kadamur, G.; Ross, E.M. Mammalian Phospholipase C. Annu. Rev. Physiol. 2013, 75, 127–154. [Google Scholar] [CrossRef]
- Kruse, A.; Kobilka, B.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef]
- Neher, E. The use of Fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology 1995, 11, 1423–1442. [Google Scholar] [CrossRef]
- Saternos, H.C.; Almarghalani, D.A.; Gibson, H.M.; Meqdad, M.A.; Antypas, R.B.; Lingireddy, A.; AbouAlaiwi, W.A. Distribution and function of the muscarinic receptor subtypes in the cardiovascular system. Physiol. Genom. 2018, 50, 1–9. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kochkina, E.N.; Kopylova, E.E.; Rogachevskaja, O.A.; Kovalenko, N.P.; Kabanova, N.V.; Kotova, P.D.; Bystrova, M.F.; Kolesnikov, S.S. Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform. Cells 2024, 13, 562. https://doi.org/10.3390/cells13070562
Kochkina EN, Kopylova EE, Rogachevskaja OA, Kovalenko NP, Kabanova NV, Kotova PD, Bystrova MF, Kolesnikov SS. Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform. Cells. 2024; 13(7):562. https://doi.org/10.3390/cells13070562
Chicago/Turabian StyleKochkina, Ekaterina N., Elizaveta E. Kopylova, Olga A. Rogachevskaja, Nina P. Kovalenko, Natalia V. Kabanova, Polina D. Kotova, Marina F. Bystrova, and Stanislav S. Kolesnikov. 2024. "Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform" Cells 13, no. 7: 562. https://doi.org/10.3390/cells13070562
APA StyleKochkina, E. N., Kopylova, E. E., Rogachevskaja, O. A., Kovalenko, N. P., Kabanova, N. V., Kotova, P. D., Bystrova, M. F., & Kolesnikov, S. S. (2024). Agonist-Induced Ca2+ Signaling in HEK-293-Derived Cells Expressing a Single IP3 Receptor Isoform. Cells, 13(7), 562. https://doi.org/10.3390/cells13070562