
LRRK2 G2019S Promotes Colon Cancer Potentially via LRRK2–GSDMD Axis-Mediated Gut Inflammation


 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DSS-Induced Acute Colitis
2.3. Disease Activity Index (DAI)
2.4. AOM/DSS-Induced Colon Cancer
2.5. LRRK2 and GSDMD Inhibitor Administration
2.6. Immunoblot
2.7. ELISA
2.8. Intestinal Permeability Analysis
2.9. Isolation of Intestinal Epithelial Cells (IECs)
2.10. Total ROS and Mitochondrial ROS Measurement
2.11. Colonic Explants
2.12. Quantitative PCR
2.13. Immunohistochemistry
2.14. Histological Analysis
2.15. Subcellular Fractionation Protocol
2.16. Statistical Analysis
3. Results
3.1. LRRK2 G2019S Promotes the Pathogenesis of Colitis-Associated Cancer
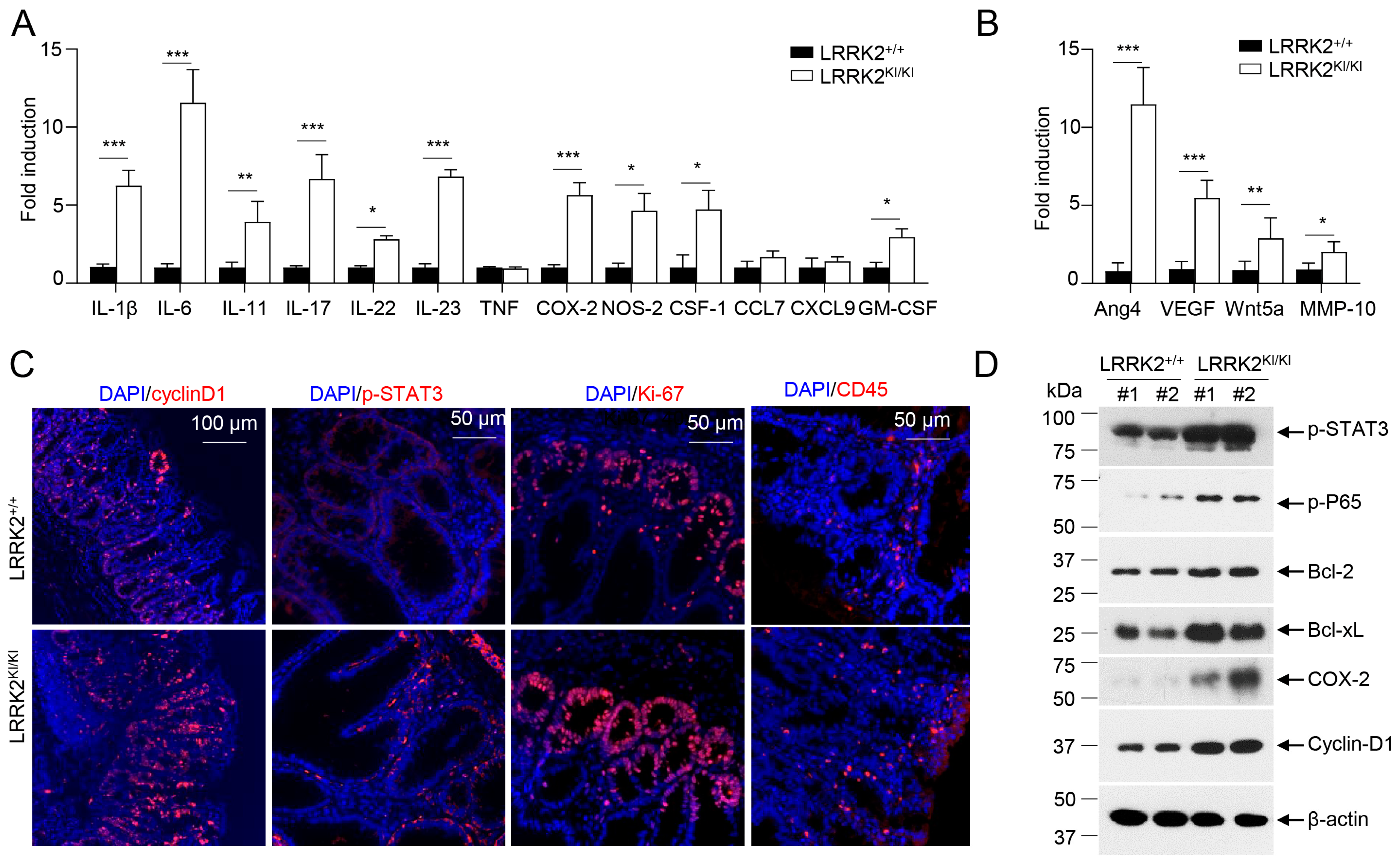
3.2. LRRK2 G2019S Promotes Inflammation and Cell Proliferation in Tumors
3.3. LRRK2 G2019S KI Mice Are Highly Susceptible to DSS-Induced Colitis
3.4. Kinase Activity of LRRK2 G2019S Is Potentially Critical for Exacerbated Colitis and CAC
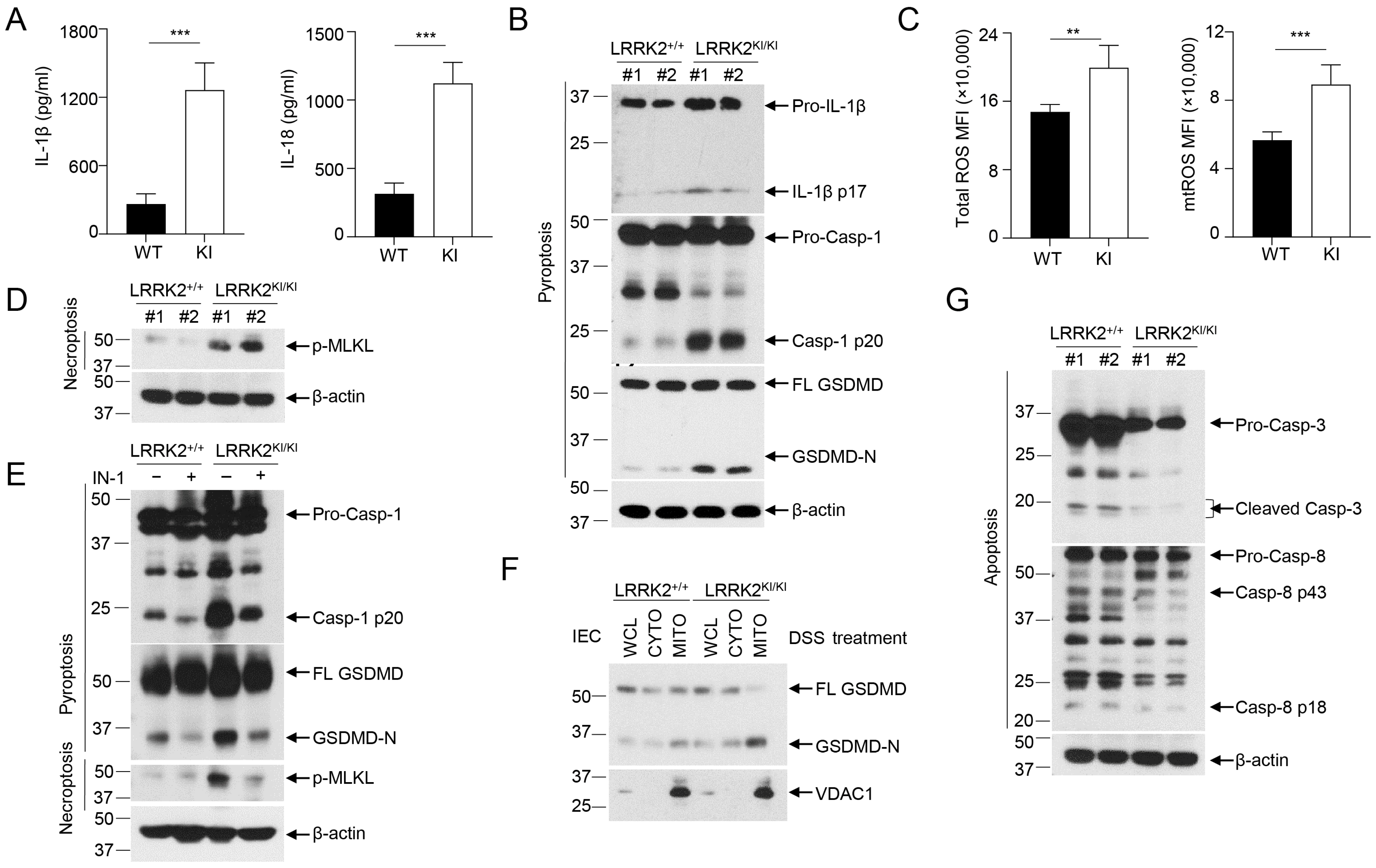
3.5. LRRK2 G2019S Promotes Inflammasome Activation and Necrosis in the Gut Epithelium
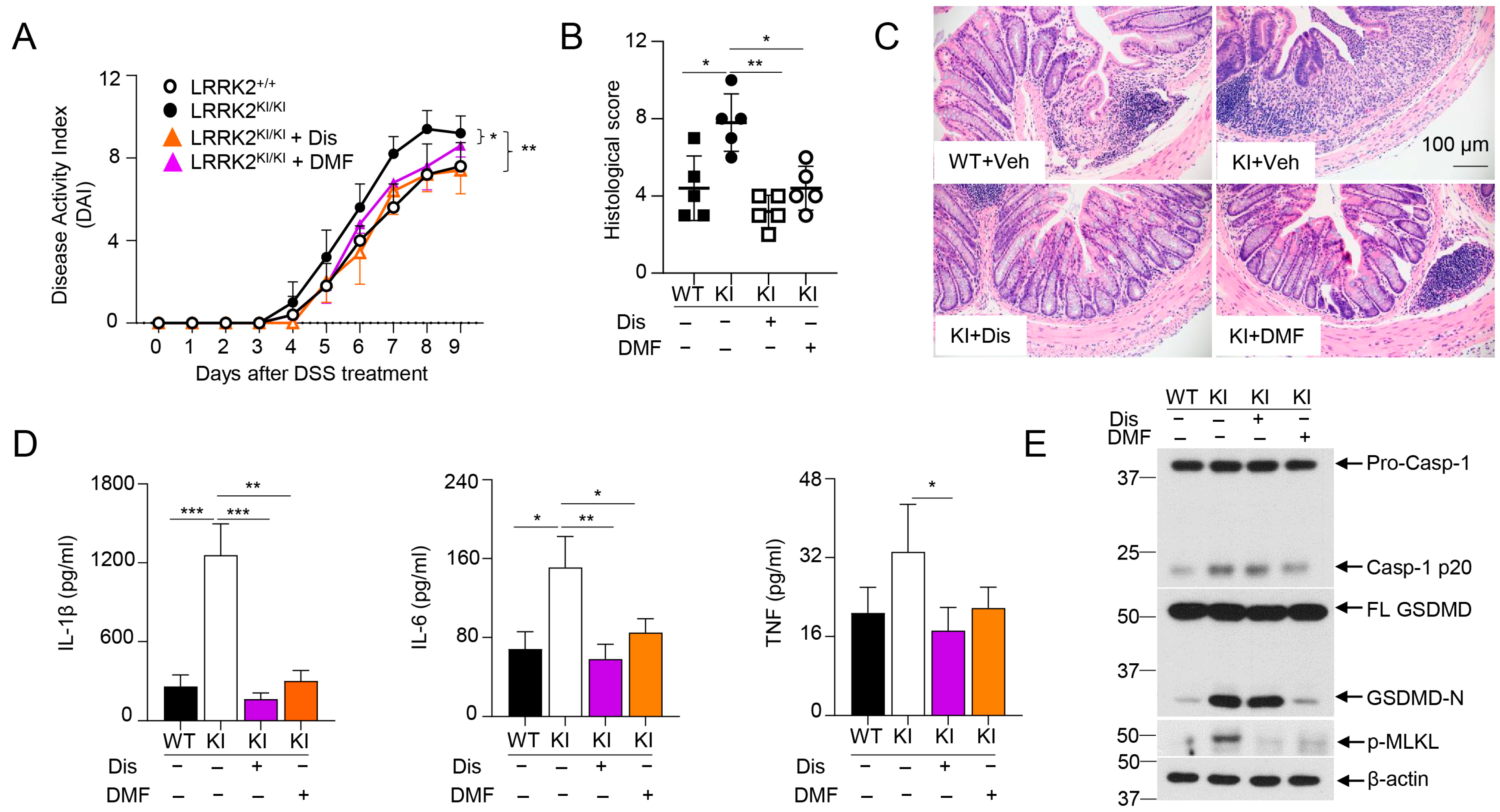
3.6. GSDMD Inhibitors Ameliorated the Severity of Colitis in LRRK2 G2019S KI Mice
3.7. LRRK2 G2019S Promotes Inflammation and Cell Proliferation during the Early Stage of Tumorigenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef]
- Usmani, A.; Shavarebi, F.; Hiniker, A. The Cell Biology of LRRK2 in Parkinson’s Disease. Mol. Cell. Biol. 2021, 41. [Google Scholar] [CrossRef]
- Erb, M.L.; Moore, D.J. LRRK2 and the Endolysosomal System in Parkinson’s Disease. J. Park. Dis. 2020, 10, 1271–1291. [Google Scholar] [CrossRef]
- Lewis, P.A. Leucine rich repeat kinase 2: A paradigm for pleiotropy. J. Physiol. 2019, 597, 3511–3521. [Google Scholar] [CrossRef]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73. [Google Scholar] [CrossRef]
- Cabezudo, D.; Baekelandt, V.; Lobbestael, E. Multiple-Hit Hypothesis in Parkinson’s Disease: LRRK2 and Inflammation. Front. Neurosci. 2020, 14, 376. [Google Scholar] [CrossRef]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson’s. Front. Neurosci. 2020, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, X.; Li, Y.; Zhao, J.; Liu, Z.; Hu, Z.; Wang, Y.; Yao, Y.; Miller, A.W.; Su, B.; et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 2017, 214, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.; Strober, W. Immunological Features of LRRK2 Function and Its Role in the Gut-Brain Axis Governing Parkinson’s Disease. J. Park. Dis. 2023, 13, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Tsafaras, G.; Baekelandt, V. The role of LRRK2 in the periphery: Link with Parkinson’s disease and inflammatory diseases. Neurobiol. Dis. 2022, 172, 105806. [Google Scholar] [CrossRef] [PubMed]
- Hulihan, M.M.; Ishihara-Paul, L.; Kachergus, J.; Warren, L.; Amouri, R.; Elango, R.; Prinjha, R.K.; Upmanyu, R.; Kefi, M.; Zouari, M.; et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: A case-control genetic study. Lancet Neurol. 2008, 7, 591–594. [Google Scholar] [CrossRef]
- Ozelius, L.J.; Senthil, G.; Saunders-Pullman, R.; Ohmann, E.; Deligtisch, A.; Tagliati, M.; Hunt, A.L.; Klein, C.; Henick, B.; Hailpern, S.M.; et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2006, 354, 424–425. [Google Scholar] [CrossRef]
- Inzelberg, R.; Cohen, O.S.; Aharon-Peretz, J.; Schlesinger, I.; Gershoni-Baruch, R.; Djaldetti, R.; Nitsan, Z.; Ephraty, L.; Tunkel, O.; Kozlova, E.; et al. The LRRK2 G2019S mutation is associated with Parkinson disease and concomitant non-skin cancers. Neurology 2012, 78, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Saunders-Pullman, R.; Barrett, M.J.; Stanley, K.M.; Luciano, M.S.; Shanker, V.; Severt, L.; Hunt, A.; Raymond, D.; Ozelius, L.J.; Bressman, S.B. LRRK2 G2019S mutations are associated with an increased cancer risk in Parkinson disease. Mov. Disord. 2010, 25, 2536–2541. [Google Scholar] [CrossRef]
- Agalliu, I.; San Luciano, M.; Mirelman, A.; Giladi, N.; Waro, B.; Aasly, J.; Inzelberg, R.; Hassin-Baer, S.; Friedman, E.; Ruiz-Martinez, J.; et al. Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: A pooled analysis. JAMA Neurol. 2015, 72, 58–65. [Google Scholar] [CrossRef]
- Agalliu, I.; Ortega, R.A.; Luciano, M.S.; Mirelman, A.; Pont-Sunyer, C.; Brockmann, K.; Vilas, D.; Tolosa, E.; Berg, D.; Waro, B.; et al. Cancer outcomes among Parkinson’s disease patients with leucine rich repeat kinase 2 mutations, idiopathic Parkinson’s disease patients, and nonaffected controls. Mov. Disord. 2019, 34, 1392–1398. [Google Scholar] [CrossRef]
- Lee, J.Y.S.; Ng, J.H.; Saffari, S.E.; Tan, E.K. Parkinson’s disease and cancer: A systematic review and meta-analysis on the influence of lifestyle habits, genetic variants, and gender. Aging 2022, 14, 2148–2173. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Olen, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sorensen, H.T.; Ludvigsson, J.F. Colorectal cancer in Crohn’s disease: A Scandinavian population-based cohort study. Lancet Gastroenterol. Hepatol. 2020, 5, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, I.S.; O’Toole, A.; Deasy, J.; McNamara, D.A.; Burke, J.P. A meta-analysis of the clinicopathological characteristics and survival outcomes of inflammatory bowel disease associated colorectal cancer. Int. J. Color. Dis. 2017, 32, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Dugum, M.; Lin, J.; Lopez, R.; Estfan, B.; Manilich, E.; Stocchi, L.; Shen, B.; Liu, X. Recurrence and survival rates of inflammatory bowel disease-associated colorectal cancer following postoperative chemotherapy: A comparative study. Gastroenterol. Rep. 2017, 5, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.V.; Ahnen, D.J.; Baron, J.A.; Campbell, P.T.; Gallinger, S.; Grady, W.M.; LeMarchand, L.; Lindor, N.M.; Potter, J.D.; Newcomb, P.A. Survival after inflammatory bowel disease-associated colorectal cancer in the Colon Cancer Family Registry. World J. Gastroenterol. 2013, 19, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Thasler, W.E.; Preissler, G.; Heide, T.; Khalil, P.N.; Mikhailov, M.; Jauch, K.W.; Kreis, M.E.; Rentsch, M.; Kleespies, A. Clinical outcome of IBD-associated versus sporadic colorectal cancer: A matched-pair analysis. J. Gastrointest. Surg. 2013, 17, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Greten, F.R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- Neufert, C.; Becker, C.; Neurath, M.F. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007, 2, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Kohno, H.; Sugie, S.; Nakagama, H.; Tanaka, T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2006, 27, 162–169. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Shutinoski, B.; Hakimi, M.; Harmsen, I.E.; Lunn, M.; Rocha, J.; Lengacher, N.; Zhou, Y.Y.; Khan, J.; Nguyen, A.; Hake-Volling, Q.; et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.A.; Kannarkat, G.T.; Cintron, A.F.; Butkovich, L.M.; Fraser, K.B.; Chang, J.; Grigoryan, N.; Factor, S.A.; West, A.B.; Boss, J.M.; et al. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef]
- Nichols, W.C.; Pankratz, N.; Hernandez, D.; Paisan-Ruiz, C.; Jain, S.; Halter, C.A.; Michaels, V.E.; Reed, T.; Rudolph, A.; Shults, C.W.; et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005, 365, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Neufert, C.; Weigmann, B.; Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2007, 2, 541–546. [Google Scholar] [CrossRef]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, e3678. [Google Scholar] [CrossRef]
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. Methods Mol. Biol. 2016, 1422, 297–307. [Google Scholar] [CrossRef]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Peter, I.; Tajima, M.; Nakamura, S.; Strober, W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Mahe, M.M.; Aihara, E.; Schumacher, M.A.; Zavros, Y.; Montrose, M.H.; Helmrath, M.A.; Sato, T.; Shroyer, N.F. Establishment of Gastrointestinal Epithelial Organoids. Curr. Protoc. Mouse Biol. 2013, 3, 217–240. [Google Scholar] [CrossRef]
- Cui, H.; Cai, Y.; Wang, L.; Jia, B.; Li, J.; Zhao, S.; Chu, X.; Lin, J.; Zhang, X.; Bian, Y.; et al. Berberine Regulates Treg/Th17 Balance to Treat Ulcerative Colitis Through Modulating the Gut Microbiota in the Colon. Front. Pharmacol. 2018, 9, 571. [Google Scholar] [CrossRef]
- Kihara, N.; de la Fuente, S.G.; Fujino, K.; Takahashi, T.; Pappas, T.N.; Mantyh, C.R. Vanilloid receptor-1 containing primary sensory neurones mediate dextran sulphate sodium induced colitis in rats. Gut 2003, 52, 713–719. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Xiao, H.; Gulen, M.F.; Qin, J.; Yao, J.; Bulek, K.; Kish, D.; Altuntas, C.Z.; Wald, D.; Ma, C.; Zhou, H.; et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007, 26, 461–475. [Google Scholar] [CrossRef]
- Parikh, A.A.; Salzman, A.L.; Kane, C.D.; Fischer, J.E.; Hasselgren, P.O. IL-6 production in human intestinal epithelial cells following stimulation with IL-1 beta is associated with activation of the transcription factor NF-kappa B. J. Surg. Res. 1997, 69, 139–144. [Google Scholar] [CrossRef]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Role of prostanoids in gastrointestinal cancer. J. Clin. Investig. 2018, 128, 2732–2742. [Google Scholar] [CrossRef]
- Sancho, E.; Batlle, E.; Clevers, H. Signaling pathways in intestinal development and cancer. Annu. Rev. Cell Dev. Biol. 2004, 20, 695–723. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Lei-Leston, A.C.; Murphy, A.G.; Maloy, K.J. Epithelial Cell Inflammasomes in Intestinal Immunity and Inflammation. Front. Immunol. 2017, 8, 1168. [Google Scholar] [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 2022, 185, 3214–3231.e23. [Google Scholar] [CrossRef]
- Bulek, K.; Zhao, J.; Liao, Y.; Rana, N.; Corridoni, D.; Antanaviciute, A.; Chen, X.; Wang, H.; Qian, W.; Miller-Little, W.A.; et al. Epithelial-derived gasdermin D mediates nonlytic IL-1beta release during experimental colitis. J. Clin. Investig. 2020, 130, 4218–4234. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrin, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef]
- Luerman, G.C.; Nguyen, C.; Samaroo, H.; Loos, P.; Xi, H.; Hurtado-Lorenzo, A.; Needle, E.; Stephen Noell, G.; Galatsis, P.; Dunlop, J.; et al. Phosphoproteomic evaluation of pharmacological inhibition of leucine-rich repeat kinase 2 reveals significant off-target effects of LRRK-2-IN-1. J. Neurochem. 2014, 128, 561–576. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef]
- Derkinderen, P.; de Guilhem de Lataillade, A.; Neunlist, M.; Rolli-Derkinderen, M. Mild Chronic Colitis Triggers Parkinsonism in LRRK2 Mutant Mice through Activating TNF-alpha Pathway. Mov. Disord. 2022, 37, 664–665. [Google Scholar] [CrossRef]
- Schonig, K.; Schwenk, F.; Rajewsky, K.; Bujard, H. Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res. 2002, 30, e134. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, J.Z.; Sakaguchi, T.; Maretzky, T.; Gurung, P.; Short, S.; Xiong, Y.; Kang, Z. LRRK2 G2019S promotes the development of colon cancer via modulating intestinal inflammation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cabezudo, D.; Tsafaras, G.; Van Acker, E.; Van den Haute, C.; Baekelandt, V. Mutant LRRK2 exacerbates immune response and neurodegeneration in a chronic model of experimental colitis. Acta Neuropathol. 2023, 146, 245–261. [Google Scholar] [CrossRef]
- Illig, D.; Kotlarz, D. Dysregulated inflammasome activity in intestinal inflammation-Insights from patients with very early onset IBD. Front. Immunol. 2022, 13, 1027289. [Google Scholar] [CrossRef]
- Gao, S.J.; Zhang, L.; Lu, W.; Wang, L.; Chen, L.; Zhu, Z.; Zhu, H.H. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to Crohn’s disease. World J. Gastroenterol. 2015, 21, 8711–8722. [Google Scholar] [CrossRef]
- Schoultz, I.; Verma, D.; Halfvarsson, J.; Torkvist, L.; Fredrikson, M.; Sjoqvist, U.; Lordal, M.; Tysk, C.; Lerm, M.; Soderkvist, P.; et al. Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn’s disease in Swedish men. Am. J. Gastroenterol. 2009, 104, 1180–1188. [Google Scholar] [CrossRef]
- Villani, A.C.; Lemire, M.; Fortin, G.; Louis, E.; Silverberg, M.S.; Collette, C.; Baba, N.; Libioulle, C.; Belaiche, J.; Bitton, A.; et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 2009, 41, 71–76. [Google Scholar] [CrossRef]
- Satsangi, J.; Wolstencroft, R.A.; Cason, J.; Ainley, C.C.; Dumonde, D.C.; Thompson, R.P. Interleukin 1 in Crohn’s disease. Clin. Exp. Immunol. 1987, 67, 594–605. [Google Scholar]
- McAlindon, M.E.; Hawkey, C.J.; Mahida, Y.R. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 1998, 42, 214–219. [Google Scholar] [CrossRef]
- Mahida, Y.R.; Wu, K.; Jewell, D.P. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut 1989, 30, 835–838. [Google Scholar] [CrossRef]
- Casini-Raggi, V.; Kam, L.; Chong, Y.J.; Fiocchi, C.; Pizarro, T.T.; Cominelli, F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J. Immunol. 1995, 154, 2434–2440. [Google Scholar] [CrossRef]
- Ludwiczek, O.; Vannier, E.; Borggraefe, I.; Kaser, A.; Siegmund, B.; Dinarello, C.A.; Tilg, H. Imbalance between interleukin-1 agonists and antagonists: Relationship to severity of inflammatory bowel disease. Clin. Exp. Immunol. 2004, 138, 323–329. [Google Scholar] [CrossRef]
- Mitsialis, V.; Wall, S.; Liu, P.; Ordovas-Montanes, J.; Parmet, T.; Vukovic, M.; Spencer, D.; Field, M.; McCourt, C.; Toothaker, J.; et al. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology 2020, 159, 591–608.e10. [Google Scholar] [CrossRef]
- Hedl, M.; Zheng, S.; Abraham, C. The IL18RAP region disease polymorphism decreases IL-18RAP/IL-18R1/IL-1R1 expression and signaling through innate receptor-initiated pathways. J. Immunol. 2014, 192, 5924–5932. [Google Scholar] [CrossRef]
- Imielinski, M.; Baldassano, R.N.; Griffiths, A.; Russell, R.K.; Annese, V.; Dubinsky, M.; Kugathasan, S.; Bradfield, J.P.; Walters, T.D.; Sleiman, P.; et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009, 41, 1335–1340. [Google Scholar] [CrossRef]
- Barnett, K.C.; Li, S.; Liang, K.; Ting, J.P. A 360 degrees view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell 2023, 186, 2288–2312. [Google Scholar] [CrossRef]
- Chen, W.; Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.D. Innate immune inflammatory cell death: PANoptosis and PANoptosomes in host defense and disease. Eur. J. Immunol. 2023, 53, e2250235. [Google Scholar] [CrossRef]
- Pierdomenico, M.; Negroni, A.; Stronati, L.; Vitali, R.; Prete, E.; Bertin, J.; Gough, P.J.; Aloi, M.; Cucchiara, S. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am. J. Gastroenterol. 2014, 109, 279–287. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef]
- Parker, A.; Vaux, L.; Patterson, A.M.; Modasia, A.; Muraro, D.; Fletcher, A.G.; Byrne, H.M.; Maini, P.K.; Watson, A.J.M.; Pin, C. Elevated apoptosis impairs epithelial cell turnover and shortens villi in TNF-driven intestinal inflammation. Cell Death Dis. 2019, 10, 108. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef]
- Chan, F.K. Fueling the flames: Mammalian programmed necrosis in inflammatory diseases. Cold Spring Harb. Perspect. Biol. 2012, 4, a008805. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, Y.; Yao, J.; Li, D.F.; Wang, L.S. Role of Pyroptosis in Inflammatory Bowel Disease (IBD): From Gasdermins to DAMPs. Front. Pharmacol. 2022, 13, 833588. [Google Scholar] [CrossRef]
- Canli, O.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Lower, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host-microbiota maladaptation in colorectal cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Dorr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Gao, J.Z.; Sakaguchi, T.; Maretzky, T.; Gurung, P.; Narayanan, N.S.; Short, S.; Xiong, Y.; Kang, Z. LRRK2 G2019S Promotes Colon Cancer Potentially via LRRK2–GSDMD Axis-Mediated Gut Inflammation. Cells 2024, 13, 565. https://doi.org/10.3390/cells13070565
Wang Y, Gao JZ, Sakaguchi T, Maretzky T, Gurung P, Narayanan NS, Short S, Xiong Y, Kang Z. LRRK2 G2019S Promotes Colon Cancer Potentially via LRRK2–GSDMD Axis-Mediated Gut Inflammation. Cells. 2024; 13(7):565. https://doi.org/10.3390/cells13070565
Chicago/Turabian StyleWang, Yuhang, Joyce Z. Gao, Taylor Sakaguchi, Thorsten Maretzky, Prajwal Gurung, Nandakumar S. Narayanan, Sarah Short, Yiqin Xiong, and Zizhen Kang. 2024. "LRRK2 G2019S Promotes Colon Cancer Potentially via LRRK2–GSDMD Axis-Mediated Gut Inflammation" Cells 13, no. 7: 565. https://doi.org/10.3390/cells13070565
APA StyleWang, Y., Gao, J. Z., Sakaguchi, T., Maretzky, T., Gurung, P., Narayanan, N. S., Short, S., Xiong, Y., & Kang, Z. (2024). LRRK2 G2019S Promotes Colon Cancer Potentially via LRRK2–GSDMD Axis-Mediated Gut Inflammation. Cells, 13(7), 565. https://doi.org/10.3390/cells13070565