
Spotlight on New Hallmarks of Drug-Resistance towards Personalized Care for Epithelial Ovarian Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. What Is the Roadmap of Multilayered Heterogeneity in EOC?
3.1.1. Clinical and Histopathological Heterogeneity of EOC across the Main Subtypes
3.1.2. Developmental Heterogeneity of EOC
3.1.3. Cellular Heterogeneity of EOC
3.1.4. Microenvironmental Heterogeneity in EOC
3.1.5. Heterogeneity of Molecular Milieu in EOC
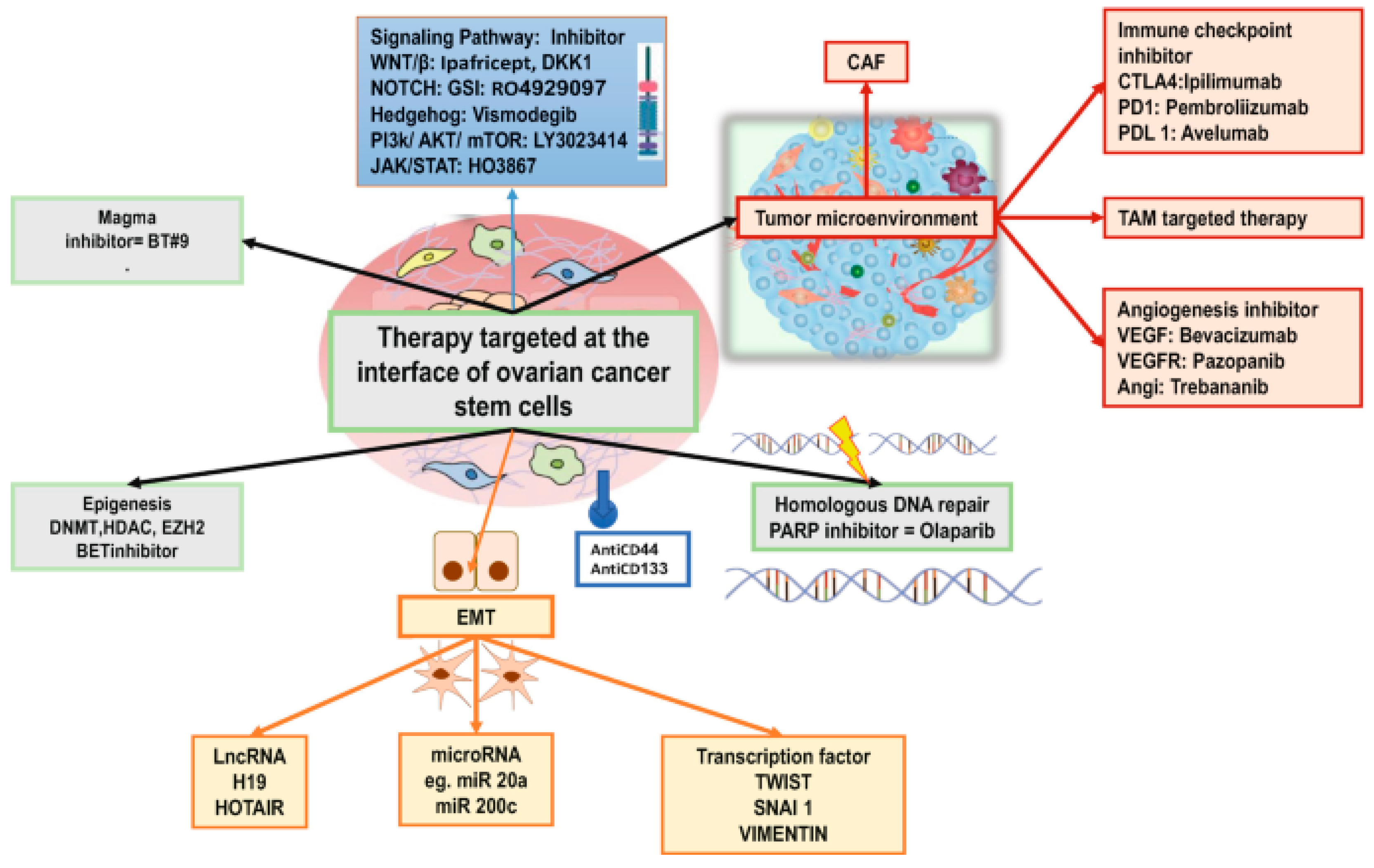
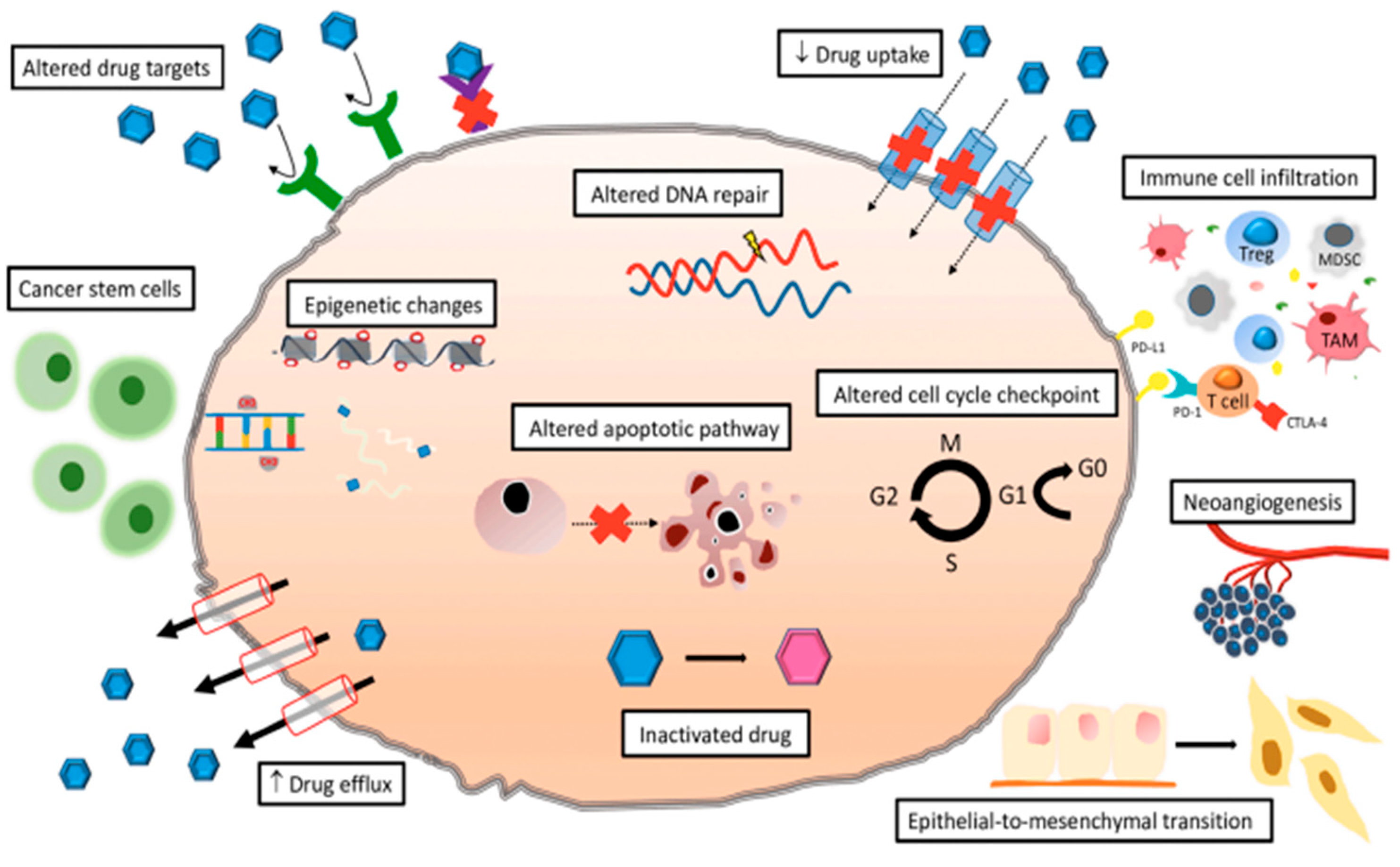
3.2. Any Viable Options and Challenges in Targeting Drug Resistant CSCs?
3.3. Are There Any Candidate Biomarkers for Prognosis and Outcome Prediction?
3.3.1. miRNA, Exosomes, and Chemoresistance
3.3.2. Liquid Biopsy-Based Biomarkers in EOC
4. Discussion and Future Challenges
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teeuwssen, M.; Fodde, R. Wnt signaling in ovarian cancer stemness, EMT, and therapy resistance. J. Clin. Med. 2019, 8, 1658. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Matias-Guiu, X.; Amant, F.; Concin, N.; Davidson, B.; Fotopoulou, C.; González-Martin, A.; Gourley, C.; Leary, A.; Lorusso, D.; et al. ESGO–ESMO–ESP consensus conference recommendations on ovarian cancer: Pathology and molecular biology and early, advanced, and recurrent disease. Ann. Oncol. 2024, 35, 248–266. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Ovarian cancer including fallopian tube cancer and primary peritoneal cancer. In NCCN Practice Guidelines; Version 1; NCCN: Plymouth Meeting, PA, USA, 2023; p. 19462. [Google Scholar]
- O’Malley, D.M.; Krivak, T.C.; Kabil, N.; Munley, J.; Moore, K.N. PARP inhibitors in ovarian cancer: A review. Target. Oncol. 2023, 18, 471–503. [Google Scholar] [CrossRef]
- Coughlan, A.Y.; Testa, G. Exploiting epigenetic dependencies in ovarian cancer therapy. Int. J. Cancer 2021, 149, 1732–1743. [Google Scholar] [CrossRef]
- Marchetti, C.; De Felice, F.; Romito, A.; Iacobelli, V.; Sassu, C.M.; Corrado, G.; Ricci, C.; Scambia, G.; Fagotti, A. Chemotherapy resistance in epithelial ovarian cancer: Mechanisms and emerging treatments. Semin. Cancer Biol. 2021, 77, 144–166. [Google Scholar] [CrossRef]
- Roy, L.; Cowden Dahl, K. Can stemness and chemoresistance be therapeutically targeted via signaling pathways in ovarian cancer? Cancers 2018, 10, 241. [Google Scholar] [CrossRef]
- Hatina, J.; Boesch, M.; Sopper, S.; Kripnerova, M.; Wolf, D.; Reimer, D.; Marth, C.; Zeimet, A.G. Ovarian cancer stem cell heterogeneity. In Stem Cells Heterogeneity in Cancer; Birbrair, A., Ed.; Springer Nature: Cham, Switzerland, 2019; pp. 201–216. [Google Scholar]
- Lee, M.; Chang, M.Y.; Yoo, H.; Lee, K.E.; Chay, D.B.; Cho, H.; Kim, S.; Kim, Y.T.; Kim, J.-H. Clinical significance of CA125 level after the first cycle of chemotherapy on survival of patients with advanced ovarian cancer. Yonsei Med. J. 2016, 57, 580–587. [Google Scholar] [CrossRef]
- Horowitz, M.; Esakov, E.; Rose, P.; Reizes, O. Signaling within the epithelial ovarian cancer tumor microenvironment: The challenge of tumor heterogeneity. Ann. Transl. Med. 2020, 8, 905. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor microenvironment in ovarian cancer: Function and therapeutic strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef] [PubMed]
- Jou, H.-J.; Ling, P.-Y.; Hsu, H.-T. Circulating tumor cells as a “real-time liquid biopsy”: Recent advances and the application in ovarian cancer. Taiwan. J. Obs. Gynecol. 2022, 61, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Besharat, Z.M.; Farooqi, A.A.; Khalid, S.; Taghavi, K.; Besharat, R.A.; Sabato, C.; Papadia, A.; Panici, P.B.; Mueller, M.D.; et al. MiRNAs and their interplay with PI3K/AKT/mTOR pathway in ovarian cancer cells: A potential role in platinum resistance. J. Cancer Res. Clin. Oncol. 2018, 144, 2313–2318. [Google Scholar] [CrossRef]
- Chebouti, I.; Kuhlmann, J.D.; Buderath, P.; Wimberger, P.; Bokeloh, Y.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. ERCC1-expressing circulating tumor cells as a potential diagnostic tool for monitoring response to platinum-based chemotherapy and for predicting post-therapeutic outcome of ovarian cancer. Oncotarget 2017, 8, 24303–24313. [Google Scholar] [CrossRef]
- Stieg, D.C.; Wang, Y.; Liu, L.-Z.; Jiang, B.-H. ROS and miRNA dysregulation in ovarian cancer development, angiogenesis, and therapeutic resistance. Int. J. Mol. Sci. 2022, 23, 6702. [Google Scholar] [CrossRef] [PubMed]
- Balla, A.; Bhak, J.; Biró, O. The application of circulating tumor cell and cell-free DNA liquid biopsies in ovarian cancer. Mol. Cell. Probes 2022, 66, 101871. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Vikramdeo, K.S.; Sudan, S.K.; Singh, S.; Wilhite, A.; Dasgupta, S.; Rocconi, R.P.; Singh, A.P. Platinum-resistant ovarian cancer: From drug resistance mechanisms to liquid biopsy-based biomarkers for disease management. Semin. Cancer Biol. 2021, 77, 99–109. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian cancers: Genetic abnormalities, tumor heterogeneity and progression, clonal evolution, and cancer stem cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.L.; MacLaughlin, D.T.; Donahoe, P.K. Somatic stem cells of the ovary and their relationship to human ovarian cancers. In StemBook; Harvard Stem Cell Institute: Cambridge, UK, 2008. [Google Scholar]
- Soliman, A.A.; Elzarkaa, A.A.; Malik, E. Epithelial ovarian cancer, and cancer stem cells. In Ovarian Cancer: Molecular & Diagnostic Imaging and Treatment Strategies; Schatten, H., Ed.; Advances in Experimental Medicine and Biology; Springer Nature: Cham, Switzerland, 2021; Volume 1330, pp. 1–19. [Google Scholar]
- Al-Alem, L.F.; Pandya, U.M.; Baker, A.T.; Bellio, C.; Zarrella, B.D.; Clark, J.; DiGloria, C.M.; Rueda, B.R. Ovarian cancer stem cells: What progress have we made? Int. J. Biochem. Cell Biol. 2019, 107, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Padilla, M.A.A.; Binju, M.; Wan, G.; Rahmanto, Y.S.; Kaur, P.; Yu, Y. Relationship between ovarian cancer stem cells, epithelial mesenchymal transition, and tumour recurrence. Cancer Drug Resist. 2019, 2, 1127–1135. [Google Scholar] [CrossRef]
- Miree, O.; Srivastava, S.K.; Dasgupta, S.; Singh, S.; Rocconi, R.; Singh, A.P. Current and Futuristic Roadmap of Ovarian Cancer Management: An Overview. In Ovarian Cancer: Molecular & Diagnostic Imaging and Treatment Strategies; Schatten, H., Ed.; Advances in Experimental Medicine and Biology; Springer Nature: Cham, Switzerland, 2022; Volume 1330, pp. 1–19. [Google Scholar]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Daniele, G.; Raspagliesi, F.; Scambia, G.; Pisano, C.; Colombo, N.; Frezzini, S.; Tognon, G.; Artioli, G.; Gadducci, A.; Lauria, R.; et al. Bevacizumab, carboplatin, and paclitaxel in the first line treatment of advanced ovarian cancer patients: The phase IV MITO-16A/MaNGO-OV2A study. Int. J. Gynecol. Cancer: Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Haunschild, C.E.; Tewari, K.S. Bevacizumab use in the frontline, maintenance, and recurrent settings for ovarian cancer. Future Oncol. 2020, 16, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Cordani, N.; Bianchi, T.; Ammoni, L.C.; Cortinovis, D.L.; Cazzaniga, M.E.; Lissoni, A.A.; Landoni, F.; Canova, S. An overview of PARP resistance in ovarian cancer from a molecular and clinical perspective. Int. J. Mol. Sci. 2023, 24, 11890. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; González-Martín, A.; Ray-Coquard, I.; Harter, P.; Colombo, N.; Pujol, P.; Lorusso, D.; Mirza, M.R.; Brasiuniene, B.; Madry, R.; et al. European experts consensus: BRCA/homologous recombination deficiency testing in first-line ovarian cancer. Ann. Oncol. 2022, 33, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Le Page, C.; Amuzu, S.; Rahimi, K.; Gotlieb, W.; Ragoussis, J.; Tonin, P.N. Lessons learned from understanding chemotherapy resistance in epithelial tubo-ovarian carcinoma from BRCA1and BRCA2 mutation carriers. Semin. Cancer Biol. 2021, 77, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, B. microRNAs as biomarkers of ovarian cancer. Expert. Rev. Anticancer Ther. 2020, 20, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature Reviews. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Saha, S.; Parte, S.; Roy, P.; Kakar, S.S. Ovarian cancer stem cells: Characterization and role in tumorigenesis. Adv. Exp. Med. Biol. 2021, 1330, 151–169. [Google Scholar] [CrossRef]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; De Jesus Acosta, A.; Doi, T.; Longo, F.; et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: Updated analysis from the phase II KEYNOTE-158 study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef]
- Akbarzadeh, M.; Akbarzadeh, S.; Majidinia, M. Targeting Notch signaling pathway as an effective strategy in overcoming drug resistance in ovarian cancer. Pathol. Res. Pract. 2020, 216, 153158. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kim, B.-G.; Kim, J.-W.; Lee, J.B.; Park, E.; Joung, J.-G.; Kim, S.; Choi, C.H.; Kim, H.S.; Korean Gynecologic Oncology Group (KGOG) Investigators. Biomarker-guided targeted therapy in platinum-resistant ovarian cancer (AMBITION; KGOG 3045): A multicentre, open-label, five-arm, uncontrolled, umbrella trial. J. Gynecol. Oncol. 2022, 33, e45. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, Y.-L.; Yu, C.; Chang, T.; Fan, H.-Y. YAP/TEAD co-activator regulated pluripotency and chemoresistance in ovarian cancer initiated cells. PLoS ONE 2014, 9, e109575. [Google Scholar] [CrossRef]
- Borneman, R.M.; Gavin, E.; Musiyenko, A.; Richter, W.; Lee, K.J.; Crossman, D.K.; Andrews, J.F.; Wilhite, A.M.; McClellan, S.; Aragon, I.; et al. Phosphodiesterase 10A (PDE10A) as a novel target to suppress β-catenin and RAS signaling in epithelial ovarian cancer. J. Ovarian Res. 2022, 15, 120. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frezzini, S.; Lonardi, S. Spotlight on New Hallmarks of Drug-Resistance towards Personalized Care for Epithelial Ovarian Cancer. Cells 2024, 13, 611. https://doi.org/10.3390/cells13070611
Frezzini S, Lonardi S. Spotlight on New Hallmarks of Drug-Resistance towards Personalized Care for Epithelial Ovarian Cancer. Cells. 2024; 13(7):611. https://doi.org/10.3390/cells13070611
Chicago/Turabian StyleFrezzini, Simona, and Sara Lonardi. 2024. "Spotlight on New Hallmarks of Drug-Resistance towards Personalized Care for Epithelial Ovarian Cancer" Cells 13, no. 7: 611. https://doi.org/10.3390/cells13070611
APA StyleFrezzini, S., & Lonardi, S. (2024). Spotlight on New Hallmarks of Drug-Resistance towards Personalized Care for Epithelial Ovarian Cancer. Cells, 13(7), 611. https://doi.org/10.3390/cells13070611