
Nanoquercetin and Extracellular Vesicles as Potential Anticancer Therapeutics in Hepatocellular Carcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
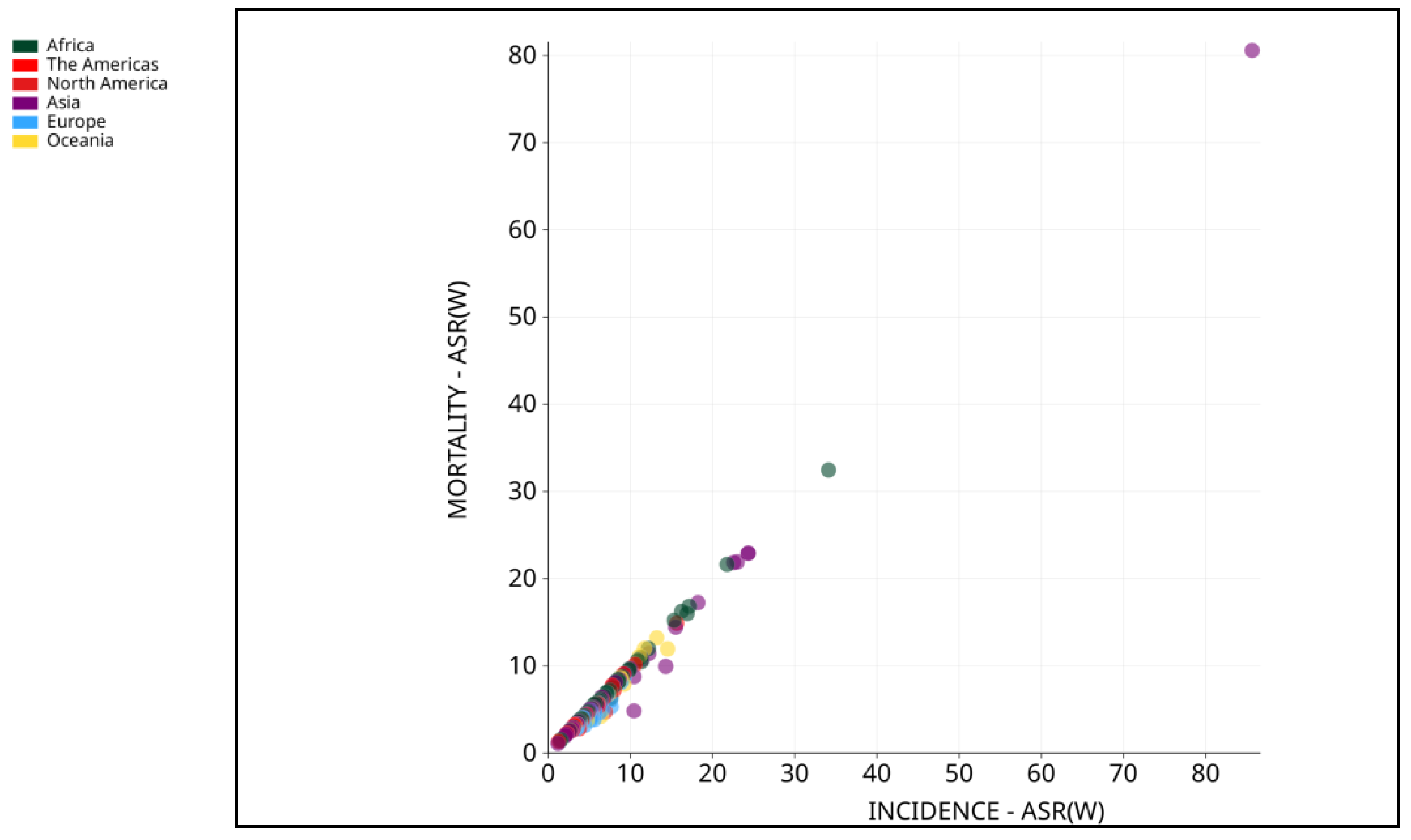
2. Pathophysiology of HCC
3. Molecular Triggers of Hepatocellular Carcinoma
4. Checkpoint Targets of HCC
4.1. Wnt–β-Catenin Signaling
4.1.1. CTNNB1
4.1.2. Adenomatous Polyposis Coli (APC)
4.1.3. AXIN1
4.2. Telomere Maintenance
TERT
4.3. Cell Cycle Regulation
4.3.1. TP53
4.3.2. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL)/DR4/DR5
4.3.3. CDKN2A, CCND1, FGF3, FGF4, or FGF1
4.4. Oxidative Stress
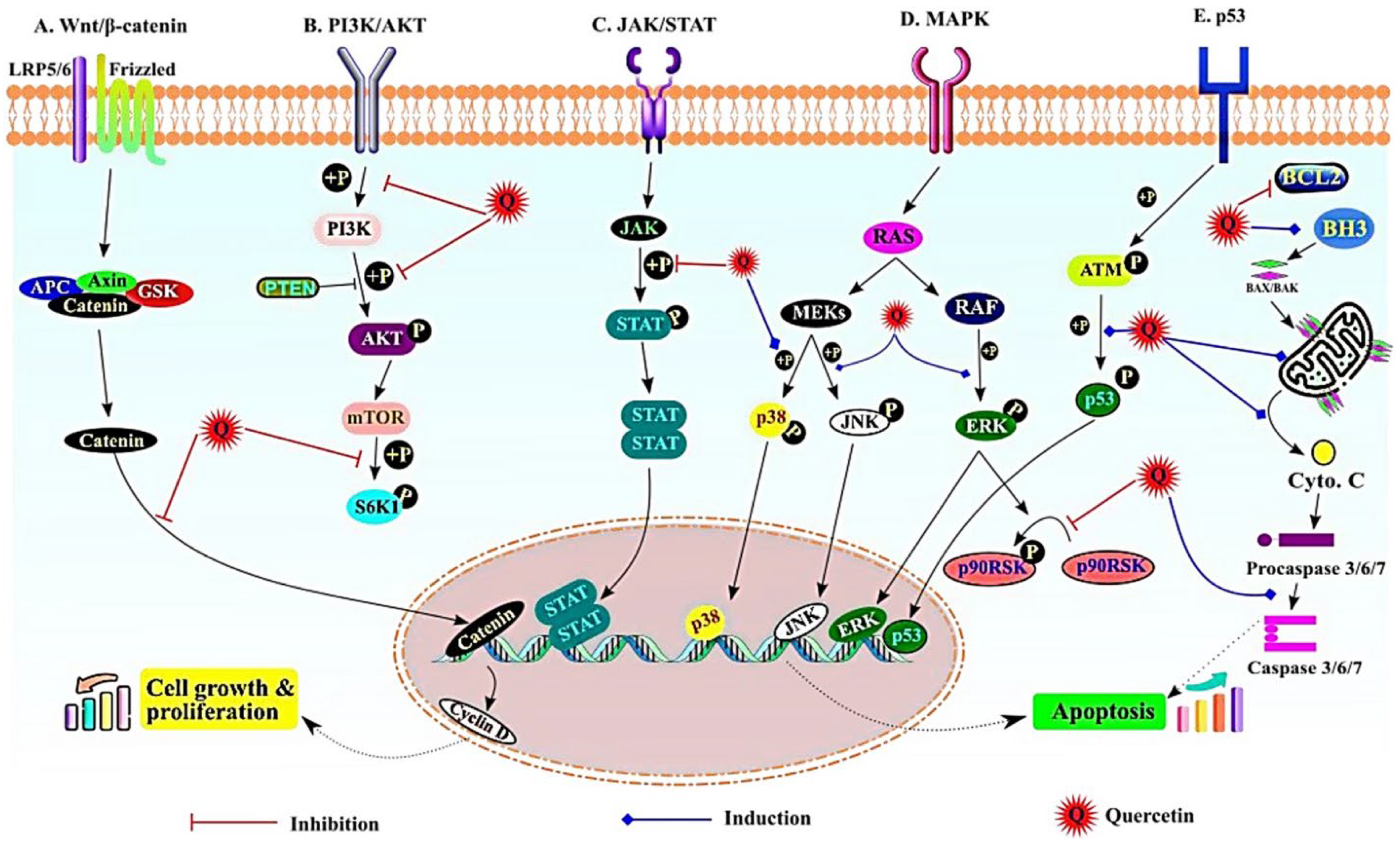
5. Potential Anticancer Mechanism of Nanoquercetin in HCC
5.1. Wnt/β-Catenin Signaling Pathway
5.2. PI3K/AKT Pathway
5.3. JAK/STAT Signaling
5.4. MAPK Signaling
5.5. NF-ĸB, p53, and Apoptotic Signaling
6. Therapeutic Profile of Other Natural Anticancer Compounds
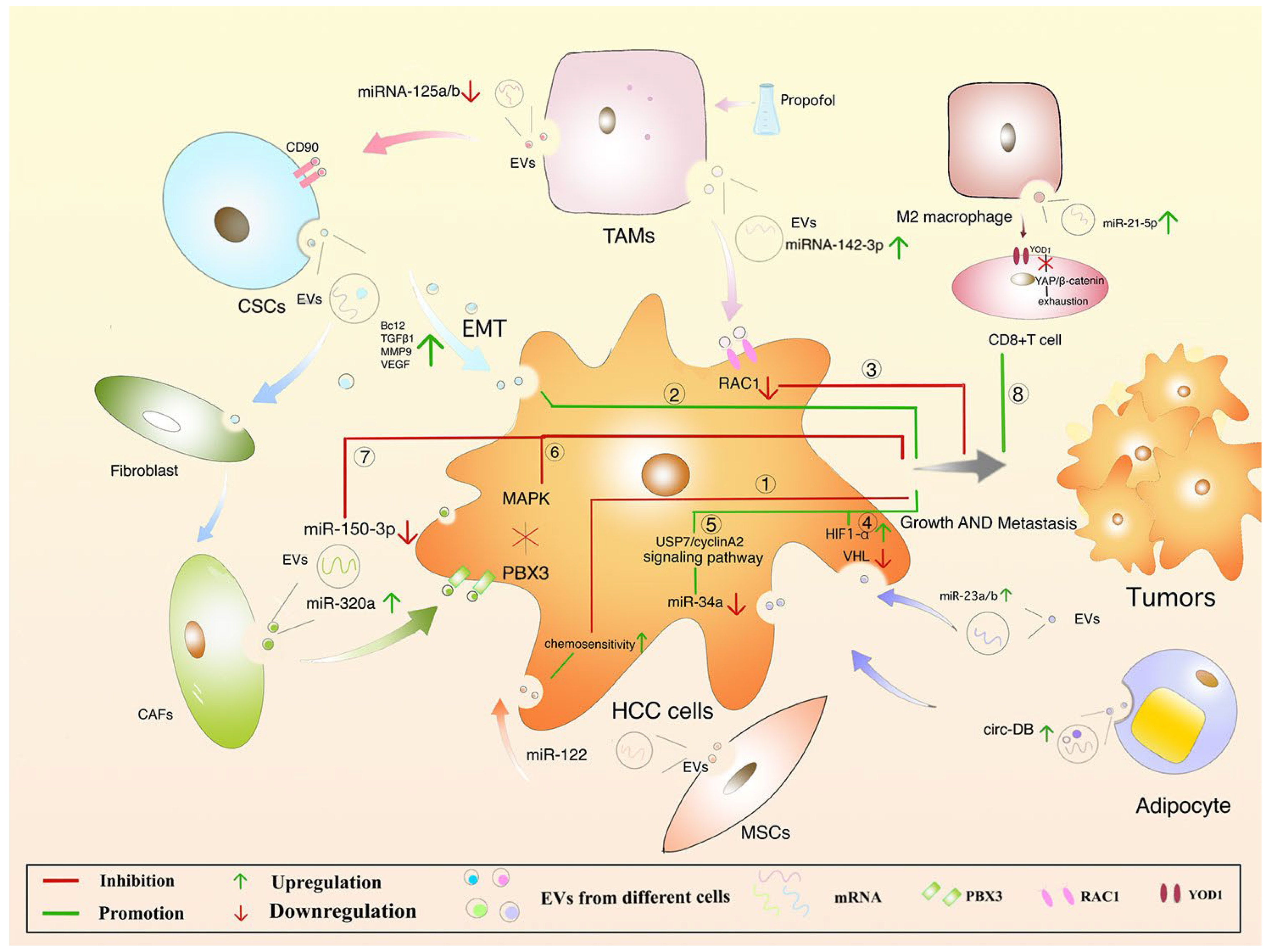
7. Protective Mechanism of EVs in HCC
8. Challenges and Perspectives of Anticancer EVs Biopharmaceuticals
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| UVR | Unrelenting virological response |
| NASH | Non-alcoholic steatohepatitis |
| NAFLD | Non-alcoholic fatty liver disease |
| EVs | Extracellular vesicles |
| AAPC | Average annual percent change |
| HKLC | Hong Kong Liver Cancer |
| CLIP | Cancer of the Liver Italian Program |
| BCLC | Barcelona Clinic Liver Cancer |
| AASLD | American Association for the Study of Liver Diseases |
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548, Erratum in JAMA Oncol. 2017, 3, 418. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. GLOBOCAN 2018. IARC. Available online: https://gco.iarc.fr/today/online-analysis-map?v=2020&mode=population&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=11&type=0&statistic=5&prevalence=0&population_groupearth&color_palette=default&map_scale=quantile&map_nb_colors=5&continent=0&rotate=%255B10%252C0%255D (accessed on 3 March 2023).
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [PubMed]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1. [Google Scholar] [CrossRef] [PubMed]
- Murag, S.; Ahmed, A.; Kim, D. Recent Epidemiology of Nonalcoholic Fatty Liver Disease. Gut and Liver 2021, 15, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N. Metabolic disease as a risk of hepatocellular carcinoma. Clin. Mol. Hepatol. 2021, 27, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular car-cinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Zhao, L. The gut microbiota and obesity: From correlation to causality. Nat. Rev. Microbiol. 2013, 11, 639–647. [Google Scholar] [CrossRef] [PubMed]
- WHO. Data Visualization Tools for Exploring the Global Cancer Burden in 2020. 2020. Available online: https://gco.iarc.fr/today/ (accessed on 3 March 2022).
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Cheng, Y.; Zhang, S.; Fan, J.; Gao, Q. Changing epidemiology of hepatocellular carcinoma in Asia. Liver Int. 2022, 42, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Tellapuri, S.; Sutphin, P.D.; Beg, M.S.; Singal, A.G.; Kalva, S.P. Staging systems of hepatocellular carcinoma: A review. Indian J. Gastroenterol. 2018, 37, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, H.; Wang, M.; Zhao, Y.; Cai, G.; Qi, X.; Han, G. Combination Therapy of Sorafenib and TACE for Unresectable HCC: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e91124. [Google Scholar] [CrossRef] [PubMed]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hoffmann, K.; Schemmer, P. Treatment of Hepatocellular Carcinoma: A Systematic Review. Liver Cancer 2012, 1, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, W.; Wang, B.; Gao, Y.; Song, Z.; Zheng, Q.C. Ligand-based targeted therapy: A novel strategy for hepatocellular carcinoma. Int. J. Nanomed. 2016, 11, 5645–5669. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Shafizadeh, N.; Kakar, S. Diagnosis of Well-differentiated Hepatocellular Lesions: Role of immunohistochemistry and other an-cillary techniques. Adv. Anat. Pathol. 2011, 18, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Herrera Sanchez, M.B.; Chiabotto, G.; Fonsato, V.; Navarro-Tableros, V.; Pasquino, C.; Tapparo, M.; Camussi, G. Human Liver Stem Cells: A Liver-Derived Mesenchymal Stromal Cell-Like Population with Pro-regenerative Properties. Front. Cell Dev. Biol. 2021, 9, 644088. [Google Scholar] [CrossRef]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.-C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Gellert-Kristensen, H.; Richardson, T.G.; Smith, G.D.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Stender, S. Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 2020, 72, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Letouzé, E. Mutational Processes in Hepatocellular Carcinoma: The Story of Aristolochic Acid. Semin. Liver Dis. 2019, 39, 334–340. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Dang, H.T.; Wang, X.W. Identification of Drivers from Cancer Genome Diversity in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2014, 15, 11142–11160. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Jang, S.J.; Shim, J.H.; Kim, D.; Hong, S.M.; Sung, C.O.; Baek, D.; Haq, F.; Ansari, A.A.; Lee, S.Y.; et al. Genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014, 60, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Doderer, M.S.; Anguiano, Z.; Suresh, U.; Dashnamoorthy, R.; Bishop, A.J.; Chen, Y. Pathway Distiller - multisource biological pathway consolidation. BMC Genomics 2012, 13 (Suppl. 6), S18. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. The Cancer Genome Atlas. Available online: http://cancergenome.nih.gov/ (accessed on 4 March 2023).
- Tao, J.; Xu, E.; Zhao, Y.; Singh, S.; Li, X.; Couchy, G.; Chen, X.; Zucman-Rossi, J.; Chikina, M.; Monga, S.P. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology 2016, 64, 1587–1605. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti–PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Krutsenko, Y.; Moghe, A.; Singh, S.; Poddar, M.; Bell, A.; Oertel, M.; Singhi, A.D.; Geller, D.; Chen, X.; et al. Nuclear factor erythroid 2–related factor 2 and β-Catenin Coactivation in Hepatocellular Cancer: Biological and Therapeutic Implications. Hepatology 2021, 74, 741–759. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Behari, J.; Cieply, B.; Michalopoulos, G.K.; Monga, S.P. Conditional Deletion of β-Catenin Reveals Its Role in Liver Growth and Regeneration. Gastroenterology 2006, 131, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Nagse, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef]
- Li, J.; Quan, H.; Liu, Q.; Si, Z.; He, Z.; Qi, H. Alterations of Axis Inhibition Protein 1 (AXIN1) in Hepatitis B Virus-Related Hepatocellular Carcinoma and Overexpression of AXIN1 Induces Apoptosis in Hepatocellular Cancer Cells. Oncol. Res. 2013, 20, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. TERTpromoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; Franze, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol Lett. 2020, 19, 2368–2374. [Google Scholar] [CrossRef] [PubMed]
- Sirma, H.; Kumar, M.; Meena, J.K.; Witt, B.; Weise, J.M.; Lechel, A.; Ande, S.; Sakk, V.; Guguen–Guillouzo, C.; Zender, L.; et al. The promoter of human telomerase reverse transcriptase is activated during liver regeneration and hepatocyte proliferation. Gastroenterology 2011, 141, 326–337.e3. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, W.; Lu, W.; Chen, M.; Luo, M.; Zhang, C.; Li, Y.; Qin, G.; Shi, D.; Xiao, B.; et al. RBFOX3 Promotes Tumor Growth and Progression via hTERT Signaling and Predicts a Poor Prognosis in Hepatocellular Carcinoma. Theranostics 2017, 7, 3138–3154. [Google Scholar] [CrossRef]
- Esopi, D.; Graham, M.K.; Brosnan-Cashman, J.A.; Meyers, J.; Vaghasia, A.; Gupta, A.; Kumar, B.; Haffner, M.C.; Heaphy, C.M.; De Marzo, A.M.; et al. Pervasive promoter hypermethylation of silenced TERT alleles in human cancers. Cell. Oncol. 2020, 43, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Hoshida, Y. Depicting the role of TP53 in hepatocellular carcinoma progression. J. Hepatol. 2011, 55, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Jeng, Y.-M.; Hsu, H.-C. Mutation of the DR5/TRAIL receptor 2 gene is infrequent in hepatocellular carcinoma. Cancer Lett. 2002, 181, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qiu, J.; He, G.; He, W.; Liu, C.; Cai, D.; Pan, H. TRAIL promotes hepatocellular carcinoma apoptosis and inhibits proliferation and migration via interacting with IER3. Cancer Cell Int. 2021, 21, 63. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Lemmers, B.; Hakem, A.; Matysiak-Zablocki, E.; Murakami, K.; Au, P.B.; Berry, D.M.; Tamblyn, L.; Shehabeldin, A.; Migon, E.; et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003, 17, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Chung, C.W.; Woo, H.-N.; Hong, G.S.; Nagata, S.; Jung, Y.-K. Reconstitution of Caspase-8 Sensitizes JB6 Cells to TRAIL. Biochem. Biophys. Res. Commun. 2000, 277, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-Dependent Recruitment of Endogenous FADD and Caspase-8 to Death Receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Cook, A.; Keane, M.M.; Kelliher, M.; Lipkowitz, S.; Liu, Z.-G. The Death Domain Kinase RIP Is Essential for TRAIL (Apo2L)-Induced Activation of IκB Kinase and c-Jun N-Terminal Kinase. Mol. Cell. Biol. 2000, 20, 6638–6645. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.-L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL Receptors 1 (DR4) and 2 (DR5) Signal FADD-Dependent Apoptosis and Activate NF-κB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Belinky, F.; Nativ, N.; Stelzer, G.; Zimmerman, S.; Stein, T.I.; Safran, M.; Lancet, D. PathCards: Multi-source consolidation of human biological pathways. Database 2015, 2015, bav006. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Gopal, P.; Singal, A.G. The Changing Landscape of Hepatocellular Carcinoma: Etiology, genetics, and therapy. Am. J. Pathol. 2014, 184, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424, Erratum in Nat. Rev. Clin. Oncol. 2015, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a Therapeutic Strategy Targeting Amplified FGF19 in Liver Cancer by Oncogenomic Screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yada, N.; Hagiwara, S.; Sakurai, T.; Kitano, M.; Kudo, M. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 1646–1653. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wong, C.C.; Fu, L.; Chen, H.; Zhao, L.; Li, C.; Zhou, Y.; Zhang, Y.; Xu, W.; Yang, Y.; et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 2018, 10, eaap9840, Erratum in Sci. Transl. Med. 2018, 10, eaav4522. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Yen, G.C. Phenolic compounds: Evidence for inhibitory effects against obesity and their underlying molecular signaling mechanisms. Mol. Nutr. Food Res. 2008, 52, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-X.; Jiang, S.-S.; Zhang, X.-F.; Zhou, Z.-Q.; Pan, Q.-Z.; Chen, C.-L.; Zhao, J.-J.; Tang, Y.; Xia, J.-C.; Weng, D.-S. Protein kinase CK2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget 2015, 6, 34800–34817. [Google Scholar] [CrossRef]
- Tawfic, S.; Yu, S.; Wang, H.; Faust, R.; Davis, A.; Ahmed, K. Protein kinase CK2 signal in neoplasia. Histol. Histopathol. 2001, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Yenice, S.; Davis, A.T.; Goueli, S.A.; Akdas, A.; Limas, C.; Ahmed, K. Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994, 24, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Croquelois, A.; Blindenbacher, A.; Terracciano, L.; Wang, X.; Langer, I.; Radtke, F.; Heim, M.H. Inducible inactivation ofNotch1 causes nodular regenerative hyperplasia in mice. Hepatology 2005, 41, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Mojsin, M.; Vicentic, J.M.; Schwirtlich, M.; Topalovic, V.; Stevanovic, M. Quercetin reduces pluripotency, migration and adhesion of human teratocarcinoma cell line NT2/D1 by inhibiting Wnt/β-catenin signaling. Food Funct. 2014, 5, 2564–2573. [Google Scholar] [CrossRef]
- Kim, H.; Seo, E.-M.; Sharma, A.R.; Ganbold, B.; Park, J.; Sharma, G.; Kang, Y.-H.; Song, D.-K.; Lee, S.-S.; Nam, J.-S. Regulation of Wnt signaling activity for growth suppression induced by quercetin in 4T1 murine mammary cancer cells. Int. J. Oncol. 2013, 43, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Fang, Y.; Wang, S.-X. Quercetin suppresses HeLa cells by blocking PI3K/Akt pathway. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Laudet, B.; Zohrabian, V.M.; Murali, R.; Jhanwar-Uniyal, M. The antiproliferative effect of Quercetin in cancer cells is mediated via inhibition of the PI3K-Akt/PKB pathway. Anticancer Res. 2006, 26, 1177–1181. [Google Scholar] [PubMed]
- Tan, H.K.; Moad, A.I.H.; Tan, M.L. The mTOR Signalling Pathway in Cancer and the Potential mTOR Inhibitory Activities of Natural Phytochemicals. Asian Pac. J. Cancer Prev. 2014, 15, 6463–6475. [Google Scholar] [CrossRef] [PubMed]
- Fyffe, C.; Falasca, M. 3-Phosphoinositide-dependent protein kinase-1 as an emerging target in the management of breast cancer. Cancer Manag. Res. 2013, 5, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Maurya, A.K.; Vinayak, M. PI-103 and Quercetin Attenuate PI3K-AKT Signaling Pathway in T- Cell Lymphoma Exposed to Hydrogen Peroxide. PLoS ONE 2016, 11, e0160686. [Google Scholar] [CrossRef]
- Navarro-Núñez, L.; Lozano, M.L.; Martínez, C.; Vicente, V.; Rivera, J. Effect of quercetin on platelet spreading on collagen and fibrinogen and on multiple platelet kinases. Fitoterapia 2010, 81, 75–80. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; He, L.-Y.; Chen, Y.; Wang, W.-Y.; Zhao, X.-H.; Wu, M.-Y. Quercetin affects leptin and its receptor in human gastric cancer MGC-803 cells and JAK-STAT pathway. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2012, 28, 12–16. [Google Scholar] [PubMed]
- Michaud-Levesque, J.; Bousquet-Gagnon, N.; Béliveau, R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp. Cell Res. 2012, 318, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-S.; Ku, J.M.; Choi, H.-S.; Choi, Y.K.; Woo, J.-K.; Kim, M.; Kim, I.; Na, C.H.; Hur, H.; Jang, B.-H.; et al. Quercetin induces caspase-dependent extrinsic apoptosis through inhibition of signal transducer and activator of transcription 3 signaling in HER2-overexpressing BT-474 breast cancer cells. Oncol. Rep. 2016, 36, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.-L.; Liu, Y.-Q.; Wang, P.; Song, C.-H.; Wang, K.-J.; Dai, L.-P.; Zhang, J.-Y.; Ye, H. The effect of quercetin nanoparticle on cervical cancer progression by inducing apoptosis, autophagy and anti-proliferation via JAK2 suppression. Biomed. Pharmacother. 2016, 82, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, J.; Liu, T.; Li, S.; Feng, J.; Yu, Q.; Zhang, J.; Chen, J.; Zhou, Y.; Ji, J.; et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019, 8, 4806–4820. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakopoulou, M.; Lendenmann, T.; Pramotton, F.M.; Giampietro, C.; Stefopoulos, G.; Poulikakos, D.; Ferrari, A.; Leal-Egaña, A.; Letort, G.; Martiel, J.-L.; et al. Cell cycle–dependent force transmission in cancer cells. Mol. Biol. Cell 2018, 29, 2528–2539. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, H.; Wang, Y.; Song, F.; Yuan, Y. Inhibitory effects of quercetin on the progression of liver fibrosis through the regulation of NF-кB/IкBα, p38 MAPK, and Bcl-2/Bax signaling. Int. Immunopharmacol. 2017, 47, 126–133. [Google Scholar] [CrossRef]
- Asgharian, P.; Tazekand, A.P.; Hosseini, K.; Forouhandeh, H.; Ghasemnejad, T.; Ranjbar, M.; Hasan, M.; Kumar, M.; Beirami, S.M.; Tarhriz, V.; et al. Potential mechanisms of quercetin in cancer prevention: Focus on cellular and molecular targets. Cancer Cell Int. 2022, 22, 257. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.P.; Lima, C.F.; Rohde, M.; Pereira-Wilson, C. Quercetin enhances 5-fluorouracil-induced apoptosis in MSI colorectal cancer cells through p53 modulation. Cancer Chemother. Pharmacol. 2011, 68, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J.; Liu, L.; Sharma, S.; Dong, Q. Quercetin Potentiates Doxorubicin Mediated Antitumor Effects against Liver Cancer through p53/Bcl-xl. PLoS ONE 2012, 7, e51764. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, K.; Ghosh, S.; Mukherjee, A.; Sadhukhan, R.; Mondal, J.; Khuda-Bukhsh, A.R. Quercetin induces cytochrome-c release and ROS accumulation to promote apoptosis and arrest the cell cycle in G2/M, in cervical carcinoma: Signal cascade and drug-DNA interaction. Cell Prolif. 2013, 46, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, K.; Khuda-Bukhsh, A.R.; Huh, S.-O. PLGA-Loaded Gold-Nanoparticles Precipitated with Quercetin Downregulate HDAC-Akt Activities Controlling Proliferation and Activate p53-ROS Crosstalk to Induce Apoptosis in Hepatocarcinoma Cells. Mol. Cells 2015, 38, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Lee, Y.-H.; Sharma, A.R.; Park, J.-B.; Jagga, S.; Sharma, G.; Lee, S.-S.; Nam, J.-S. Quercetin induces apoptosis and cell cycle arrest in triple-negative breast cancer cells through modulation of Foxo3a activity. Korean J. Physiol. Pharmacol. 2017, 21, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; De Amicis, F.; Sirianni, R.; Sinicropi, M.S.; Puoci, F.; Casaburi, I.; Saturnino, C.; Pezzi, V. Progress to Improve Oral Bioavailability and Beneficial Effects of Resveratrol. Int. J. Mol. Sci. 2019, 20, 1381. [Google Scholar] [CrossRef]
- Pannu, N.; Bhatnagar, A. Resveratrol: From enhanced biosynthesis and bioavailability to multitargeting chronic diseases. Biomed. Pharmacother. 2019, 109, 2237–2251. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.-J.; Wei, Q.-Y.; Fang, J.-G.; Yang, L.; Liu, Z.-L.; Wyche, J.H.; Han, Z. The 3,4-dihydroxyl groups are important for trans-resveratrol analogs to exhibit enhanced antioxidant and apoptotic activities. Anticancer Res. 2004, 24, 999–1002. [Google Scholar]
- Colin, D.; Lancon, A.; Delmas, D.; Lizard, G.; Abrossinow, J.; Kahn, E.; Jannin, B.; Latruffe, N. Antiproliferative activities of resveratrol and related compounds in human hepatocyte derived HepG2 cells are associated with biochemical cell disturbance revealed by fluorescence analyses. Biochimie 2008, 90, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.X.; Lv, L.H.; Wan, Y.L.; Cao, Y.; Li, G.L.; Lin, H.M.; Zhou, R.; Shang, C.Z.; Cao, J.; He, H.; et al. Vps4A functions as a tumor suppressor by regulating the secretion and uptake of exosomal microRNAs in human hepatoma cells. Hepatology 2015, 61, 1284–1294. [Google Scholar] [CrossRef]
- Giordano, S.; Columbano, A. MicroRNAs: New tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology 2012, 57, 840–847. [Google Scholar] [CrossRef]
- Wang, J.; Pu, J.; Zhang, Y.; Yao, T.; Luo, Z.; Li, W.; Xu, G.; Liu, J.; Wei, W.; Deng, Y. Exosome-transmitted long non-coding RNA SENP3-EIF4A1 suppresses the progression of hepatocellular carcinoma. Aging 2020, 12, 11550–11567. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Xie, B.; Yang, X.; Liang, H.; Jiang, X.; Zhang, D.; Xue, P.; Chen, D.; Shao, Z. MiR-324-5p Suppresses Hepatocellular Carcinoma Cell Invasion by Counteracting ECM Degradation through Post-Transcriptionally Downregulating ETS1 and SP1. PLoS ONE 2015, 10, e0133074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Sun, W.; Yue, S.; Yang, J.; Li, J.; Ma, B.; Wang, J.; Yang, X.; Pu, M.; et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017, 397, 33–42. [Google Scholar] [CrossRef]
- Jiang, W.; Tan, Y.; Cai, M.; Zhao, T.; Mao, F.; Zhang, X.; Xu, W.; Yan, Z.; Qian, H.; Yan, Y. Human Umbilical Cord MSC-Derived Exosomes Suppress the Development of CCl4-Induced Liver Injury through Antioxidant Effect. Stem Cells Int. 2018, 2018, 6079642. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, X.; Zhang, X.; Shao, T.; Luo, Y.; Wang, W.; Han, Y. Extracellular Vesicles and Hepatocellular Carcinoma: Opportunities and Challenges. Front. Oncol. 2022, 12, 884369. [Google Scholar] [CrossRef] [PubMed]
- Yugawa, K.; Yoshizumi, T.; Mano, Y.; Itoh, S.; Harada, N.; Ikegami, T.; Kohashi, K.; Oda, Y.; Mori, M. Cancer-associated fibroblasts promote hepatocellular carcinoma progression through downregulation of exosomal miR-150-3p. Eur. J. Surg. Oncol. 2021, 47, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zhu, Y.; Xiong, Y.; Ge, Y.-Y.; Yun, J.-P.; Zhuang, S.-M. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology 2009, 50, 113–121. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, N.; Li, S.; Fang, J.-H.; Chen, M.-X.; Yang, J.; Jia, W.-H.; Yuan, Y.; Zhuang, S.-M. MicroRNA-195 Suppresses Angiogenesis and Metastasis of Hepatocellular Carcinoma by Inhibiting the Expression of VEGF, VAV2, and CDC42. Hepatology 2013, 58, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Quan, Y.; Fan, S.; Wang, H.; Liang, J.; Huang, L.; Chen, L.; Liu, Q.; He, P.; Ye, Y. Exosome-transmitted circular RNA hsa_circ_0051443 suppresses hepatocellular carcinoma progression. Cancer Lett. 2020, 475, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry, PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing and Quality Assurance; FDA: Silver Spring, MD, USA, 2004. Available online: http://www.fda.gov/cder/guidance/published.html (accessed on 8 March 2023).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raghav, A.; Jeong, G.B. Nanoquercetin and Extracellular Vesicles as Potential Anticancer Therapeutics in Hepatocellular Carcinoma. Cells 2024, 13, 638. https://doi.org/10.3390/cells13070638
Raghav A, Jeong GB. Nanoquercetin and Extracellular Vesicles as Potential Anticancer Therapeutics in Hepatocellular Carcinoma. Cells. 2024; 13(7):638. https://doi.org/10.3390/cells13070638
Chicago/Turabian StyleRaghav, Alok, and Goo Bo Jeong. 2024. "Nanoquercetin and Extracellular Vesicles as Potential Anticancer Therapeutics in Hepatocellular Carcinoma" Cells 13, no. 7: 638. https://doi.org/10.3390/cells13070638
APA StyleRaghav, A., & Jeong, G. B. (2024). Nanoquercetin and Extracellular Vesicles as Potential Anticancer Therapeutics in Hepatocellular Carcinoma. Cells, 13(7), 638. https://doi.org/10.3390/cells13070638