
The Temporal Relationship between Blood–Brain Barrier Integrity and Microglial Response following Neonatal Hypoxia Ischemia

 , , ,
, , , 
Abstract
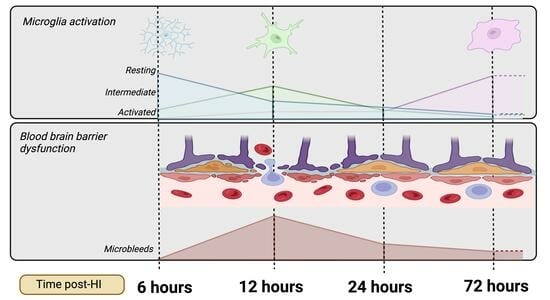
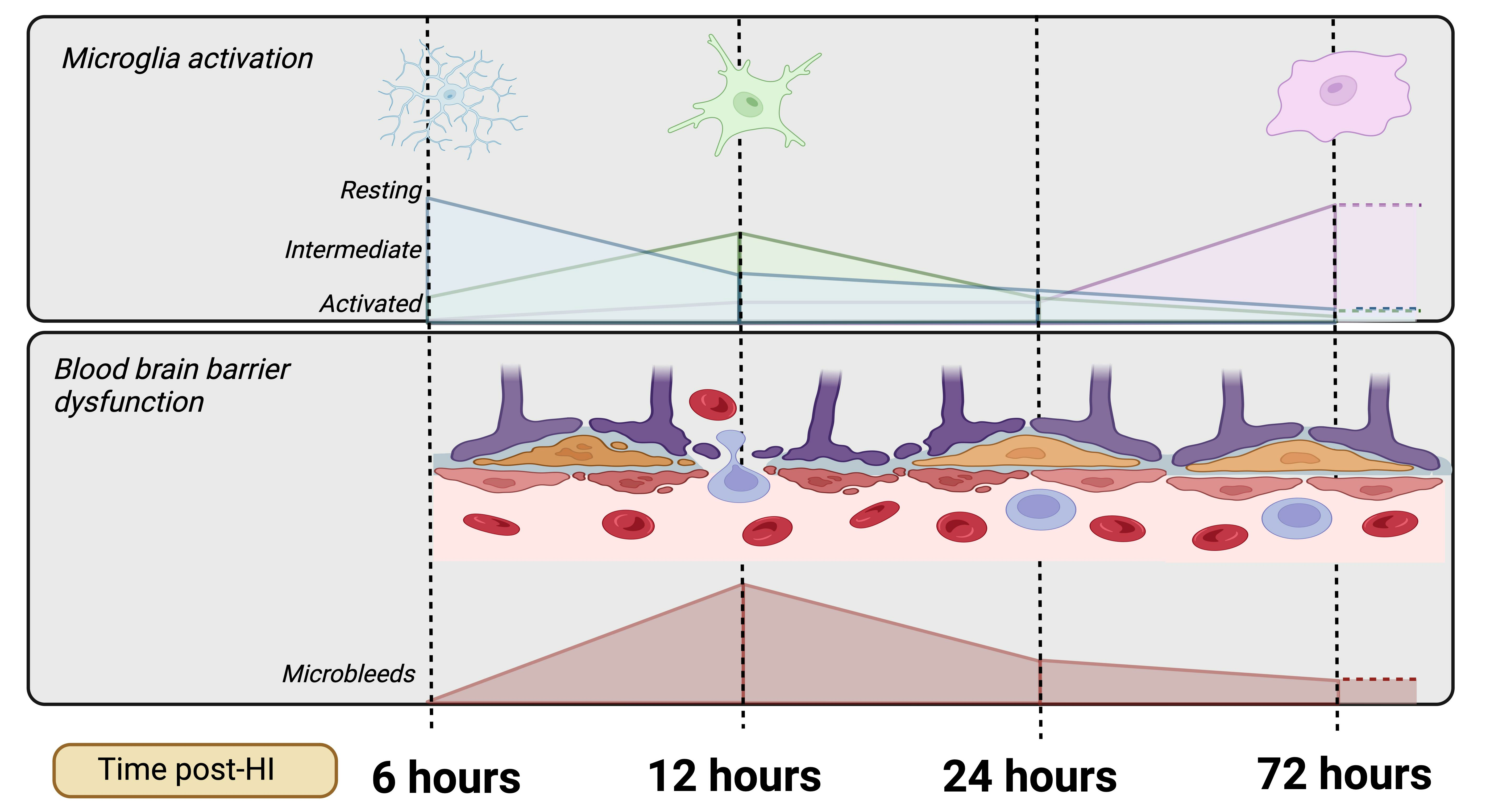
:
1. Introduction
2. Materials and Methods
- Ethics approval:
- Animals:
- Animal source and housing
- Surgery protocol: modified Rice–Vannucci model to induce HI injury:
- Post-mortem and brain processing
- Immunohistochemistry
- Immunohistological assessment:
area of the contralateral hemisphere
- Microbleed criteria
- (a).
- Non-linear red blood cells.
- (b).
- Red blood cells surrounded by parenchyma.
- (c).
- Erythrocytes not contained within a vessel.
- (d).
- A total of >5 cells clustered together, <10 mm.
- (e).
- Exclusion of red blood cells from the choroid plexus and ventricles.
- Statistical analysis:
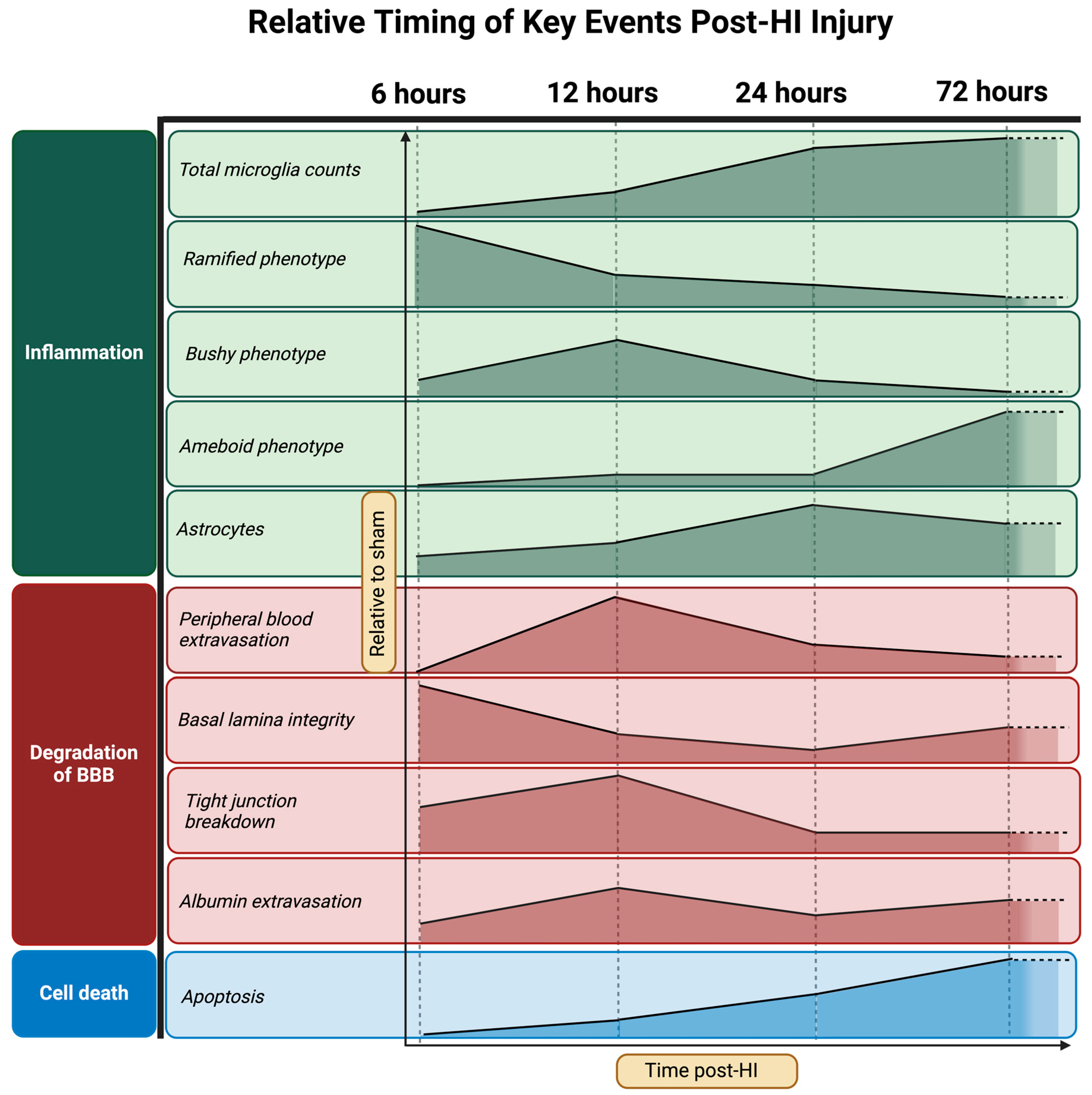
3. Results
- Animal Cohort Characteristics
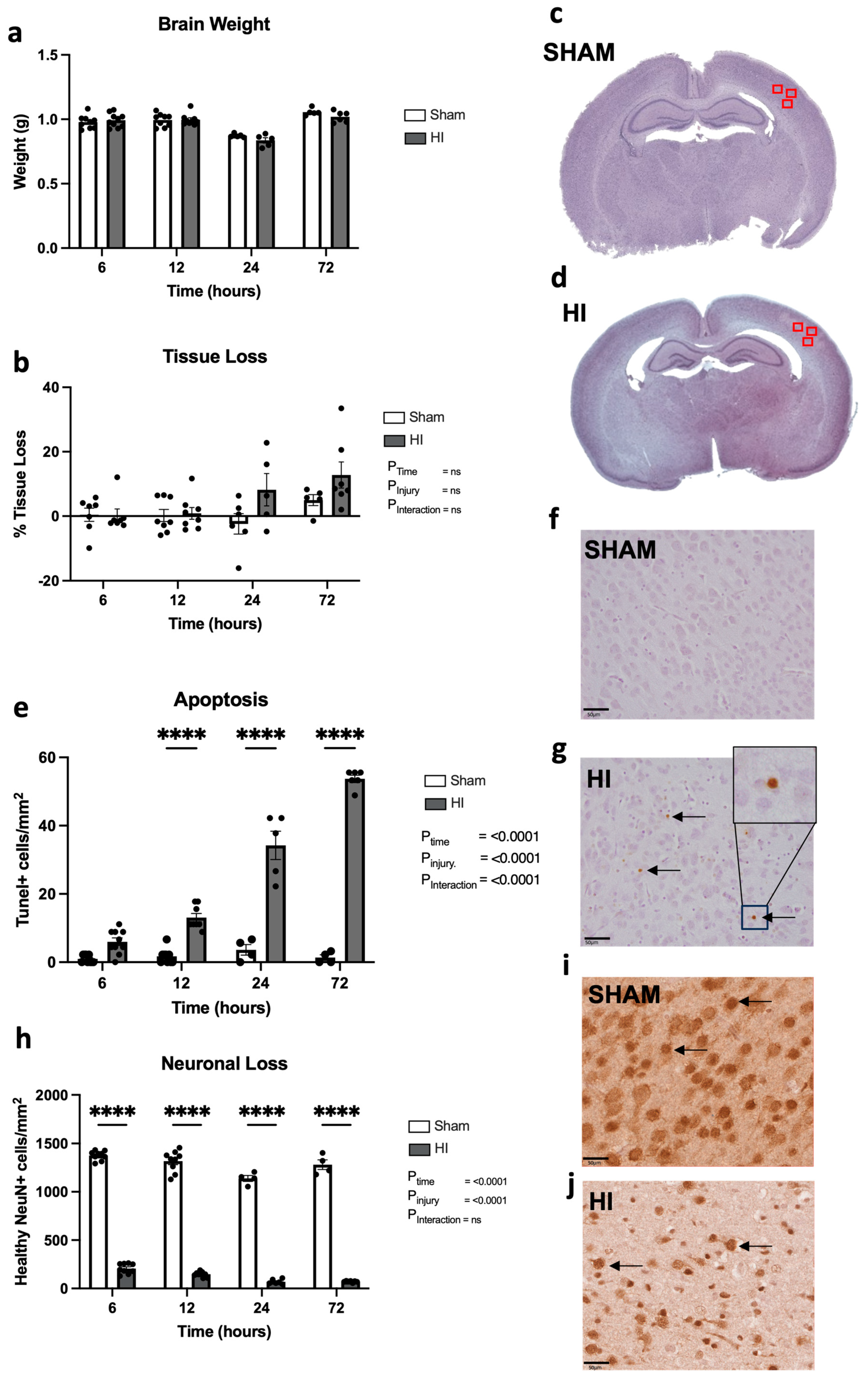
- Progression of neuropathology over time following perinatal HI.
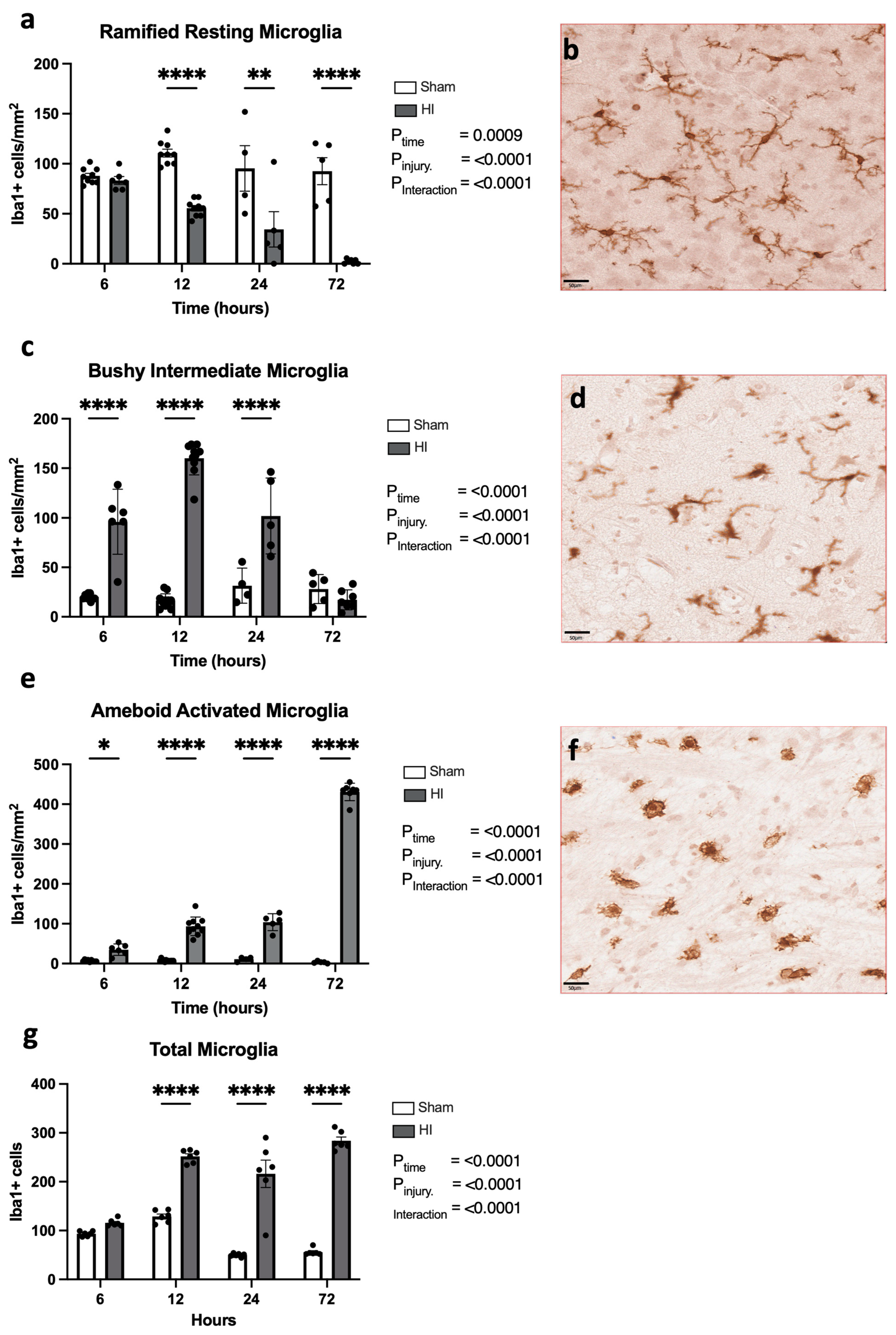
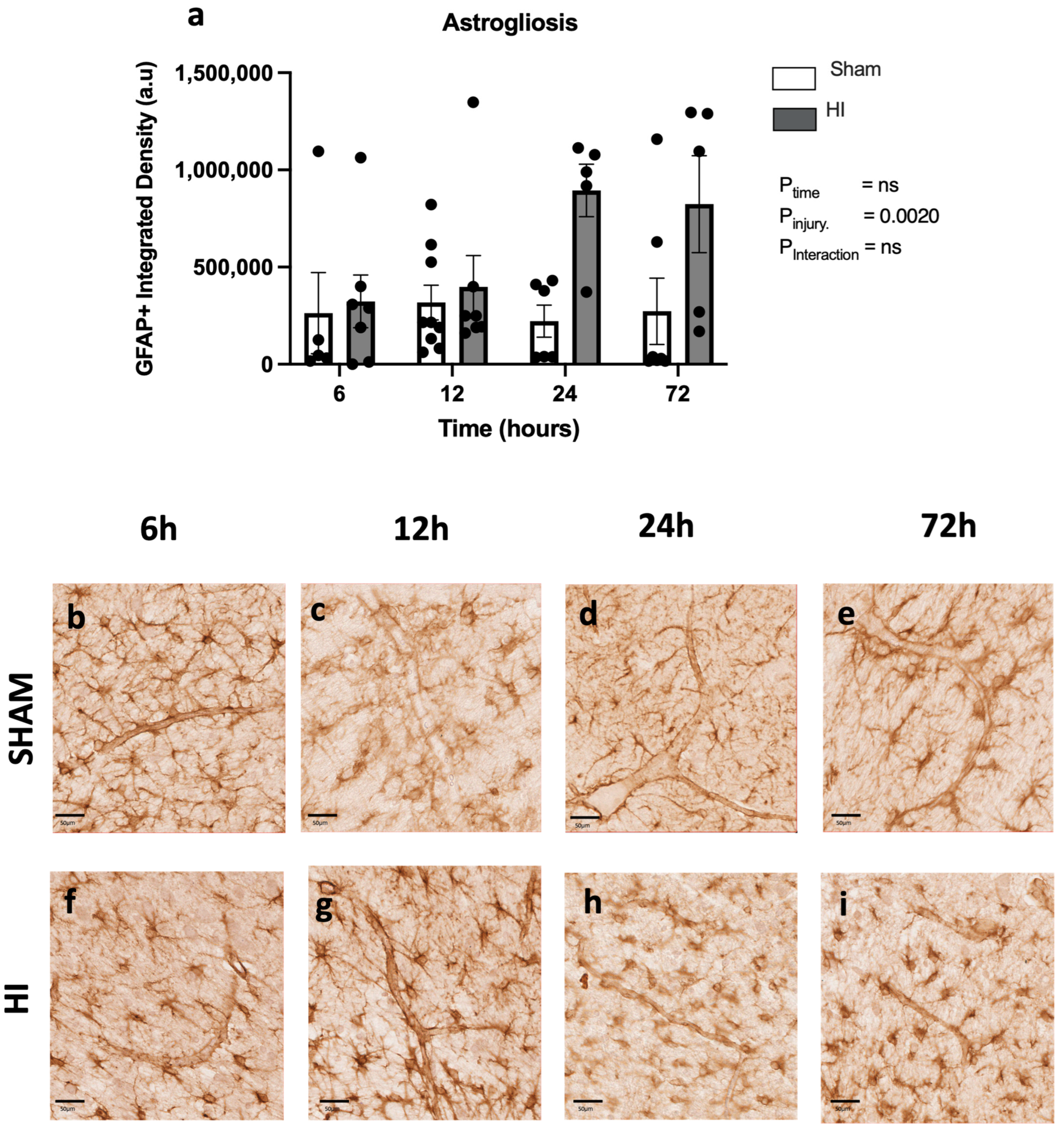
- Progression of microglial activation in the brain over time following perinatal HI
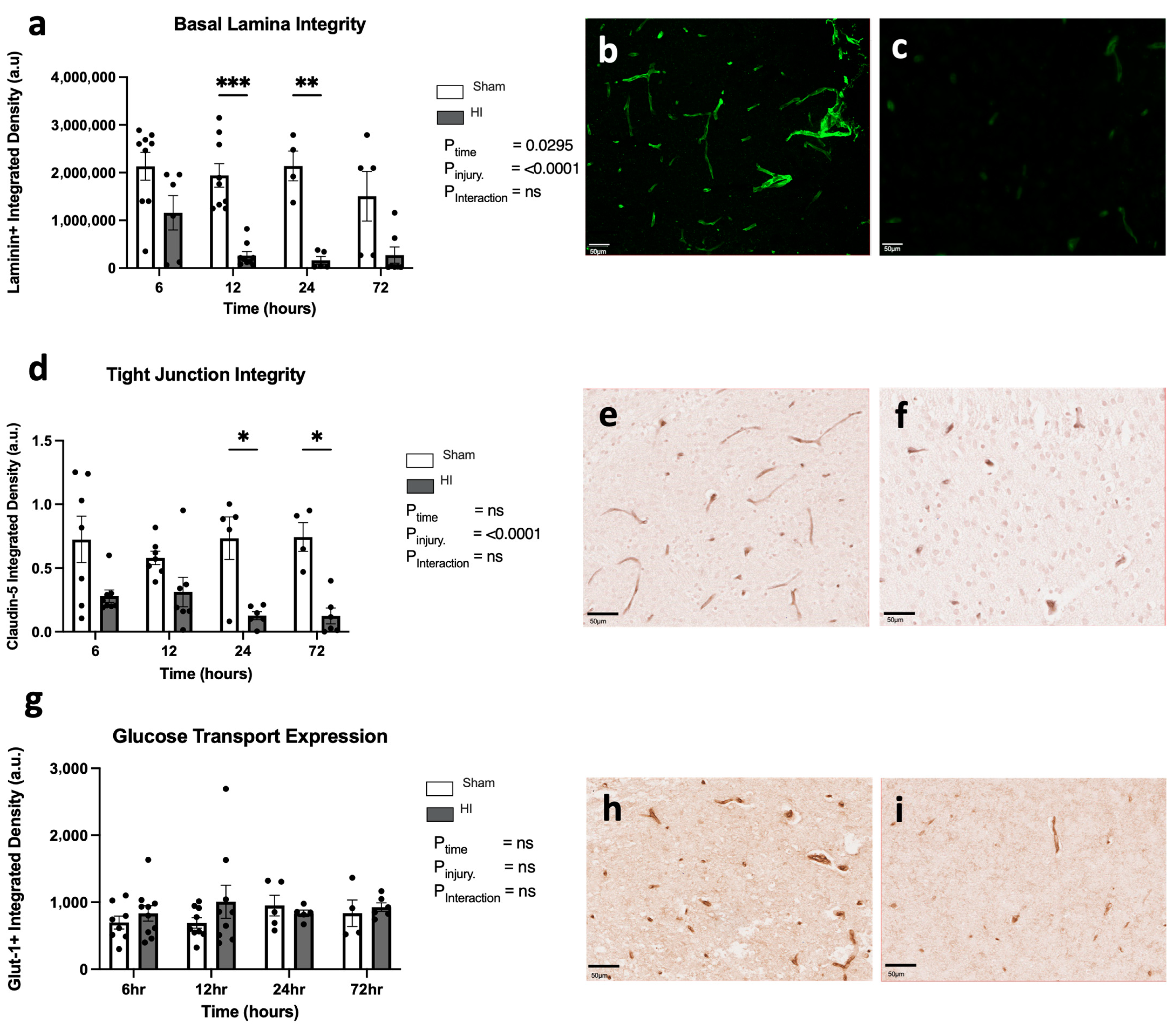
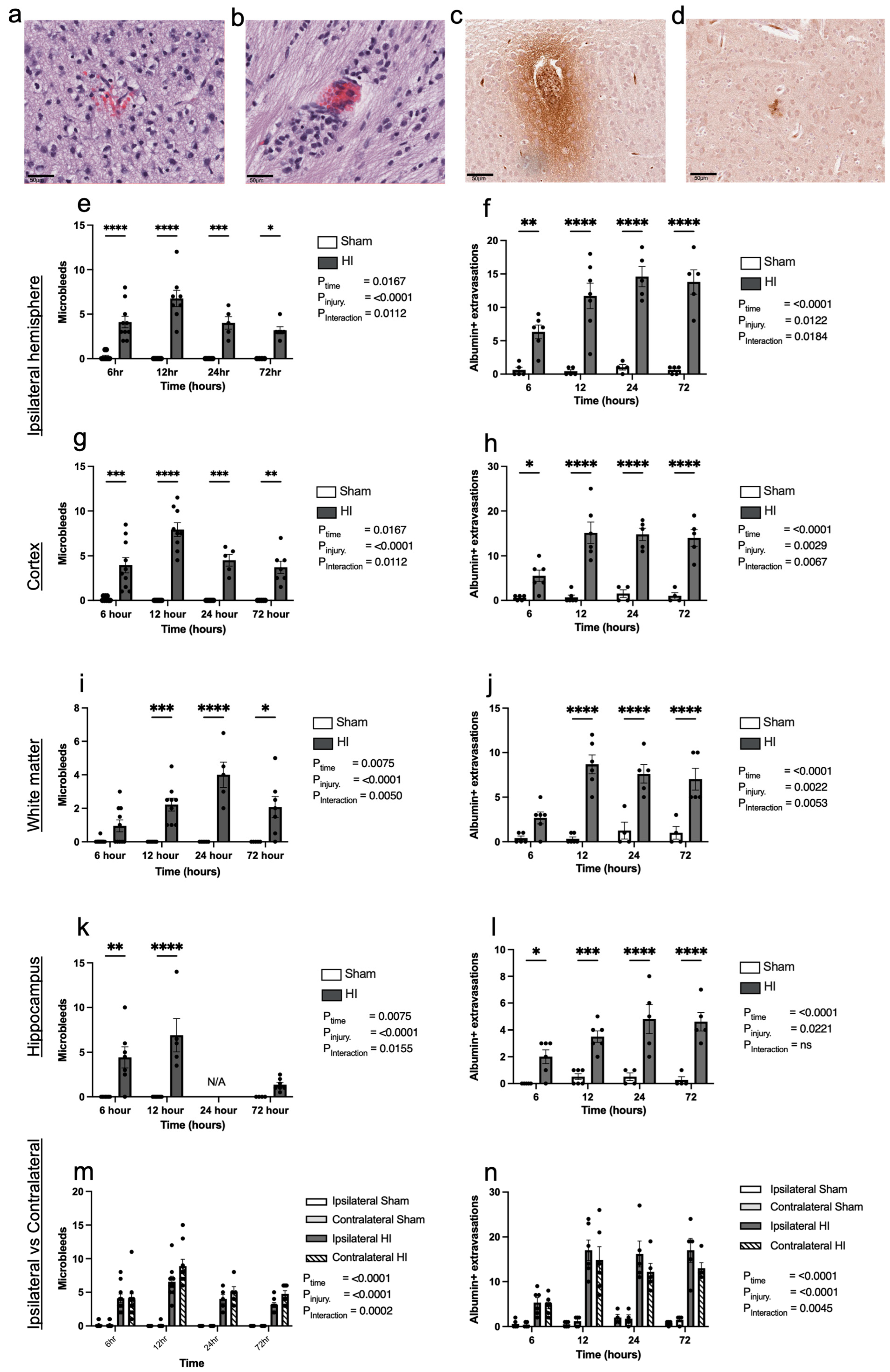
- Progression of HI-mediated BBB breakdown: Extravasation of peripheral blood
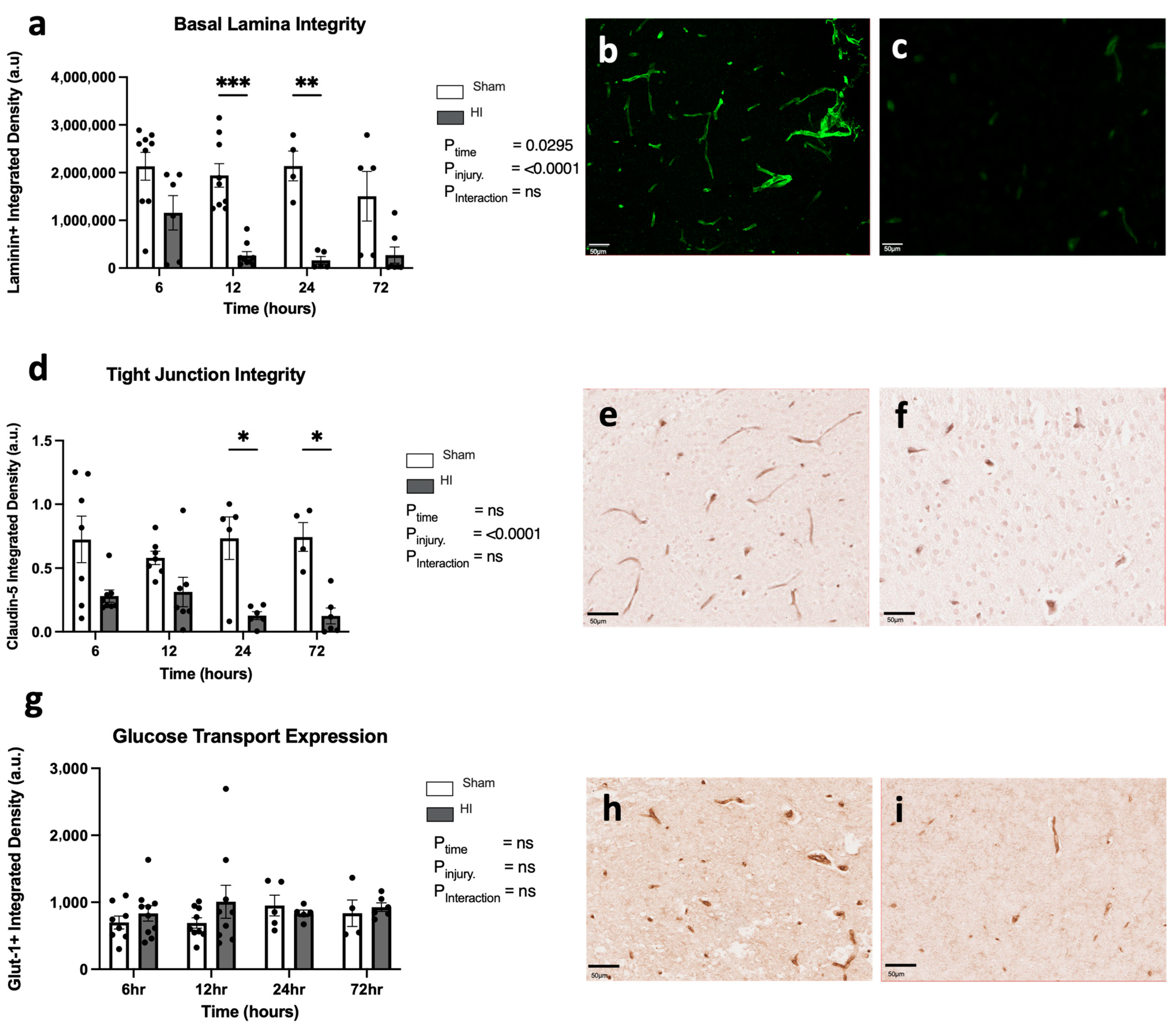
- Progression of HI-mediated BBB breakdown: Structural integrity
- Microbleed correlation analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Butt, A.M. Chapter 3—Physiology of the Blood–Brain Barrier and Mechanisms of Transport across the BBB. In Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors; Kesharwani, P., Gupta, U., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 49–74. [Google Scholar]
- Amarenco, P.; Bogousslavsky, J.; Caplan, L.R.; Donnan, G.A.; Hennerici, M.G. Classification of stroke subtypes. Cerebrovasc. Dis. 2009, 27, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, E.; Chevignard, M.; Vuillerot, C.; Tiberghien, A.; Godard, I. Pediatric stroke rehabilitation: A review of techniques facilitating motor recovery. Ann. Phys. Rehabil. Med. 2016, 59, e2. [Google Scholar] [CrossRef]
- Shipley, L.; Gale, C.; Sharkey, D. Trends in the incidence and management of hypoxic-ischaemic encephalopathy in the therapeutic hypothermia era: A national population study. Arch. Dis. Child Fetal Neonatal Ed. 2021, 106, 529–534. [Google Scholar] [CrossRef]
- Deveber, G.; Andrew, M.; Adams, C.; Bjornson, B.; Booth, F.; Buckley, D.J.; Camfield, C.S.; David, M.; Humphreys, P.; Langevin, P.; et al. Cerebral sinovenous thrombosis in children. N. Engl. J. Med. 2001, 345, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Pin, T.W.; Eldridge, B.; Galea, M.P. A review of developmental outcomes of term infants with post-asphyxia neonatal encephalopathy. Eur. J. Paediatr. Neurol. 2009, 13, 224–234. [Google Scholar] [CrossRef]
- Basto, F.M.; Lyden, P. Hypothermia in acute ischemic stroke therapy. Handb. Clin. Neurol. 2018, 157, 823–837. [Google Scholar] [PubMed]
- Zhou, K.Q.; Bennet, L.; Wassink, G.; McDouall, A.; Curtis, M.A.; Highet, B.; Stevenson, T.J.; Gunn, A.J.; Davidson, J.O. Persistent cortical and white matter inflammation after therapeutic hypothermia for ischemia in near-term fetal sheep. J. Neuroinflamm. 2022, 19, 139. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.; Zhou, K. Targeting neuroinflammation after therapeutic hypothermia for perinatal hypoxic-ischemic brain injury. Neural Regen. Res. 2023, 18, 1261–1262. [Google Scholar] [CrossRef]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The yin and yang of microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef]
- McDonald, C.A.; Penny, T.R.; Paton, M.C.B.; Sutherland, A.E.; Nekkanti, L.; Yawno, T.; Castillo-Melendez, M.; Fahey, M.C.; Jones, N.M.; Jenkin, G.; et al. Effects of umbilical cord blood cells, and subtypes, to reduce neuroinflammation following perinatal hypoxic-ischemic brain injury. J. Neuroinflamm. 2018, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Salman, R.A.-S.; Warach, S.; Launer, L.J.; Van Buchem, M.; Breteler, M.M. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.M.; Chaudhary, U.J.; Brown, M.M.; Yousry, T.A.; Kallis, C.; Jager, H.R.; Werring, D.J. The Microbleed Anatomical Rating Scale (MARS). Reliability of a tool to map brain microbleeds. Neurology 2009, 73, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, C.; Salman, R.A.-S.; Wardlaw, J. Spontaneous brain microbleeds: Systematic review, subgroup analyses and standards for study design and reporting. Brain 2007, 130 Pt 8, 1988–2003. [Google Scholar] [CrossRef]
- Rocha, A.K.A.d.A.; Allison, B.J.; Yawno, T.; Polglase, G.R.; Sutherland, A.E.; Malhotra, A.; Jenkin, G.; Castillo-Melendez, M.; Miller, S.L. Early- versus Late-Onset Fetal Growth Restriction Differentially Affects the Development of the Fetal Sheep Brain. Dev. Neurosci. 2017, 39, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Adelson, P.D.; Lifshitz, J. Microglia: Dismantling and rebuilding circuits after acute neurological injury. Metab. Brain Dis. 2015, 30, 393–400. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Bell, A.; Watt, A.P.; Dudink, I.; Pham, Y.; Sutherland, A.E.; Allison, B.J.; McDonald, C.A.; Castillo-Melendez, M.; Jenkin, G.; Malhotra, A.; et al. Endothelial colony forming cell administration promotes neurovascular unit development in growth restricted and appropriately grown fetal lambs. Stem Cell Res. Ther. 2023, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Melendez, M.; Baburamani, A.A.; Cabalag, C.; Yawno, T.; Witjaksono, A.; Miller, S.L.; Walker, D.W. Experimental Modelling of the Consequences of Brief Late Gestation Asphyxia on Newborn Lamb Behaviour and Brain Structure. PLoS ONE 2013, 8, e77377. [Google Scholar] [CrossRef]
- Mallard, C.; Ek, C.J.; Vexler, Z.S. The myth of the immature barrier systems in the developing brain: Role in perinatal brain injury. J. Physiol. 2018, 596, 5655–5664. [Google Scholar] [CrossRef]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier mechanisms in the developing brain. Front. Pharmacol. 2012, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, K.; Fukuda, A.; Togari, H.; Wada, Y.; Nishino, H. Vulnerability to Cerebral Hypoxic-Ischemic Insult in Neonatal but Not in Adult Rats Is in Parallel with Disruption of the Blood-Brain Barrier. Stroke 1997, 28, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Ek, C.J.; D’Angelo, B.; Baburamani, A.; Lehner, C.; Leverin, A.-L.; Smith, P.L.; Nilsson, H.; Svedin, P.; Hagberg, H.; Mallard, C. Brain Barrier Properties and Cerebral Blood Flow in Neonatal Mice Exposed to Cerebral Hypoxia-Ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Aridas, J.D.; Yawno, T.; Sutherland, A.E.; Nitsos, I.; Ditchfield, M.; Wong, F.Y.; Fahey, M.C.; Malhotra, A.; Wallace, E.M.; Jenkin, G.; et al. Detecting brain injury in neonatal hypoxic ischemic encephalopathy: Closing the gap between experimental and clinical research. Exp. Neurol. 2014, 261, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.C.; Nesic, O.B.; Perez-Polo, J.R. Oxygen resuscitation does not ameliorate neonatal hypoxia/ischemia-induced cerebral edema. J. Neurosci. Res. 2010, 88, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J.P.; Drukarch, B.; Van Dam, A.-M. (Peri)vascular production and action of pro-inflammatory cytokines in brain pathology. Clin. Sci. 2007, 112, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The Neurovascular Unit: Effects of Brain Insults During the Perinatal Period. Front. Neurosci. 2019, 13, 1452. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A critical role in ischemic stroke. CNS Neurosci. Ther. 2021, 27, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.C. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann. Neurol. 2008, 63, 272–287. [Google Scholar] [CrossRef]
- Jiao, H.; Wang, Z.; Liu, Y.; Wang, P.; Xue, Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J. Mol. Neurosci. 2011, 44, 130–139. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol.-Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, S.J.; Seaman, L.B.; Vannucci, R.C. Effects of Hypoxia-Ischemia on GLUT1 and GLUT3 Glucose Transporters in Immature Rat Brain. J. Cereb. Blood Flow Metab. 1996, 16, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Melendez, M.; Yawno, T.; Allison, B.J.; Jenkin, G.; Wallace, E.M.; Miller, S.L. Cerebrovascular adaptations to chronic hypoxia in the growth restricted lamb. Int. J. Dev. Neurosci. 2015, 45, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood–Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Gamdzyk, M.; Lenahan, C.; Tang, J.; Tan, S.; Zhang, J.H. The Dual Role of Microglia in Blood-Brain Barrier Dysfunction after Stroke. Curr. Neuropharmacol. 2020, 18, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood–brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef]
- Hedtjärn, M.; Mallard, C.; Hagber, H.G. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2004, 24, 1333–1351. [Google Scholar] [CrossRef]
- Melani, A.; Dettori, I.; Corti, F.; Cellai, L.; Pedata, F. Time-course of protection by the selective A2A receptor antagonist SCH58261 after transient focal cerebral ischemia. Neurol. Sci. 2015, 36, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Constanzo, J.; Midavaine, É.; Fouquet, J.; Lepage, M.; Descoteaux, M.; Kirby, K.; Tremblay, L.; Masson-Côté, L.; Geha, S.; Longpré, J.-M.; et al. Brain irradiation leads to persistent neuroinflammation and long-term neurocognitive dysfunction in a region-specific manner. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 102, 109954. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Vetreno, R.P. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 2015, 233, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Karperien, A.L.; Jelinek, H.F.; Buchan, A.M. Box-Counting Analysis of Microglia Form in Schizophrenia, Alzheimer’s Disease and Affective Disorder. Fractals 2008, 16, 103–107. [Google Scholar] [CrossRef]
- Izzy, S.; Liu, Q.; Fang, Z.; Lule, S.; Wu, L.; Chung, J.Y.; Sarro-Schwartz, A.; Brown-Whalen, A.; Perner, C.; Hickman, S.E.; et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Front. Cell. Neurosci. 2019, 13, 307. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, A.E.; Salm, A.K. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J. Neurosci. 2003, 23, 7759–7766. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. The multifaceted profile of activated microglia. Mol. Neurobiol. 2009, 40, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Baburamani, A.A.; Supramaniam, V.G.; Hagberg, H.; Mallard, C. Microglia toxicity in preterm brain injury. Reprod. Toxicol. 2014, 48, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D. Ameboid microglia as effectors of inflammation in the central nervous system. J. Neurosci. Res. 1987, 18, 155–171. [Google Scholar] [CrossRef]
- Hellström Erkenstam, N.; Smith, P.L.P.; Fleiss, B.; Nair, S.; Svedin, P.; Wang, W.; Boström, M.; Gressens, P.; Hagberg, H.; Brown, K.L.; et al. Temporal Characterization of Microglia/Macrophage Phenotypes in a Mouse Model of Neonatal Hypoxic-Ischemic Brain Injury. Front. Cell. Neurosci. 2016, 10, 286. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/Macrophage Polarization Dynamics Reveal Novel Mechanism of Injury Expansion After Focal Cerebral Ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, L.; Wang, X.; Jiang, R.; Bai, Q.; Wang, G. Microglia: A Double-Edged Sword in Intracerebral Hemorrhage from Basic Mechanisms to Clinical Research. Front. Immunol. 2021, 12, 675660. [Google Scholar] [CrossRef] [PubMed]
- Penny, T.R.; Pham, Y.; Sutherland, A.E.; Lee, J.; Jenkin, G.; Fahey, M.C.; Miller, S.L.; McDonald, C.A. Umbilical cord blood therapy modulates neonatal hypoxic ischemic brain injury in both females and males. Sci. Rep. 2021, 11, 15788. [Google Scholar] [CrossRef] [PubMed]
- Hoesch, R.E.; Geocadin, R.G. Therapeutic hypothermia for global and focal ischemic brain injury—A cool way to improve neurologic outcomes. Neurologist 2007, 13, 331–342. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 h | 12 h | 24 h | 72 h | |
|---|---|---|---|---|
| Number (n) | ||||
| Sham | 9 | 9 | 6 | 5 |
| HI | 10 | 9 | 5 | 7 |
| Sex ratio (F/M) | ||||
| Sham | 6/3 | 5/4 | 1/5 | 2/3 |
| HI | 5/5 | 6/3 | 1/4 | 4/3 |
| Brain weight (g) | ||||
| Sham | 0.97 ± 0.018 | 0.994 ± 0.016 | 0.87 ± 0.005 | 1.05 ± 0.012 |
| HI | 0.997 ± 0.015 | 1.006 ± 0.017 | 0.835 ± 0.021 | 1.018 ± 0.017 |
| Pup mortality | ||||
| Sham | 0 | 0 | 0 | 0 |
| HI | 0 | 0 | 0 | 0 |
| Antibody | Target | Antigen Retrieval | Block Endogenous Peroxidases | Protein Block | Primary Antibody | Primary Antibody Concentration | Secondary Antibody |
|---|---|---|---|---|---|---|---|
| NeuN (Merck Millipore, Burlington, MA, USA; cat#MAB377) | Neurons | Heated in 0.1 M of citric acid buffer (pH 6) for 15 min and left to cool for 20 min | 3% H202 in PBS for 10 min | 5% NGS +1% BSA in PBS for 30 min | Mouse anti-NueN in 1% BSA + 0.3% TX-PBS | 1:1000 | Goat anti-mouse, biotinylated IgG |
| Iba-1 (Wako Pure Chemical Industries, Osaka, Japan; cat#019-19741) | Microglia | Heated in 0.01 M of citric buffer (pH 6) for 15 min and left to cool for 20 min | 3% H202 in PBS for 30 min | 10% NGS in 0.1 M PBS for 30 min | Rabbit anti-Iba-1 in 0.2% TX-PBS | 1:1000 | Goat anti-rabbit, biotinylated IgG |
| Glut-1 (Sigma-Aldrich, St. Louis, MI, USA; cat#ABI4683) | Glucose transporters | Heated in 0.01 M of citric buffer (pH 6) for 20 mis and left to cool for 20 min | 0.3% H202 in PBS for 15 min | DAKO protein-free blocker for 1 h | Rabbit anti- Glut-1 in PBS | 1:200 | Goat anti-rabbit, biotinylated IgG |
| Glial fibrillary acidic protein (GFAP) (Sigma-Aldrich, St. Louis, MI, USA; cat#G3893) | Reactive astrocytes | Heated in 0.01 M of citric acid buffer (pH 6) for 9 min and left to cool for 15 min | 0.3% H202 in 50% methanol for 20 min | 5% NGS in PBS for 30 min | Mouse anti-GFAP in 0.3% in TX-PBS | 1:400 | Goat anti-mouse, biotinylated IgG |
| Laminin (Sigma-Aldrich, St. Louis, MI, USA; cat#NB300-144) | Basal lamina | 1:500 proteinase K for 30 min at 37 degrees, humidified | N/A | 10% NGS in 0.1% TX-PBS for 30 min | Rabbit anti-laminin in 0.1% PBST and 2% NGS | 1:200 | Goat anti-rabbit-488 |
| Albumin (Waltham, MA, USA; A110-134A) | Albumin | N/A | 3% hydrogen peroxide in 50% methanol for 10 min | 5% NRS + 3% BSA in PBS-TX for 90 min | Sheep anti-rat albumin in 0.1% TX-PBS and 0.5% fish gelatin | 1:2000 | Rabbit anti-sheep |
| Outcome | R2 Value | p-Value | Confidence Interval |
|---|---|---|---|
| Tissue Loss | |||
| 6 | 0.003856 | 0.7948 | −1.415, 1.099 |
| 12 | 0.008832 | 0.7107 | −0.7536, 0.5258 |
| 24 | 0.5758 | 0.0178 | 0.5229, 3.969 |
| 72 | 0.03043 | 0.5877 | −2.060, 3.444 |
| Apoptosis | |||
| 6 | 0.6381 | <0.0001 | 0.05811, 0.1353 |
| 12 | 0.2987 | 0.0127 | 0.01686, 0.1229 |
| 24 | 0.5821 | 0.0168 | 0.08507, 0.6160 |
| 72 | 0.5488 | 0.0058 | 0.2593, 1.177 |
| Neuronal Loss | |||
| 6 | 0.4963 | 0.0008 | −21.35, −6.823 |
| 12 | 0.8546 | <0.0001 | −13.09, −8.392 |
| 24 | 0.7372 | 0.0030 | −18.51, −5.629 |
| 72 | 0.5729 | 0.0044 | −27.99, −6.816 |
| Resting Microglia | |||
| 6 | 0.1253 | 0.1257 | −0.3446, 0.04604 |
| 12 | 0.7597 | <0.0001 | −0.6013, −0.3252 |
| 24 | 0.3116 | 0.1183 | −1.668, 0.2353 |
| 72 | 0.5207 | 0.0081 | −2.228, −0.4307 |
| Intermediate Microglia | |||
| 6 | 0.4138 | 0.0022 | 0.4358, 1.687 |
| 12 | 0.8465 | <0.0001 | 1.030, 1.630 |
| 24 | 0.1907 | 0.2399 | −0.4374, 1.477 |
| 72 | 0.1855 | 0.1622 | −0.4843, 0.09319 |
| Activated Microglia | |||
| 6 | 0.3543 | 0.0056 | 0.2127, 1.070 |
| 12 | 0.8247 | <0.0001 | 0.5544, 0.9131 |
| 24 | 0.5861 | 0.0162 | 0.2473, 1.739 |
| 72 | 0.5959 | 0.0033 | 2.594, 9.764 |
| Total Microglia | |||
| 6 | 0.2622 | 0.0210 | 0.2590, 2.799 |
| 12 | 0.7849 | <0.0001 | 1.722, 3.045 |
| 24 | 0.1232 | 0.3543 | −1.239, 3.029 |
| 72 | 0.5158 | 0.0085 | 1.514, 8.025 |
| Basal Lamina Integrity | |||
| 6 | 0.1603 | 0.0803 | −1437, 89.98 |
| 12 | 0.4751 | 0.0016 | −2097, −596.6 |
| 24 | 0.3064 | 0.1220 | −1825, 268.3 |
| 72 | 0.3470 | 0.0439 | −2330, −39.61 |
| Tight-Junction Integrity | |||
| 6 | 0.07908 | 0.2297 | −0.1054, 0.02702 |
| 12 | 0.2830 | 0.0231 | −0.05113, −0.004337 |
| 24 | 0.4918 | 0.0353 | −0.1409, −0.006748 |
| 72 | 0.6159 | 0.0025 | −0.1883, −0.05368 |
| Astrogliosis | |||
| 6 | 0.000642 | 0.9155 | −97,873, 88,340 |
| 12 | 0.0173 | 0.6028 | −36,306, 60,560 |
| 24 | 0.511 | 0.0302 | 12,014, 177,005 |
| 72 | 0.01398 | 0.7143 | −143,535, 201,924 |
| Glucose Transport Expression | |||
| 6 | 0.01526 | 0.6039 | −47.72, 79.76 |
| 12 | 0.03846 | 0.4354 | −39.92, 88.32 |
| 24 | 0.01302 | 0.7701 | −24.01, 31.09 |
| 72 | 0.01084 | 0.7474 | −41.57, 30.81 |
| Albumin Extravasation | |||
| 6 | 0.5837 | 0.0006 | 0.5245, 1.509 |
| 12 | 0.5975 | 0.0012 | 0.6644, 2.082 |
| 24 | 0.4710 | 0.0412 | 0.07557, 2.784 |
| 72 | 0.6678 | 0.0012 | 1.183, 3.520 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jithoo, A.; Penny, T.R.; Pham, Y.; Sutherland, A.E.; Smith, M.J.; Petraki, M.; Fahey, M.C.; Jenkin, G.; Malhotra, A.; Miller, S.L.; et al. The Temporal Relationship between Blood–Brain Barrier Integrity and Microglial Response following Neonatal Hypoxia Ischemia. Cells 2024, 13, 660. https://doi.org/10.3390/cells13080660
Jithoo A, Penny TR, Pham Y, Sutherland AE, Smith MJ, Petraki M, Fahey MC, Jenkin G, Malhotra A, Miller SL, et al. The Temporal Relationship between Blood–Brain Barrier Integrity and Microglial Response following Neonatal Hypoxia Ischemia. Cells. 2024; 13(8):660. https://doi.org/10.3390/cells13080660
Chicago/Turabian StyleJithoo, Arya, Tayla R. Penny, Yen Pham, Amy E. Sutherland, Madeleine J. Smith, Maria Petraki, Michael C. Fahey, Graham Jenkin, Atul Malhotra, Suzanne L. Miller, and et al. 2024. "The Temporal Relationship between Blood–Brain Barrier Integrity and Microglial Response following Neonatal Hypoxia Ischemia" Cells 13, no. 8: 660. https://doi.org/10.3390/cells13080660
APA StyleJithoo, A., Penny, T. R., Pham, Y., Sutherland, A. E., Smith, M. J., Petraki, M., Fahey, M. C., Jenkin, G., Malhotra, A., Miller, S. L., & McDonald, C. A. (2024). The Temporal Relationship between Blood–Brain Barrier Integrity and Microglial Response following Neonatal Hypoxia Ischemia. Cells, 13(8), 660. https://doi.org/10.3390/cells13080660