
Development and Characterisation of a New Patient-Derived Xenograft Model of AR-Negative Metastatic Castration-Resistant Prostate Cancer

 , , , ,
, , , , 
Abstract
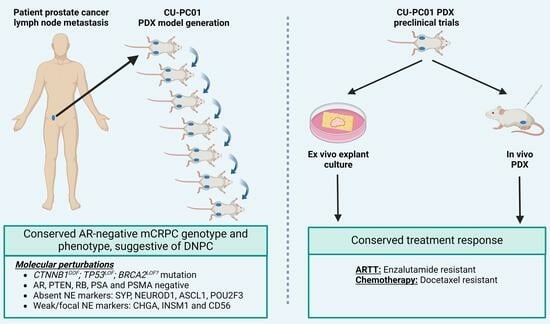
1. Introduction
2. Materials and Methods
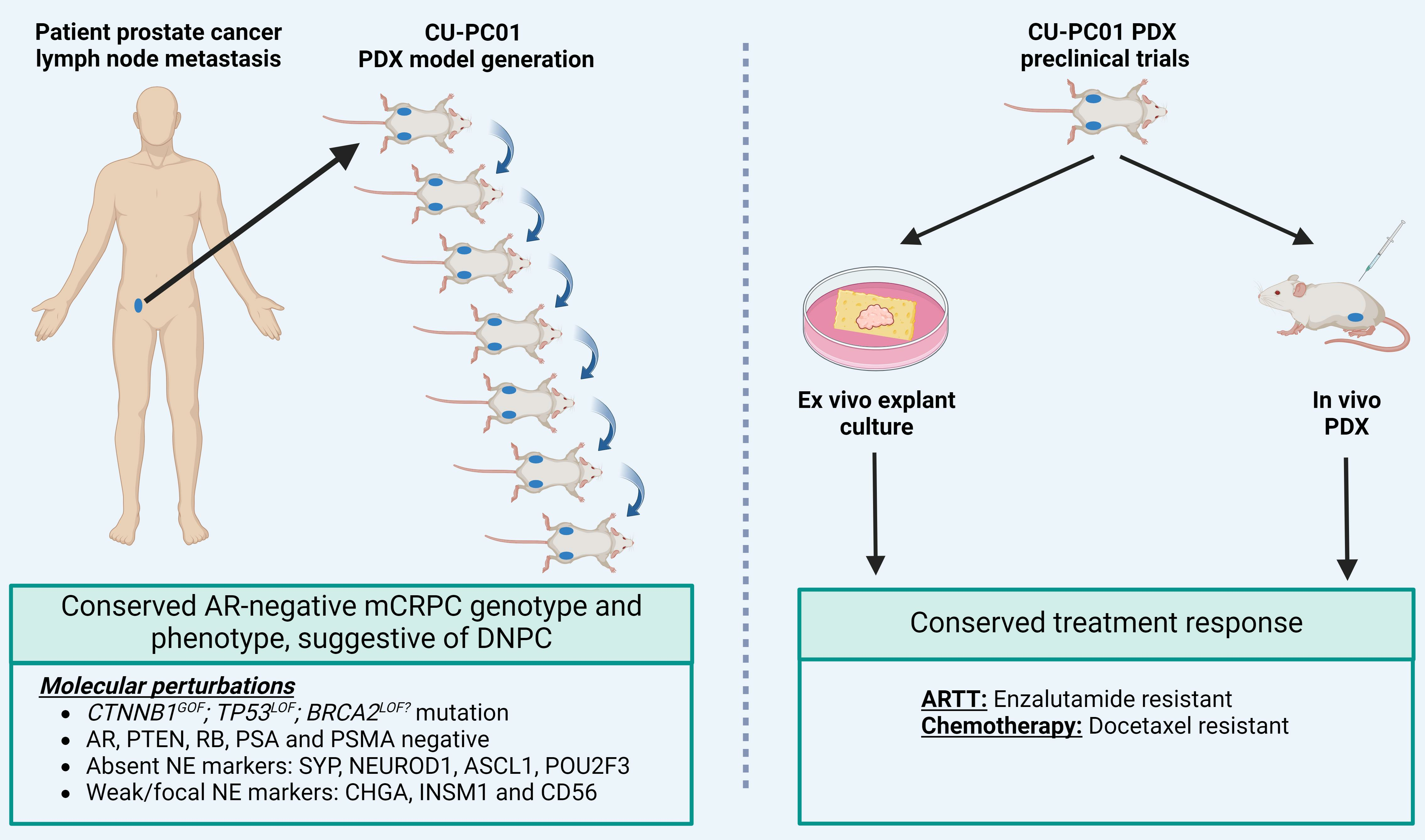
2.1. Patient Sample Collection and Processing
2.2. CU-PC01 PDX Model Generation and Cryopreservation
2.3. DNA Extraction and Whole Exome Sequencing
2.4. Gene Signature Enrichment
2.5. Epstein–Barr Virus (EBV) Detection
2.6. Immunohistochemistry
2.7. Protein Isolation and Western Blotting
2.8. RNA Isolation and QRT-PCR
2.9. Ex Vivo Explant Preclinical Trials
2.10. In Vivo Preclinical Trials
2.11. Statistical Analysis
3. Results
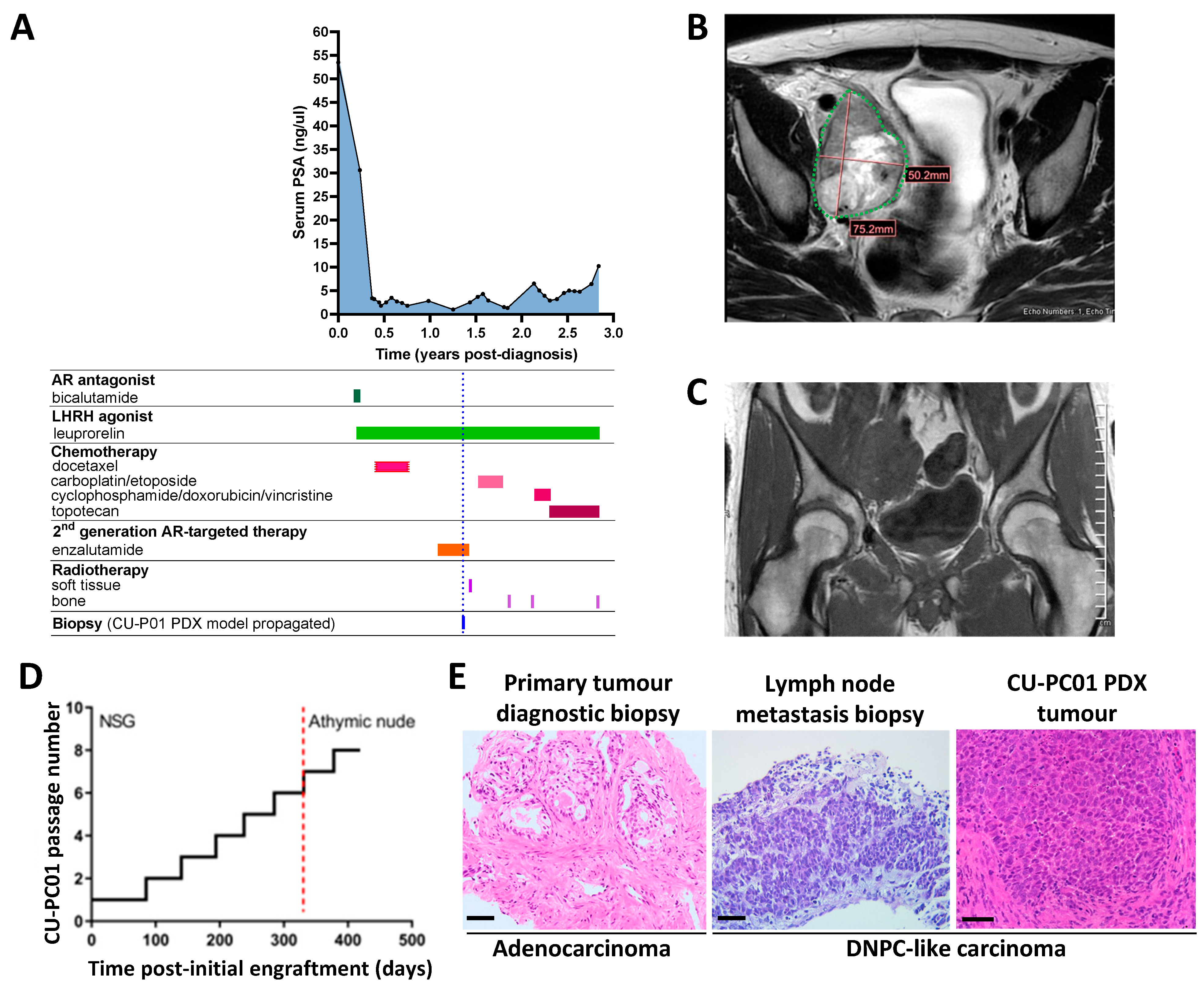
3.1. Propagation of the CU-PC01 PDX Model
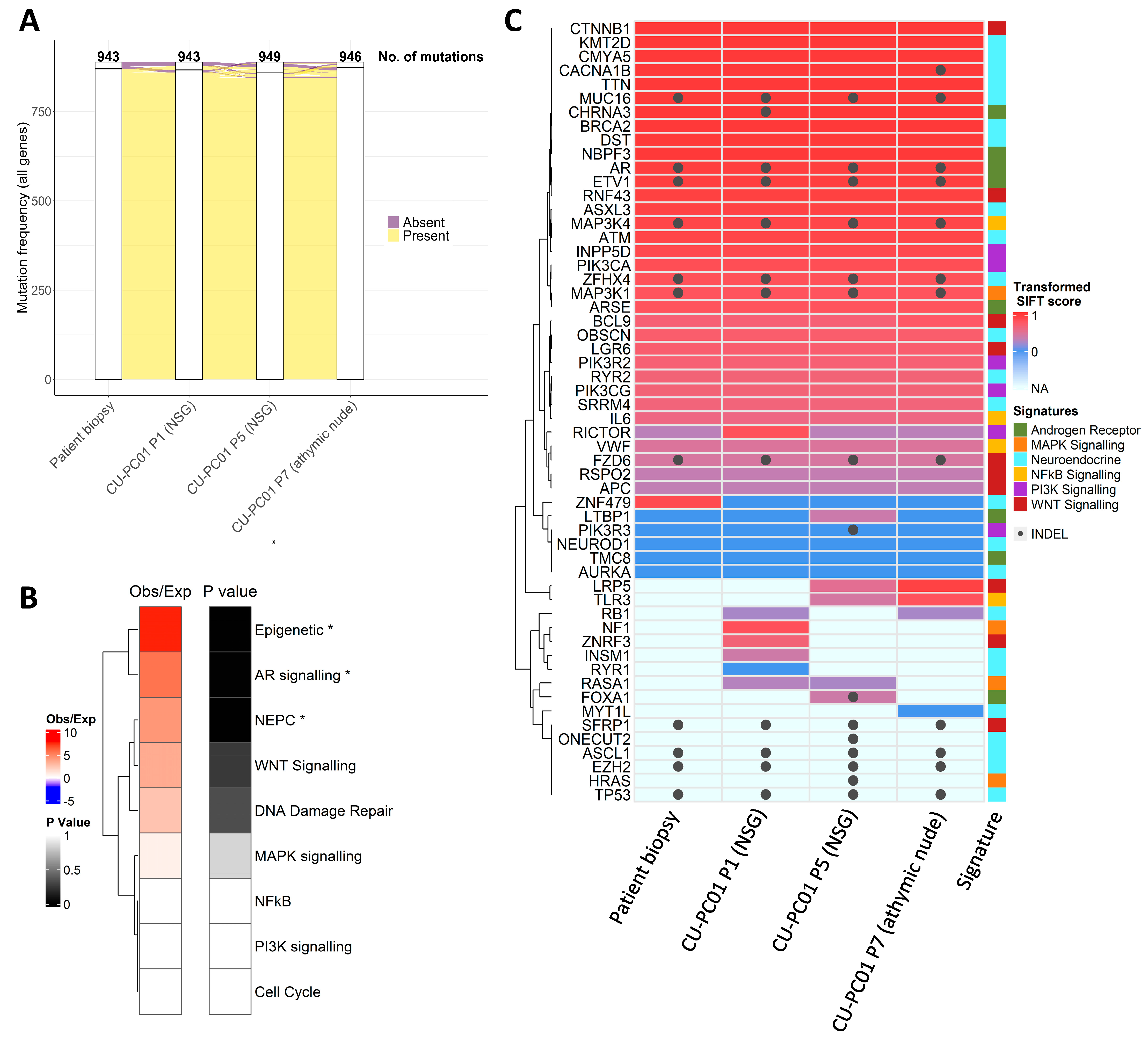
3.2. The CU-PC01 PDX Model Retains the Mutational Landscape of the Donor Patient
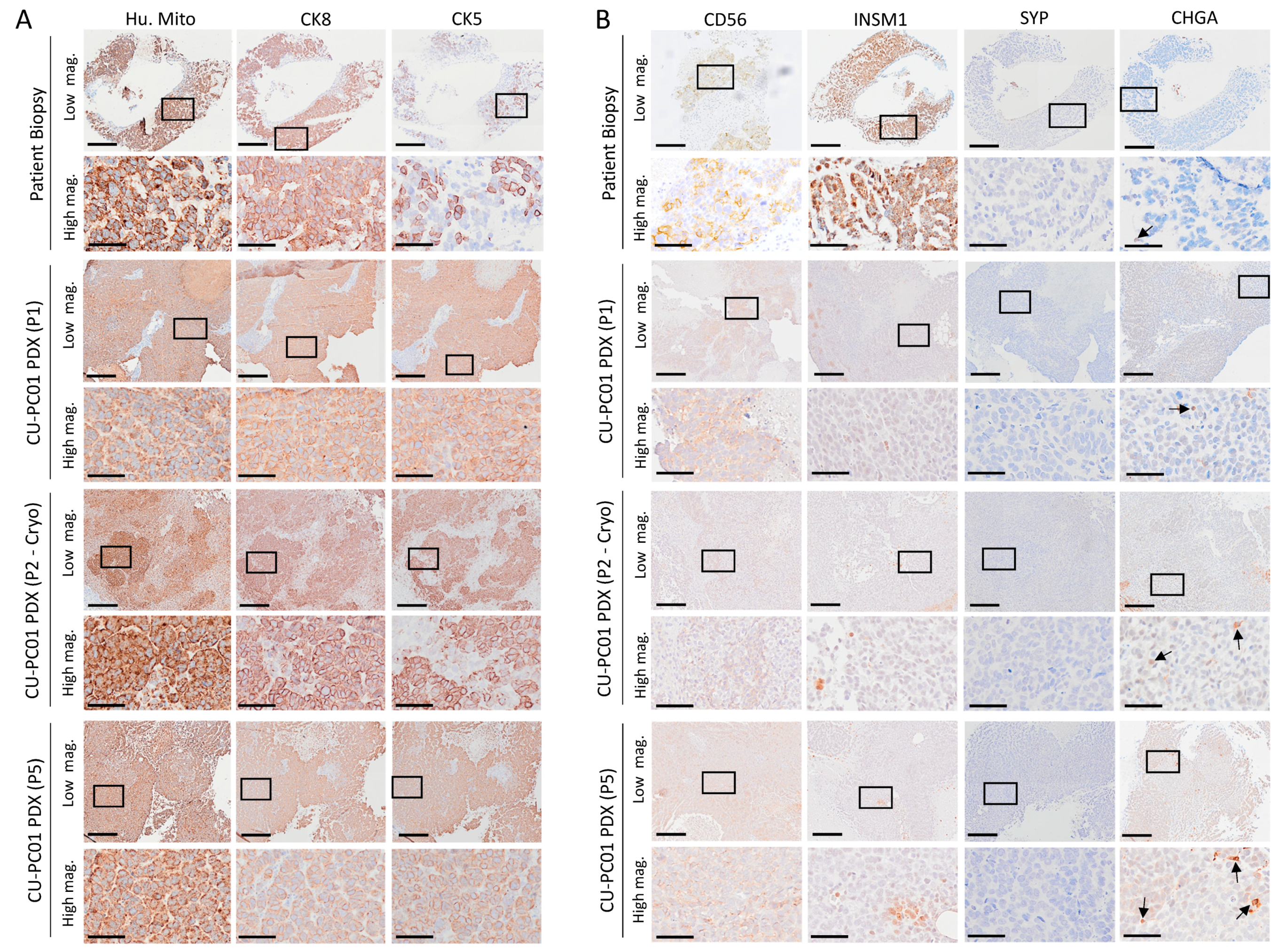
3.3. The CU-PC01 PDX Model Expresses Both Basal and Luminal Prostate Epithelial Markers, Whereas NE Markers Are Absent or Weakly Expressed
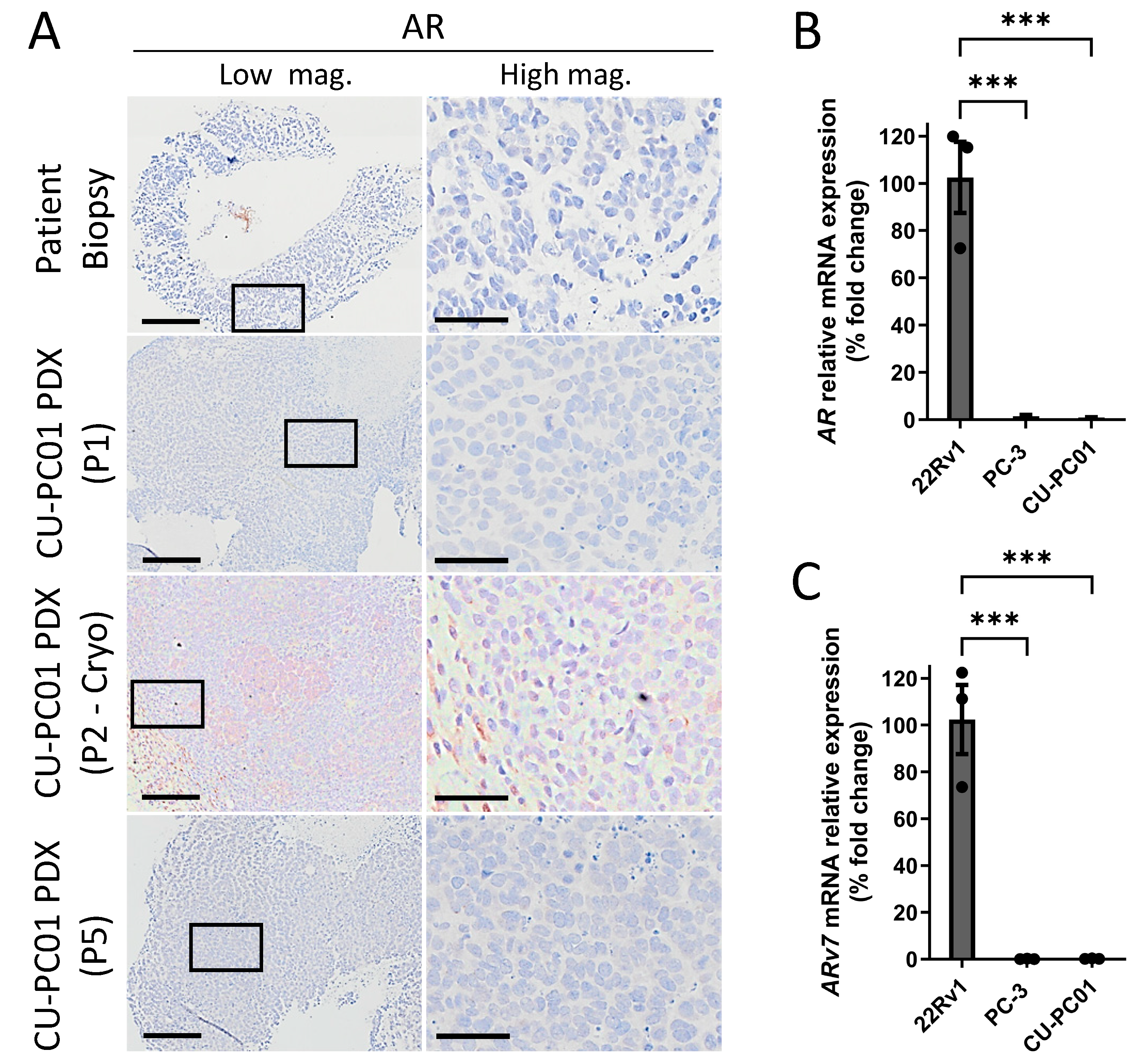
3.4. The CU-PC01 mCRPC PDX Model Is AR Negative
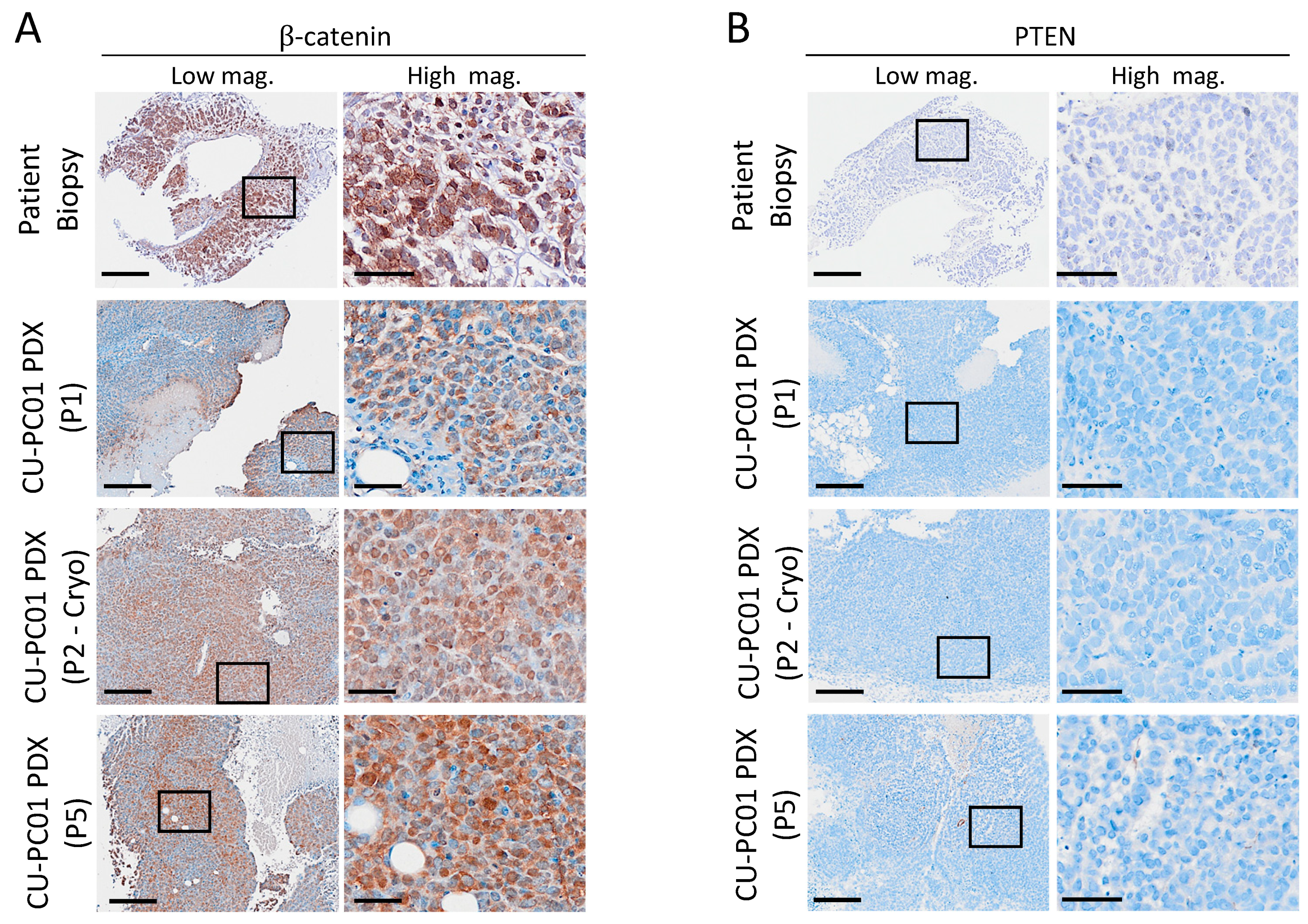
3.5. Characterisation of Wnt and PI3K Signalling Status in the CU-PC01 PDX Model
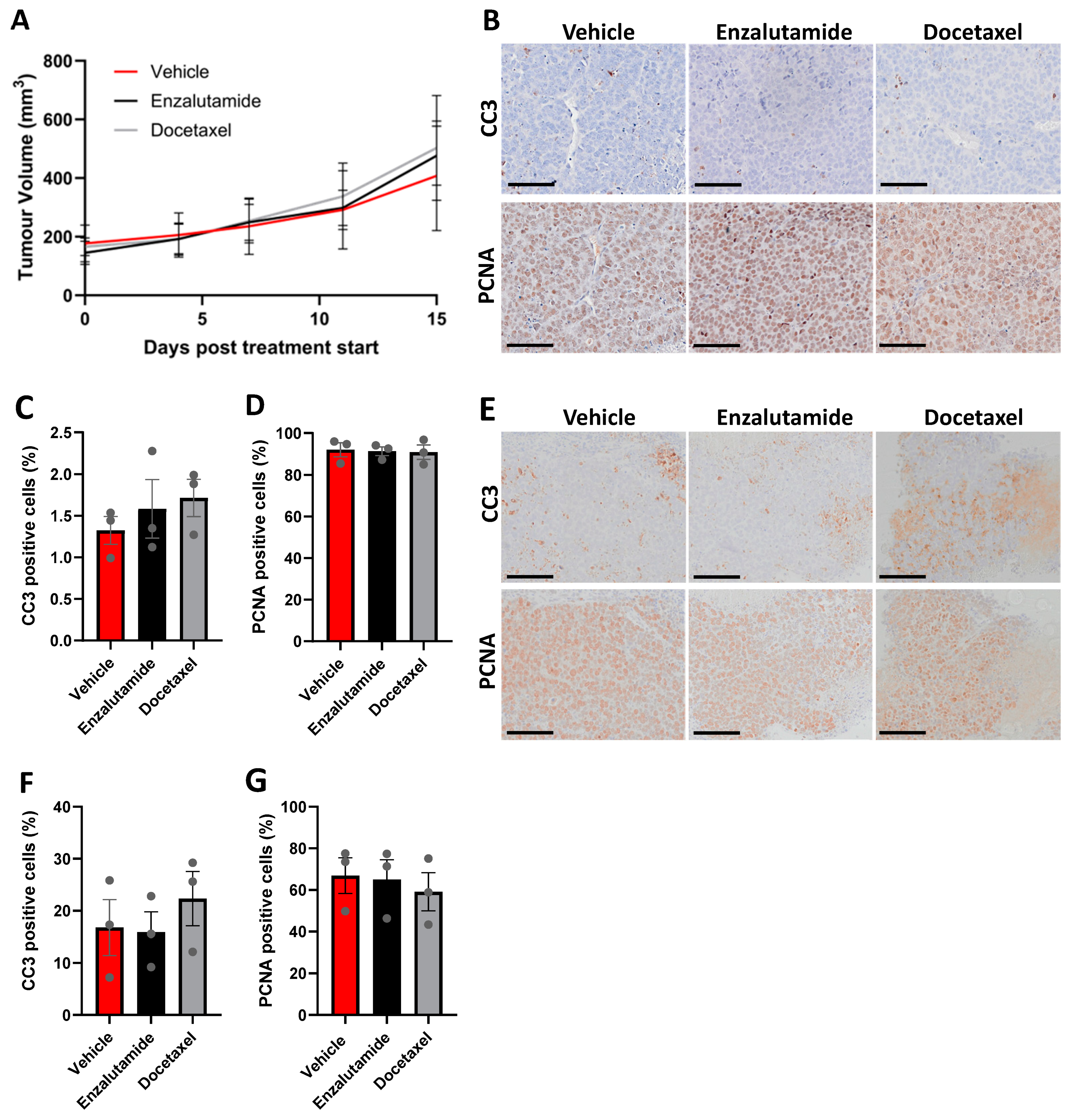
3.6. CU-PC01 PDX Tumours Are Resistant to Docetaxel and Enzalutamide Monotherapy
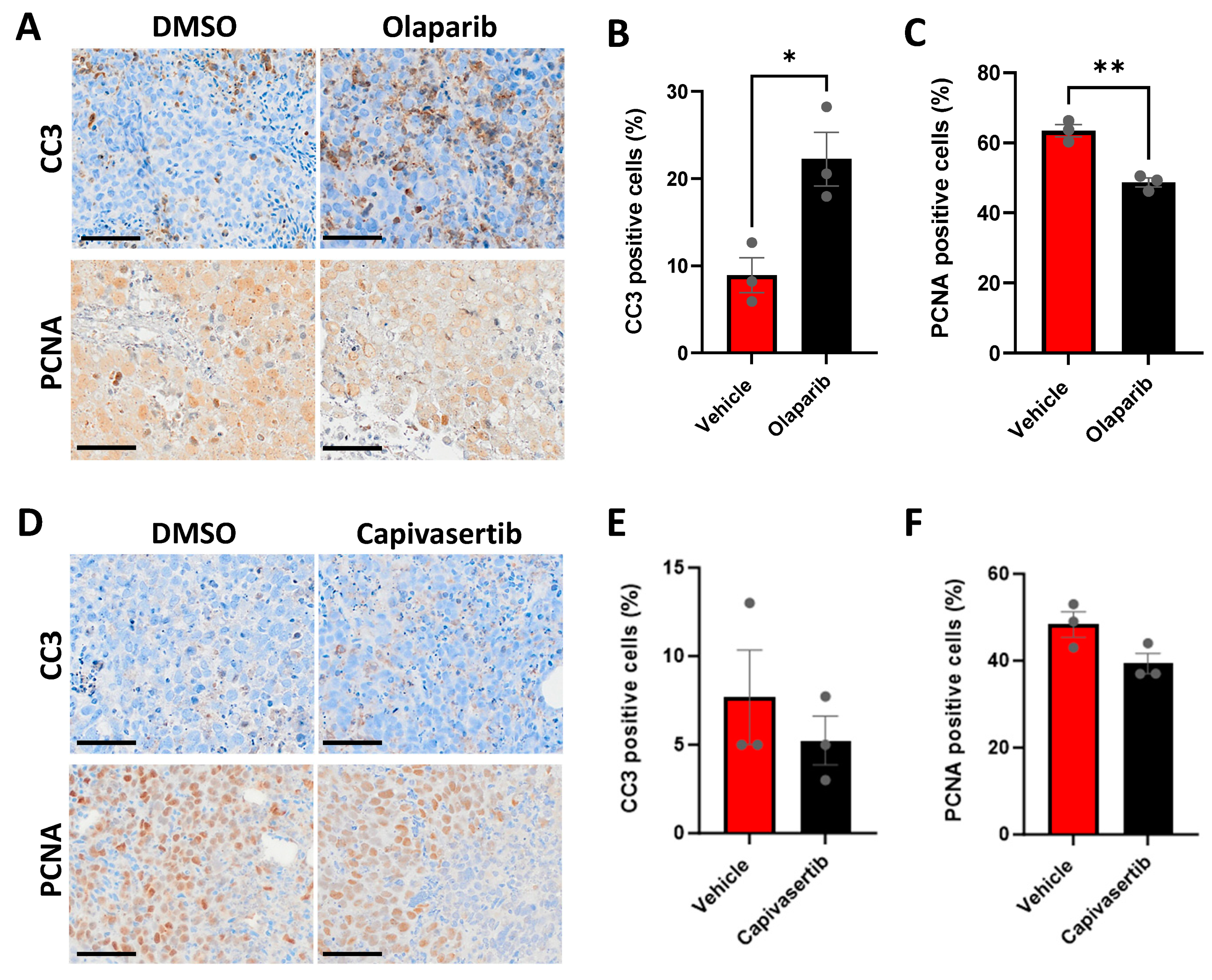
3.7. CU-PC01 PDX Ex Vivo Explants Are Sensitive to the PARP Inhibitor Olaparib
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Oh, W.K.; Miao, R.; Vekeman, F.; Sung, J.; Cheng, W.Y.; Gauthier-Loiselle, M.; Dhawan, R.; Duh, M.S. Real-world Characteristics and Outcomes of Patients With Metastatic Castration-resistant Prostate Cancer Receiving Chemotherapy Versus Androgen Receptor-targeted Therapy After Failure of First-line Androgen Receptor-targeted Therapy in the Community Setting. Clin. Genitourin. Cancer 2017, 16, 50–57. [Google Scholar] [CrossRef]
- Beltran, H.; Demichelis, F. Therapy considerations in neuroendocrine prostate cancer: What next? Endocr. Relat. Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e476. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.-M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed Morphologic Classification of Prostate Cancer With Neuroendocrine Differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Zhang, T.; Small, E.J.; Armstrong, A.J. Neuroendocrine Prostate Cancer: Subtypes, Biology, and Clinical Outcomes. J. Natl. Compr. Cancer Netw. 2014, 12, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Alabi, B.R.; Liu, S.; Stoyanova, T. Current and emerging therapies for neuroendocrine prostate cancer. Pharmacol. Ther. 2022, 238, 108255. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.K.; Yamada, Y.; Ku, S.-Y.; Venkadakrishnan, V.B.; Korsen, J.A.; Kalidindi, T.M.; Mizuno, K.; Ahn, S.H.; Seo, J.-H.; Garcia, M.M.; et al. Landscape of prostate-specific membrane antigen heterogeneity and regulation in AR-positive and AR-negative metastatic prostate cancer. Nat. Cancer 2023, 4, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Korsen, J.A.; Gutierrez, J.A.; Tully, K.M.; Carter, L.M.; Samuels, Z.V.; Khitrov, S.; Poirier, J.T.; Rudin, C.M.; Chen, Y.; Morris, M.J.; et al. Delta-like ligand 3-targeted radioimmunotherapy for neuroendocrine prostate cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2203820119. [Google Scholar] [CrossRef]
- Puca, L.; Gavyert, K.; Sailer, V.; Conteduca, V.; Dardenne, E.; Sigouros, M.; Isse, K.; Kearney, M.; Vosoughi, A.; Fernandez, L.; et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci. Transl. Med. 2019, 11, eaav0891. [Google Scholar] [CrossRef] [PubMed]
- Sayar, E.; Patel, R.A.; Coleman, I.M.; Roudier, M.P.; Zhang, A.; Mustafi, P.; Low, J.-Y.; Hanratty, B.; Ang, L.S.; Bhatia, V.; et al. Reversible epigenetic alterations mediate PSMA expression heterogeneity in advanced metastatic prostate cancer. JCI Insight 2023, 8, 162907. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Dong, X.; Gleave, M. Molecular model for neuroendocrine prostate cancer progression. BJU Int. 2018, 122, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Nguyen, H.M.; Corey, E. Generation of Prostate Cancer Patient-Derived Xenografts to Investigate Mechanisms of Novel Treatments and Treatment Resistance. Methods Mol. Biol. 2018, 1786, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Clark, A.K.; Porter, L.H.; Toivanen, R.; Bakshi, A.; Lister, N.L.; Pook, D.; Pezaro, C.J.; Sandhu, S.; Keerthikumar, S.; et al. The MURAL collection of prostate cancer patient-derived xenografts enables discovery through preclinical models of uro-oncology. Nat. Commun. 2021, 12, 5049. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.-M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, N.; Yang, J.; Shepherd, P.D.A.; Li-Ning-Tapia, E.M.; Labanca, E.; Manyam, G.C.; Ravoori, M.K.; Kundra, V.; Araujo, J.C.; Efstathiou, E.; et al. The MD Anderson Prostate Cancer Patient-derived Xenograft Series (MDA PCa PDX) Captures the Molecular Landscape of Prostate Cancer and Facilitates Marker-driven Therapy Development. Clin. Cancer Res. 2020, 26, 4933–4946. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Zhu, Y.; Coleman, I.M.; Dalrymple, S.L.; Antony, L.; Patel, R.A.; Hanratty, B.; Chikarmane, R.; Meeker, A.K.; Zheng, S.L.; et al. Resistance to androgen receptor signaling inhibition does not necessitate development of neuroendocrine prostate cancer. JCI Insight 2021, 6, 146827. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Taylor, R.A.; Toivanen, R.; Pedersen, J.; Norden, S.; Pook, D.W.; Frydenberg, M.; Papargiris, M.M.; Niranjan, B.; Richards, M.G.; et al. A preclinical xenograft model of prostate cancer using human tumors. Nat. Protoc. 2013, 8, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Parry-Jones, A.; Spary, L.K. The Wales Cancer Bank (WCB). Open J. Bioresour. 2018, 5, 10. [Google Scholar] [CrossRef]
- Porter, L.H.; Lawrence, M.G.; Wang, H.; Clark, A.K.; Bakshi, A.; Obinata, D.; Goode, D.; Papargiris, M.; Mural; Clouston, D.; et al. Establishing a cryopreservation protocol for patient-derived xenografts of prostate cancer. Prostate 2019, 79, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [CrossRef] [PubMed]
- Hieronymus, H.; Lamb, J.; Ross, K.N.; Peng, X.P.; Clement, C.; Rodina, A.; Nieto, M.; Du, J.; Stegmaier, K.; Raj, S.M.; et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006, 10, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, S.; Nevedomskaya, E.; van der Poel, H.G.; de Jong, J.; van Leenders, G.J.; Jenster, G.; Wessels, L.F.; Bergman, A.M.; Zwart, W. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol. Med. 2015, 7, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Gilmore, T.D. Mutations in the NF-κB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [PubMed]
- Biocarta. Nf-Kb Signaling Pathway Gene Set. 2023. Available online: https://maayanlab.cloud/Harmonizome/gene_set/nf-kb+signaling+pathway/Biocarta+Pathways (accessed on 1 December 2023).
- Koushyar, S.; Meniel, V.S.; Phesse, T.J.; Pearson, H.B. Exploring the Wnt Pathway as a Therapeutic Target for Prostate Cancer. Biomolecules 2022, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shi, M.; Chuen Choi, S.Y.; Wang, Y.; Lin, D.; Zeng, H.; Wang, Y. Genomic alterations in neuroendocrine prostate cancer: A systematic review and meta-analysis. BJUI Compass 2023, 4, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 15. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; White, L.R.; Stefanoff, C.G.; de Oliveira, D.E.; Felisbino, F.E.; Klumb, C.E.; Bacchi, C.E.; Seuánez, H.N.; Zalcberg, I.R. Epstein-Barr virus (EBV) detection and typing by PCR: A contribution to diagnostic screening of EBV-positive Burkitt’s lymphoma. Diagn. Pathol. 2006, 1, 17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pearson, H.B.; McCarthy, A.; Collins, C.M.P.; Ashworth, A.; Clarke, A.R. Lkb1 Deficiency Causes Prostate Neoplasia in the Mouse. Cancer Res. 2008, 68, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Perez-Mancera, P.A.; Dow, L.E.; Ryan, A.; Tennstedt, P.; Bogani, D.; Elsum, I.; Greenfield, A.; Tuveson, D.A.; Simon, R.; et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin. Investig. 2011, 121, 4257–4267. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhang, Z.; Kimball, H.; Qu, F.; Berlind, K.; Stopsack, K.H.; Lee, G.M.; Choueiri, T.K.; Kantoff, P.W. Abiraterone Acetate Induces CREB1 Phosphorylation and Enhances the Function of the CBP-p300 Complex, Leading to Resistance in Prostate Cancer Cells. Clin. Cancer. Res. 2021, 27, 2087–2099. [Google Scholar] [CrossRef] [PubMed]
- Thejer, B.M.; Adhikary, P.P.; Teakel, S.L.; Fang, J.; Weston, P.A.; Gurusinghe, S.; Anwer, A.G.; Gosnell, M.; Jazayeri, J.A.; Ludescher, M.; et al. PGRMC1 effects on metabolism, genomic mutation and CpG methylation imply crucial roles in animal biology and disease. BMC Mol. Cell Biol. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.J.; Dong, M.; Gutekunst, M.; Närhi, K.; van Zoggel, H.J.A.A.; Blom, S.; Nagaraj, A.; Metsalu, T.; Oswald, E.; Erkens-Schulze, S.; et al. Capturing complex tumour biology in vitro: Histological and molecular characterisation of precision cut slices. Sci. Rep. 2015, 5, 17187. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Taurozzi, A.J.; Beekharry, R.; Wantoch, M.; Labarthe, M.C.; Walker, H.F.; Seed, R.I.; Simms, M.; Rodrigues, G.; Bradford, J.; van der Horst, G.; et al. Spontaneous development of Epstein-Barr Virus associated human lymphomas in a prostate cancer xenograft program. PLoS ONE 2017, 12, e0188228. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Rajapakshe, K.; Shah, S.S.; Shou, J.; Eedunuri, V.K.; Foley, C.; Fiskus, W.; Rajendran, M.; Chew, S.A.; Zimmermann, M.; et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014, 74, 5631–5643. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- COSMIC. Mutation COSV65953758. Available online: https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=118050141&merge=6986395 (accessed on 1 December 2023).
- Bland, T.; Wang, J.; Yin, L.; Pu, T.; Li, J.; Gao, J.; Lin, T.P.; Gao, A.C.; Wu, B.J. WLS-Wnt signaling promotes neuroendocrine prostate cancer. iScience 2021, 24, 101970. [Google Scholar] [CrossRef]
- Kim, W.K.; Buckley, A.J.; Lee, D.-H.; Hiroto, A.; Nenninger, C.H.; Olson, A.W.; Wang, J.; Li, Z.; Vikram, R.; Adzavon, Y.M.; et al. Androgen deprivation induces double-null prostate cancer via aberrant nuclear export and ribosomal biogenesis through HGF and Wnt activation. Nat. Commun. 2024, 15, 1231. [Google Scholar] [CrossRef]
- Baena, E.; Shao, Z.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fujiwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Sood, A.; Rahimi, H.A.; Wang, W.; Gupta, N.; Hicks, J.; Mosier, S.; Gocke, C.D.; Epstein, J.I.; Netto, G.J.; et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 2014, 20, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Adduri, R.S.R.; George, S.A.; Kavadipula, P.; Bashyam, M.D. SMARCD1 is a transcriptional target of specific non-hotspot mutant p53 forms. J. Cell Physiol. 2020, 235, 4559–4570. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Nandakumar, S.; Hirani, R.; Nguyen, B.; Stopsack, K.H.; Kreitzer, C.; Rajanala, S.H.; Ghale, R.; Mazzu, Y.Z.; Pillarsetty, N.V.K.; et al. The Impact of PIK3R1 Mutations and Insulin-PI3K-Glycolytic Pathway Regulation in Prostate Cancer. Clin. Cancer Res. 2022, 28, 3603–3617. [Google Scholar] [CrossRef] [PubMed]
- Symonds, L.; Konnick, E.; Vakar-Lopez, F.; Cheng, H.H.; Schweizer, M.T.; Nelson, P.S.; Pritchard, C.C.; Montgomery, B. BRCA2 Alterations in Neuroendocrine/Small-Cell Carcinoma Prostate Cancer: A Case Series. JCO Precis. Oncol. 2022, 6, e2200091. [Google Scholar] [CrossRef]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic Analysis of Metastatic Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Altuwaijri, S.; Yeh, S.; Lai, K.-P.; Yu, S.; Chuang, K.-H.; Huang, S.-P.; Lardy, H.; Chang, C. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc. Natl. Acad. Sci. USA 2008, 105, 12188–12193. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.A.; Sayar, E.; Coleman, I.; Roudier, M.P.; Hanratty, B.; Low, J.-Y.; Jaiswal, N.; Ajkunic, A.; Dumpit, R.; Ercan, C.; et al. Characterization of HOXB13 expression patterns in localized and metastatic castration-resistant prostate cancer. J. Pathol. 2024, 262, 105–120. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Hille, C.; Gorges, T.M.; Riethdorf, S.; Mazel, M.; Steuber, T.; Amsberg, G.V.; König, F.; Peine, S.; Alix-Panabières, C.; Pantel, K. Detection of Androgen Receptor Variant 7 (ARV7) mRNA Levels in EpCAM-Enriched CTC Fractions for Monitoring Response to Androgen Targeting Therapies in Prostate Cancer. Cells 2019, 8, 1067. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, Q.; Xu, H. Wnt/β-catenin signal transduction pathway in prostate cancer and associated drug resistance. Discov. Oncol 2021, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Wnt Signaling in Cancer: Not a Binary ON:OFF Switch. Cancer Res. 2019, 79, 5901–5906. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.-K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Turnham, D.J.; Bullock, N.; Dass, M.S.; Staffurth, J.N.; Pearson, H.B. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells 2020, 9, 2342. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN Tail Regulates Protein Stability and Function. Mol. Cell. Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef] [PubMed]
- Van Ree, J.H.; Jeganathan, K.B.; Fierro Velasco, R.O.; Zhang, C.; Can, I.; Hamada, M.; Li, H.; Baker, D.J.; van Deursen, J.M. Hyperphosphorylated PTEN exerts oncogenic properties. Nat. Commun. 2023, 14, 2983. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Foti, M. Non-genomic loss of PTEN function in cancer: Not in my genes. Trends Pharmacol. Sci. 2011, 32, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kránitz, N.; Szepesváry, Z.; Kocsis, K.; Kullmann, T. Neuroendocrine Cancer of the Prostate. Pathol. Oncol. Res. 2020, 26, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Liang, X.; Wu, D.; Chen, S.; Yang, B.; Mao, W.; Shen, D. Clinicopathological characteristics and survival outcomes in neuroendocrine prostate cancer: A population-based study. Medicine 2021, 100, e25237. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Zhang, Y.; Jiang, Z.; Zhao, L.; Fan, L.; Wang, Y.; Xie, S.; Shangguan, X.; Zhu, Y.; Pan, J.; et al. Insulinoma-associated protein 1 is a novel sensitive and specific marker for small cell carcinoma of the prostate. Hum. Pathol. 2018, 79, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Möller, K.; Uhlig, R.; Gorbokon, N.; Dum, D.; Menz, A.; Büscheck, F.; Luebke, A.M.; Hube-Magg, C.; Hinsch, A.; Höflmayer, D.; et al. Comparison of INSM1 immunostaining with established neuroendocrine markers synaptophysin and chromogranin A in over 14,000 neuroendocrine and non-neuroendocrine tumors. Mol. Cell. Endocrinol. 2024, 581, 112106. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.L.; Madeb, R.; Bourne, P.; Lei, J.; Yang, X.; Tickoo, S.; Liu, Z.; Tan, D.; Cheng, L.; Hatem, F.; et al. Small Cell Carcinoma of the Prostate: An Immunohistochemical Study. Am. J. Surg. Pathol. 2006, 30, 705–712. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Y.; Zhang, Y.; Liu, D.; Ming, J.; Huang, B.; Qiu, X. Treatment-Emergent Neuroendocrine Prostate Cancer: A Clinicopathological and Immunohistochemical Analysis of 94 Cases. Front. Oncol. 2020, 10, 571308. [Google Scholar] [CrossRef]
- Wang, W.; Epstein, J.I. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am. J. Surg. Pathol. 2008, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.K.; Lehrer, J.; Alshalalfa, M.; Erho, N.; Davicioni, E.; Lotan, T.L. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer 2017, 17, 759. [Google Scholar] [CrossRef] [PubMed]
- Bery, F.; Cancel, M.; Chantôme, A.; Guibon, R.; Bruyère, F.; Rozet, F.; Mahéo, K.; Fromont, G. The Calcium-Sensing Receptor is A Marker and Potential Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancers 2020, 12, 860. [Google Scholar] [CrossRef] [PubMed]
- Okasho, K.; Ogawa, O.; Akamatsu, S. Narrative review of challenges in the management of advanced neuroendocrine prostate cancer. Transl. Androl. Urol. 2021, 10, 3953–3962. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-K.; Liu, Y.; Liao, L.; Li, W.; Danielpour, D.; Xu, J. Neuroendocrine prostate carcinoma cells originate from the p63-expressing basal cells but not the pre-existing adenocarcinoma cells in mice. Cell Res. 2019, 29, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Merkens, L.; Sailer, V.; Lessel, D.; Janzen, E.; Greimeier, S.; Kirfel, J.; Perner, S.; Pantel, K.; Werner, S.; von Amsberg, G. Aggressive variants of prostate cancer: Underlying mechanisms of neuroendocrine transdifferentiation. J. Exp. Clin. Cancer Res. 2022, 41, 46. [Google Scholar] [CrossRef] [PubMed]
- Hansel, D.E.; Nakayama, M.; Luo, J.; Abukhdeir, A.M.; Park, B.H.; Bieberich, C.J.; Hicks, J.L.; Eisenberger, M.; Nelson, W.G.; Mostwin, J.L.; et al. Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate 2009, 69, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Bryce, A.H.; Piulats, J.M.; Reaume, M.N.; Ostler, P.J.; McDermott, R.S.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Abida, W.; Daugaard, G.; et al. Rucaparib for metastatic castration-resistant prostate cancer (mCRPC): TRITON3 interim overall survival and efficacy of rucaparib vs docetaxel or second-generation androgen pathway inhibitor therapy. J. Clin. Oncol. 2023, 41, 18. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.A.; Carles, J.; Fay, A.P.; Matsubara, N.; Heinrich, D.; Szczylik, C.; De Giorgi, U.; Young Joung, J.; Fong, P.C.C.; et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): A randomised, placebo-controlled, phase 3 trial. Lancet 2023, 402, 291–303. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Sandhu, S.; Smith, M.R.; Attard, G.; Saad, M.; Olmos, D.; Castro, E.; Roubaud, G.; Pereira de Santana Gomes, A.J.; Small, E.J.; et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: Second interim analysis of the randomized phase III MAGNITUDE trial. Ann. Oncol. 2023, 34, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Crabb, S.J.; Griffiths, G.; Dunkley, D.; Downs, N.; Ellis, M.; Radford, M.; Light, M.; Northey, J.; Whitehead, A.; Wilding, S.; et al. Overall Survival Update for Patients with Metastatic Castration-resistant Prostate Cancer Treated with Capivasertib and Docetaxel in the Phase 2 ProCAID Clinical Trial. Eur. Urol. 2022, 82, 512–515. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov NCT01351103. Available online: https://clinicaltrials.gov/ct2/show/NCT01351103 (accessed on 14 January 2022).
- Patel, R.; Brzezinska, E.A.; Repiscak, P.; Ahmad, I.; Mui, E.; Gao, M.; Blomme, A.; Harle, V.; Tan, E.H.; Malviya, G.; et al. Activation of β-Catenin Cooperates with Loss of Pten to Drive AR-Independent Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 576–590. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation Type | Genetic Alteration 1 | Allelic Status | Predicted Impact 2 |
|---|---|---|---|---|
| AR signalling | ||||
| AR | Missense INDEL | A646D; A114D Non-Fs: p.457_469del/p.457_469del | Hom Hom | Deleterious NR |
| ARSE | Missense | G379S; G424S; G449S | Hom | Tolerated |
| CHRNA3 | Missense | R37H | Hom | Deleterious |
| ETV1 | Missense INDEL | G56A; G74A; G96A; G114A UTR5: c.-31delT | Het Hom | Deleterious NR |
| LTBP1 | Missense | V957A; V999A; V1010A;V1052A; V1378A | Hom | Tolerated |
| NBPF3 | Missense | Y114C; Y58C | Hom | Deleterious |
| TMC8 | Missense | N306I | Hom | Tolerated |
| NF-κB signalling | ||||
| MAP3K1 | Missense INDEL | D806N Non-Fs: p.941_942del | Hom Hom | Damaging Benign |
| MAP3K4 | Missense | H906P; H359P | Het | Tolerated |
| INDEL | Non-Fs: p.P27delinsPP/p.1185_1186del | Hom | NR | |
| IL6 | Missense | D86E; D162E | Het | Tolerated |
| VWF | Missense | H484R; Q852R; T1381A; F2561Y* | Hom | Tolerated |
| Epigenetic regulators | ||||
| KMT2D | Missense | E2678Q | Het | Deleterious |
| NEPC-associated genes | ||||
| TP53 | Stop Gain INDEL | E12X*; E39X*; E132X*; E171X* UTR5 c.-179_-181delAAA | Hom Hom | LOF NR |
| TTN | Missense | R25794Q; R25919Q; R25986Q; R32291Q; R33218Q; R34859Q | Het Het | Deleterious Deleterious |
| DST | Missense | P1984H; P2310H; P2350H; P2488H | Het | Deleterious |
| MUC16 | Missense INDEL | S3337L; V3530I; T3788I; G3826E; H4166N; I4902V; V9909I; E12290K; T10155I S7019L; N13438D Fs: p.P13560fs/p.K13558fs | Hom Hom Het Het | Deleterious Deleterious Deleterious NR |
| ZFHX4 | Missense INDEL | P1273S UTR3: c.*48delA | Het Het | Possibly damaging NR |
| ZNF479 | Missense | Y135C; M369T | Het | Tolerated |
| CACNA1B | Missense | L2215R | Hom | Deleterious |
| CMYA5 | Missense | L1669S | Hom | Deleterious |
| OBSCN | Missense | A7172V; A8129V | Hom | Tolerated |
| RYR2 | Missense | G1886S | Het | Tolerated |
| AURKA | Missense | I57V | Het | Tolerated |
| ASXL3 | Missense | N954S | Hom | Tolerated |
| NEUROD1 | Missense | T45A | Hom | Tolerated |
| SRRM4 | Missense | S243N | Het | Tolerated |
| ASCL1 | INDEL | Non-Fs: p.A50delinsAQ | Het | Benign |
| EZH2 | INDEL | UTR3: c.*21delC | Hom | Benign |
| DNA damage repair | ||||
| ATM | Missense | Y1475C*; D1853N* | Hom | Possibly damaging |
| BRCA2 | Missense | A2595D* | Hom | Deleterious |
| PI3K signalling | ||||
| INPP5D | Missense | H1169Y; H1168Y | Het | Tolerated |
| PIK3CA | Missense | I391M | Het | Tolerated |
| PIK3CG | Missense | M35K; T857A | Het | Tolerated |
| PIK3R2 | Missense | S234R; S313P | Hom | Tolerated |
| PIK3R3 | Missense | N127K; N202K; N329K; N239K; N283K | Het | Tolerated |
| RICTOR | Missense | S837F; S552F | Het | Tolerated |
| Wnt signalling | ||||
| FZD6 | Missense | M40L; M345L; M313L | Hom | Tolerated |
| INDEL | UTR3: c.*202delA | Hom | NR | |
| LGR6 | Missense | V453A; V592A; V540A; V636M; V775M; V723M | Het Het | Tolerated Damaging |
| RSPO2 | Missense | L122P; L119P; L186P | Hom | Tolerated |
| SFRP1 | INDEL | Non-Fs: p.13_14del | Het | NR |
| APC | Missense | V1804D; V1822D | Hom | Tolerated |
| BCL9 | Missense | P332L | Het | Tolerated |
| CTNNB1 | Missense | S33Y; S26Y | Het | Deleterious (GOF) |
| RNF43 | Missense | I47V P104L; P231L L291M; L418M | Het Het Het | Tolerated Deleterious Possibly damaging |
| Protein Alterations | Genetic Alterations | ||
|---|---|---|---|
| Molecular Marker | CU-PC01 PDX Status | Genetic Variant | CU-PC01 PDX Status |
| Cell differentiation | Known driver mutation | ||
| CK5 | Positive | CTNNB1 (β-catenin) | Activating mutation (GOF) |
| CK8 | Positive | TP53 | Stop gain mutation (LOF) |
| AR | Negative | BRCA2 | Missense mutation (LOF?) |
| SYP | Negative | ||
| CHGA | Rare | Candidate driver mutation | |
| CD56 | Focally weak | DST | Missense mutation |
| INSM1 | Focally weak | KMT2D | Missense mutation |
| ASCL1 | Negative | MUC16 | Missense mutation |
| NEUROD1 | Negative | TTN | Missense mutation |
| HOXB13 | Weak | ZFHX4 | Missense mutation |
| Tumour suppressor | |||
| PTEN | Negative | ||
| RB | Negative | ||
| Oncogene | |||
| Nuclear β-catenin | Positive | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turnham, D.J.; Mullen, M.S.; Bullock, N.P.; Gilroy, K.L.; Richards, A.E.; Patel, R.; Quintela, M.; Meniel, V.S.; Seaton, G.; Kynaston, H.; et al. Development and Characterisation of a New Patient-Derived Xenograft Model of AR-Negative Metastatic Castration-Resistant Prostate Cancer. Cells 2024, 13, 673. https://doi.org/10.3390/cells13080673
Turnham DJ, Mullen MS, Bullock NP, Gilroy KL, Richards AE, Patel R, Quintela M, Meniel VS, Seaton G, Kynaston H, et al. Development and Characterisation of a New Patient-Derived Xenograft Model of AR-Negative Metastatic Castration-Resistant Prostate Cancer. Cells. 2024; 13(8):673. https://doi.org/10.3390/cells13080673
Chicago/Turabian StyleTurnham, Daniel J., Manisha S. Mullen, Nicholas P. Bullock, Kathryn L. Gilroy, Anna E. Richards, Radhika Patel, Marcos Quintela, Valerie S. Meniel, Gillian Seaton, Howard Kynaston, and et al. 2024. "Development and Characterisation of a New Patient-Derived Xenograft Model of AR-Negative Metastatic Castration-Resistant Prostate Cancer" Cells 13, no. 8: 673. https://doi.org/10.3390/cells13080673
APA StyleTurnham, D. J., Mullen, M. S., Bullock, N. P., Gilroy, K. L., Richards, A. E., Patel, R., Quintela, M., Meniel, V. S., Seaton, G., Kynaston, H., Clarkson, R. W. E., Phesse, T. J., Nelson, P. S., Haffner, M. C., Staffurth, J. N., & Pearson, H. B. (2024). Development and Characterisation of a New Patient-Derived Xenograft Model of AR-Negative Metastatic Castration-Resistant Prostate Cancer. Cells, 13(8), 673. https://doi.org/10.3390/cells13080673