
Crosstalk between DNA Damage Repair and Metabolic Regulation in Hematopoietic Stem Cells

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
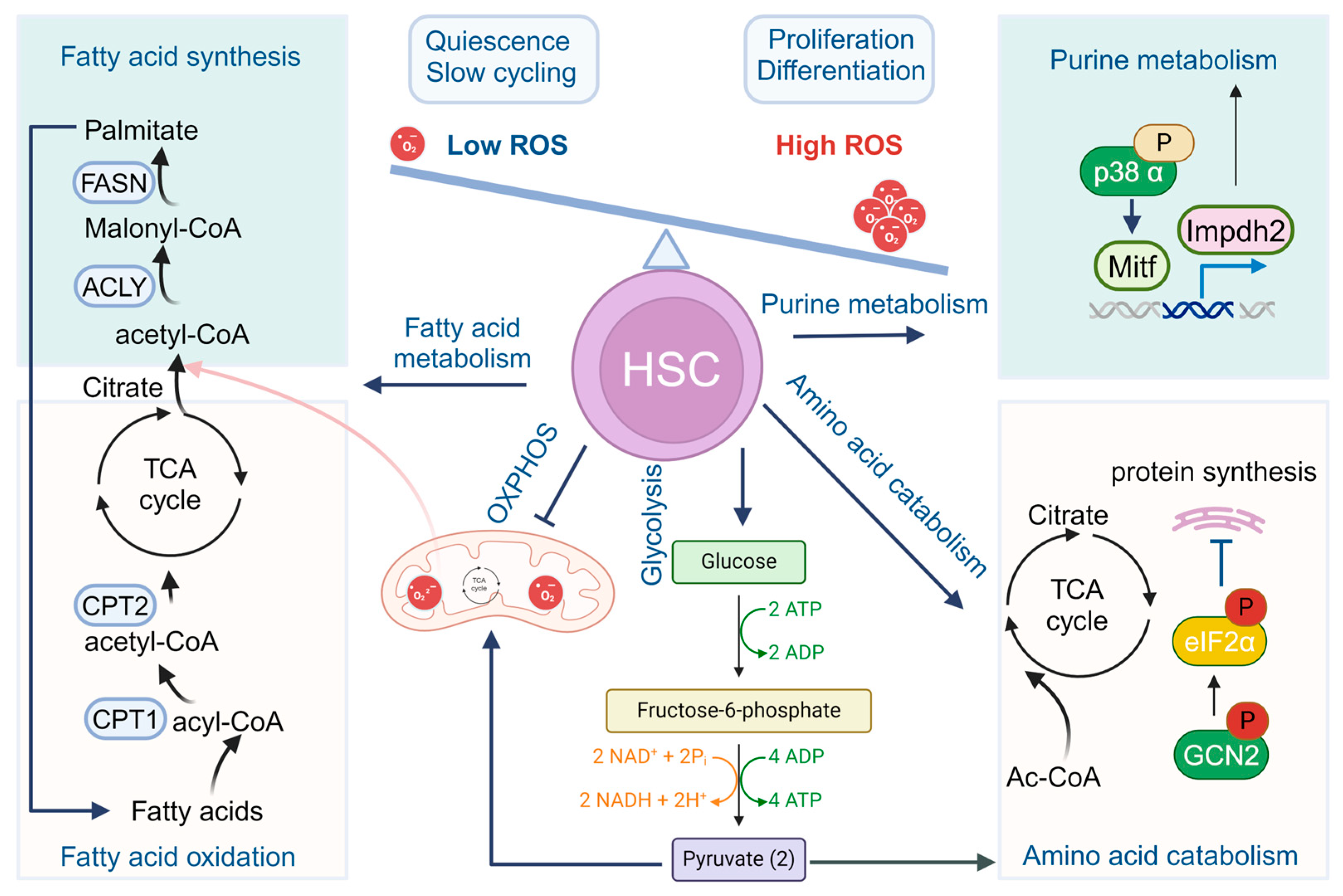
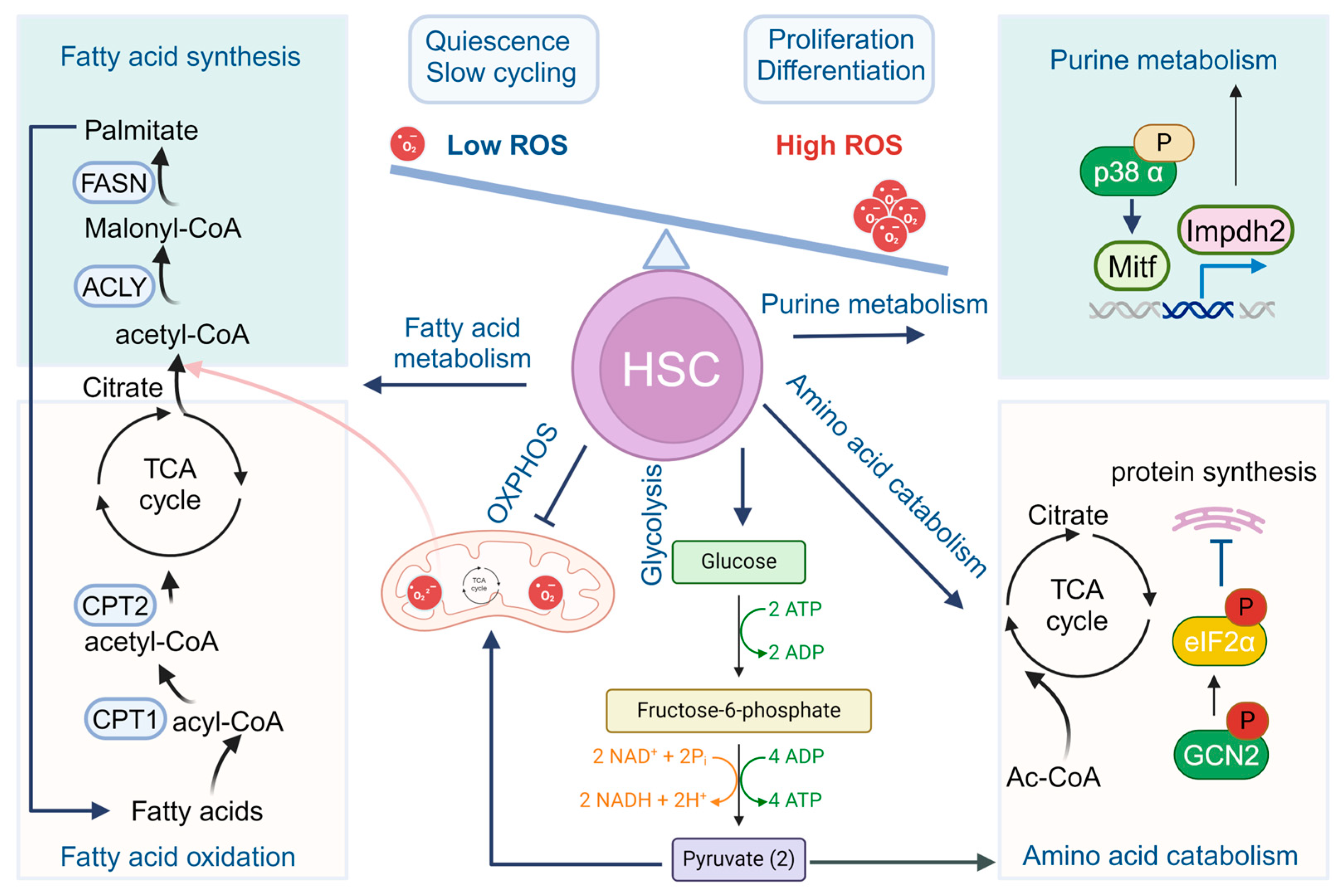
2. Metabolic Choreography in HSCs
2.1. Glycolysis and Mitochondrial Oxidative Phosphorylation
2.2. Fatty Acid Metabolism
2.3. Purine and Amino Acid Metabolism
2.4. Other Metabolic Regulation Pathways in HSC
3. Gut Microbiota and HSC Metabolism
4. Metabolism Meets HSC Genomic Integrity
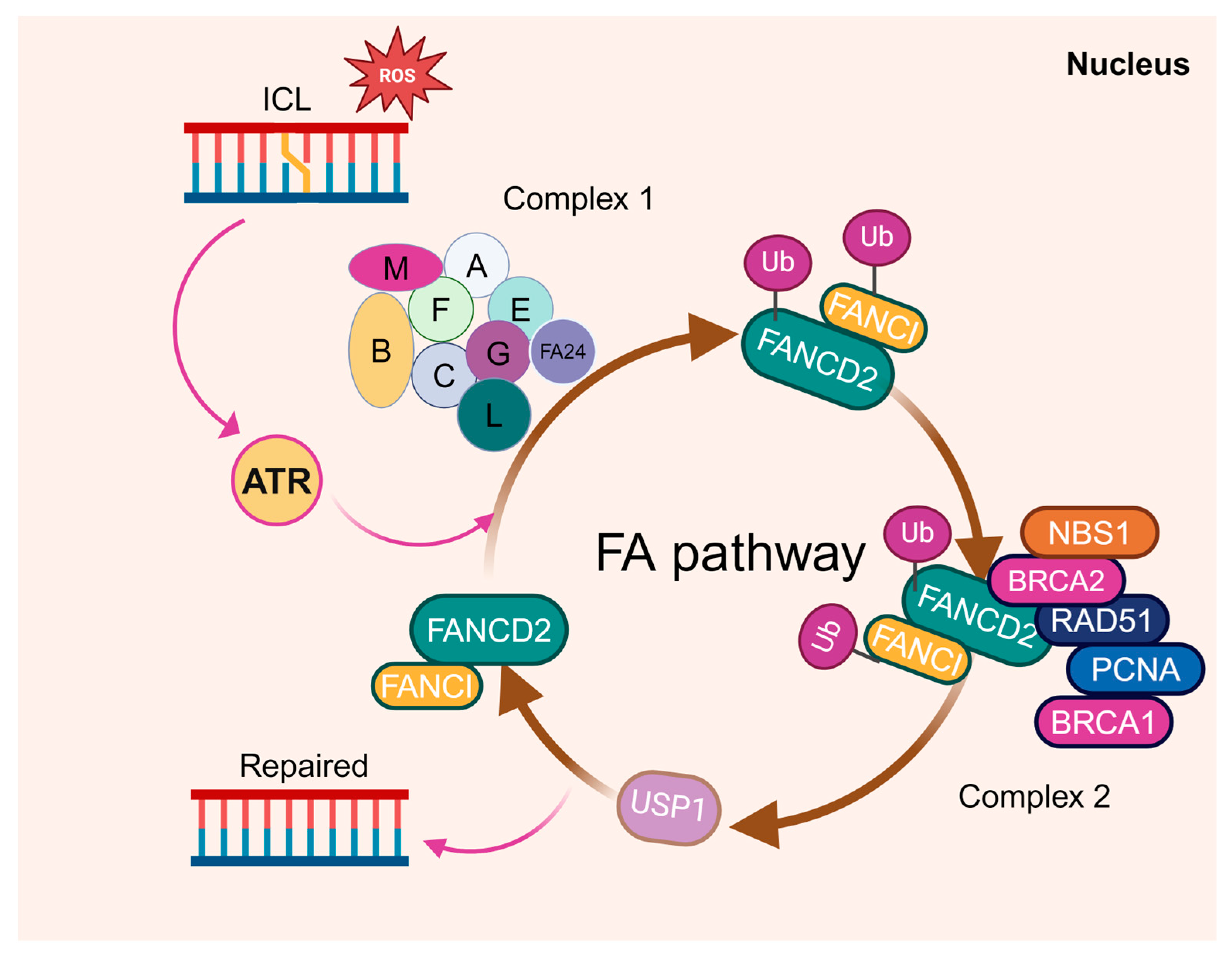
5. Fanconi Anemia, Genomic Integrity, and HSC Metabolism
5.1. FA, DNA Damage Repair, and HSC Defects
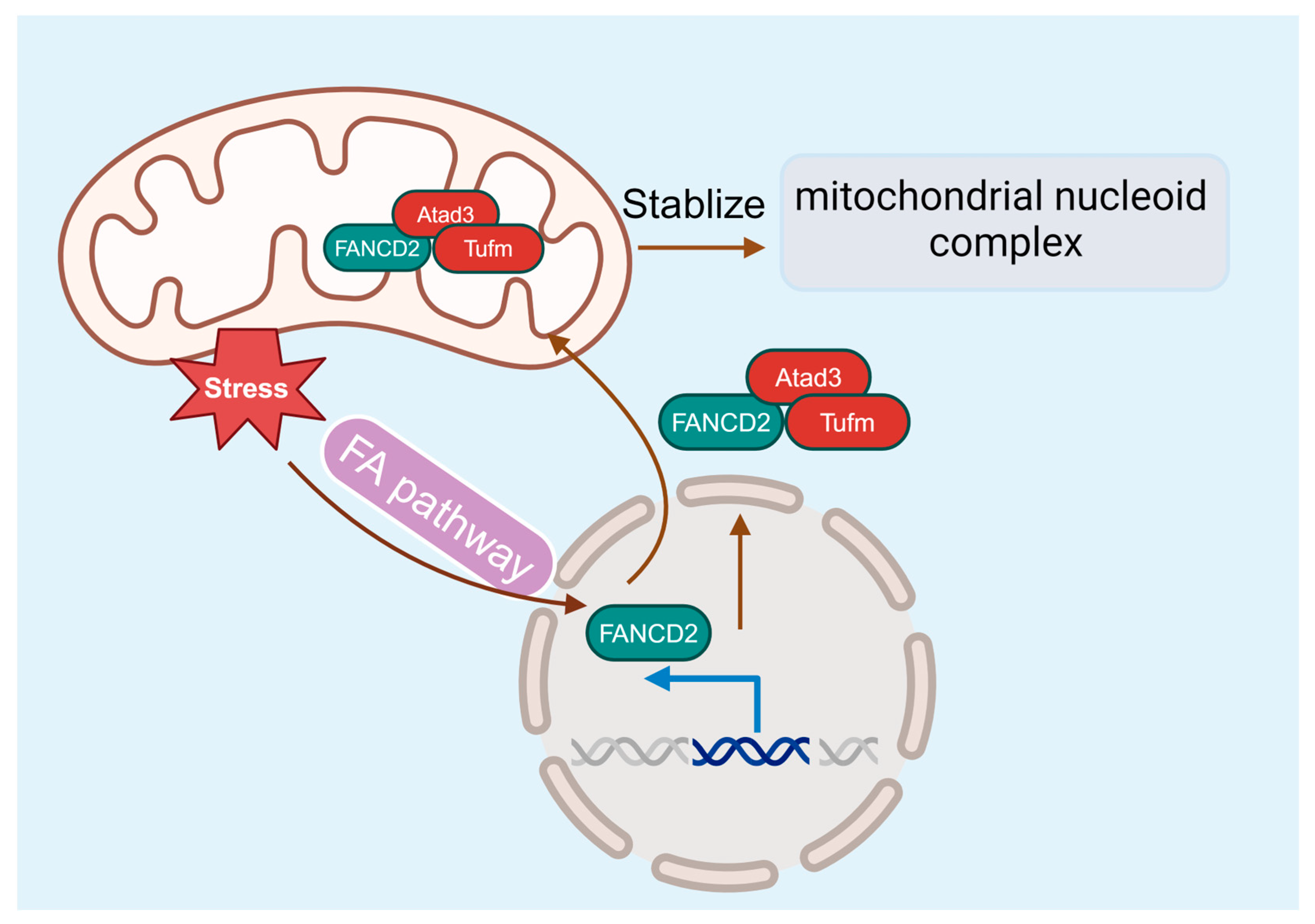
5.2. Metabolic Alterations in FA HSCs
5.3. FA and Mitochondria Dysfunction
6. Recent Technological Advances in HSC Metabolism
Author Contributions
Funding
Conflicts of Interest
References
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sipple, J.; Pang, Q.; Du, W. Salidroside stimulates DNA repair enzyme Parp-1 activity in mouse HSC maintenance. Blood 2012, 119, 4162–4173. [Google Scholar] [CrossRef] [PubMed]
- Morganti, C.; Cabezas-Wallscheid, N.; Ito, K. Metabolic Regulation of Hematopoietic Stem Cells. HemaSphere 2022, 6, e740. [Google Scholar] [CrossRef] [PubMed]
- Moretton, A.; Loizou, J.I. Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers 2020, 12, 2051. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Pierri, F. Modern management of Fanconi anemia. Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Amarachintha, S.; Wilson, A.F.; Pang, Q. SCO2 Mediates Oxidative Stress-Induced Glycolysis to Oxidative Phosphorylation Switch in Hematopoietic Stem Cells. Stem Cells 2016, 34, 960–971. [Google Scholar] [CrossRef]
- Li, X.; Wu, L.; Zopp, M.; Kopelov, S.; Du, W. p53-TP53-Induced Glycolysis Regulator Mediated Glycolytic Suppression Attenuates DNA Damage and Genomic Instability in Fanconi Anemia Hematopoietic Stem Cells. Stem Cells 2019, 37, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xu, J.; Wu, L.; Wang, J.; Lin, Q.; Chowdhury, F.A.; Mazumder, M.H.H.; Hu, G.; Li, X.; Du, W. Hes1 deficiency causes hematopoietic stem cell exhaustion. Stem Cells 2020, 38, 756–768. [Google Scholar] [CrossRef]
- Wu, L.; Li, X.; Lin, Q.; Chowdhury, F.; Mazumder, M.H.; Du, W. FANCD2 and HES1 suppress inflammation-induced PPARɣ to prevent haematopoietic stem cell exhaustion. Br. J. Haematol. 2021, 192, 652–663. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Hsu, P.; Qu, C.K. Metabolic plasticity and hematopoietic stem cell biology. Curr. Opin. Hematol. 2013, 20, 289–294. [Google Scholar] [CrossRef]
- Filippi, M.D. The multifaceted role of mitochondria in HSC fate decisions: Energy and beyond. Exp. Hematol. 2023, 128, 19–29. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Melkonian, E.A.; Schury, M.P. Biochemistry, Anaerobic Glycolysis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef]
- Yu, W.M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef]
- Piccoli, C.; Ria, R.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilio, A.; Capitanio, N. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J. Biol. Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef]
- Lonergan, T.; Bavister, B.; Brenner, C. Mitochondria in stem cells. Mitochondrion 2007, 7, 289–296. [Google Scholar] [CrossRef]
- Huang, D.; Chen, C.; Xie, L.; Yu, Z.; Zheng, J. Hematopoietic stem cell metabolism and stemness. Blood Sci. 2019, 1, 12–18. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial Role in Stemness and Differentiation of Hematopoietic Stem Cells. Stem Cells Int. 2019, 2019, 4067162. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef]
- Klimmeck, D.; Hansson, J.; Raffel, S.; Vakhrushev, S.Y.; Trumpp, A.; Krijgsveld, J. Proteomic cornerstones of hematopoietic stem cell differentiation: Distinct signatures of multipotent progenitors and myeloid committed cells. Mol. Cell. Proteom. 2012, 11, 286–302. [Google Scholar] [CrossRef]
- Kohli, L.; Passegue, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef]
- Inoue, S.; Noda, S.; Kashima, K.; Nakada, K.; Hayashi, J.; Miyoshi, H. Mitochondrial respiration defects modulate differentiation but not proliferation of hematopoietic stem and progenitor cells. FEBS Lett. 2010, 584, 3402–3409. [Google Scholar] [CrossRef]
- Howlett, M.G.; Fletcher, S.P. From autocatalysis to survival of the fittest in self-reproducing lipid systems. Nat. Rev. Chem. 2023, 7, 673–691. [Google Scholar] [CrossRef]
- Giger, S.; Kovtonyuk, L.V.; Utz, S.G.; Ramosaj, M.; Kovacs, W.J.; Schmid, E.; Ioannidis, V.; Greter, M.; Manz, M.G.; Lutolf, M.P.; et al. A Single Metabolite which Modulates Lipid Metabolism Alters Hematopoietic Stem/Progenitor Cell Behavior and Promotes Lymphoid Reconstitution. Stem Cell Rep. 2020, 15, 566–576. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef]
- Ito, K.; Ito, K. Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu. Rev. Cell Dev. Biol. 2016, 32, 399–409. [Google Scholar] [CrossRef]
- Anderson, G.A.; Rodriguez, M.; Kathrein, K.L. Regulation of stress-induced hematopoiesis. Curr. Opin. Hematol. 2020, 27, 279–287. [Google Scholar] [CrossRef]
- Karigane, D.; Kobayashi, H.; Morikawa, T.; Ootomo, Y.; Sakai, M.; Nagamatsu, G.; Kubota, Y.; Goda, N.; Matsumoto, M.; Nishimura, E.K.; et al. p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 2016, 19, 192–204. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Krall, A.S.; Mullen, P.J.; Surjono, F.; Momcilovic, M.; Schmid, E.W.; Halbrook, C.J.; Thambundit, A.; Mittelman, S.D.; Lyssiotis, C.A.; Shackelford, D.B.; et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021, 33, 1013–1026.e16. [Google Scholar] [CrossRef]
- Qi, L.; Martin-Sandoval, M.S.; Merchant, S.; Gu, W.; Eckhardt, M.; Mathews, T.P.; Zhao, Z.; Agathocleous, M.; Morrison, S.J. Aspartate availability limits hematopoietic stem cell function during hematopoietic regeneration. Cell Stem Cell 2021, 28, 1982–1999.e88. [Google Scholar] [CrossRef]
- Li, C.; Wu, B.; Li, Y.; Chen, J.; Ye, Z.; Tian, X.; Wang, J.; Xu, X.; Pan, S.; Zheng, Y.; et al. Amino acid catabolism regulates hematopoietic stem cell proteostasis via a GCN2-eIF2α axis. Cell Stem Cell 2022, 29, 1119–1134.e17. [Google Scholar] [CrossRef]
- Galán-Díez, M.; Cuesta-Domínguez, Á.; Kousteni, S. The Bone Marrow Microenvironment in Health and Myeloid Malignancy. Cold Spring Harb. Perspect. Med. 2018, 8, a031328. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Choi, S.H.; Baek, J.S.; Liu, C.; Almazan, F.; Ulrich, F.; Wiesner, P.; Taleb, A.; Deer, E.; Pattison, J.; et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013, 498, 118–122. [Google Scholar] [CrossRef]
- Gu, Q.; Yang, X.; Lv, J.; Zhang, J.; Xia, B.; Kim, J.D.; Wang, R.; Xiong, F.; Meng, S.; Clements, T.P.; et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 2019, 363, 1085–1088. [Google Scholar] [CrossRef]
- Liu, C.; Liao, W.; Chen, J.; Yu, K.; Wu, Y.; Zhang, S.; Chen, M.; Chen, F.; Wang, S.; Cheng, T.; et al. Cholesterol confers ferroptosis resistance onto myeloid-biased hematopoietic stem cells and prevents irradiation-induced myelosuppression. Redox Biol. 2023, 62, 102661. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Chen, Y.T.; Li, X.; Hsueh, P.C.; Tzeng, S.F.; Chen, H.; Shi, P.Z.; Xie, X.; Parik, S.; Planque, M.; et al. CD40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat. Immunol. 2023, 24, 452–462. [Google Scholar] [CrossRef]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef]
- Weng, H.; Huang, F.; Yu, Z.; Chen, Z.; Prince, E.; Kang, Y.; Zhou, K.; Li, W.; Hu, J.; Fu, C.; et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell 2022, 40, 1566–1582.e10. [Google Scholar] [CrossRef]
- Yang, C.; Hashimoto, M.; Lin, Q.X.X.; Tan, D.Q.; Suda, T. Sphingosine-1-phosphate signaling modulates terminal erythroid differentiation through the regulation of mitophagy. Exp. Hematol. 2019, 72, 47–59.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, B.; Li, Y.; Liu, Y.; Wang, J.; Xie, J.; Xu, X.; Tian, X.; Ye, Z.; Guan, J.; et al. Loss of sphingosine kinase 2 promotes the expansion of hematopoietic stem cells by improving their metabolic fitness. Blood 2022, 140, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Maiti, A.; Xu, P.; Qi, Q.; Kawaguchi, T.; Okano, M.; Takabe, K.; Yan, L.; Luo, C. Regulation of hypoxia-inducible factor functions in the nucleus by sphingosine-1-phosphate. FASEB J. 2020, 34, 4293–4310. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nakamura, Y.; Tanaka, S.; Kohro, T.; Li, L.X.; Huang, L.; Yao, J.; Kawamura, S.; Inoue, R.; Nishi, H.; et al. Bone marrow stromal cell antigen-1 (CD157) regulated by sphingosine kinase 2 mediates kidney fibrosis. Front. Med. 2022, 9, 993698. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, C.; Hao, X.; Su, N.; Huang, D.; Zou, Y.; Lin, S.H.; Chen, X.; Zheng, D.; Liu, L.; et al. MDH1-mediated malate-aspartate NADH shuttle maintains the activity levels of fetal liver hematopoietic stem cells. Blood 2020, 136, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Balmer, M.L.; Schurch, C.M.; Saito, Y.; Geuking, M.B.; Li, H.; Cuenca, M.; Kovtonyuk, L.V.; McCoy, K.D.; Hapfelmeier, S.; Ochsenbein, A.F.; et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J. Immunol. 2014, 193, 5273–5283. [Google Scholar] [CrossRef] [PubMed]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Walker, F.C.; Ali, A.; Han, H.; Tan, L.; Veillon, L.; Lorenzi, P.L.; Baldridge, M.T.; King, K.Y. The bacterial microbiota regulates normal hematopoiesis via metabolite-induced type 1 interferon signaling. Blood Adv. 2022, 6, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wei, Z.; Chen, J.; An, J.; Li, M.; Zhou, L.; Men, Y.; Zhao, S. Save your gut save your age: The role of the microbiome in stem cell ageing. J. Cell. Mol. Med. 2019, 23, 4866–4875. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef]
- Li, N.; Chen, H.; Wang, J. DNA damage and repair in the hematopoietic system. Acta Biochim. Biophys. Sin. 2022, 54, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidative damage to DNA in mammalian chromatin. Mutat. Res. 1992, 275, 331–342. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; Parlanti, E.; Pascucci, B.; Filomeni, G.; Mastroberardino, P.G.; Dogliotti, E. The interplay between mitochondrial functionality and genome integrity in the prevention of human neurologic diseases. Arch. Biochem. Biophys. 2021, 710, 108977. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Schönberger, K.; Cabezas-Wallscheid, N. Bidirectional interplay between metabolism and epigenetics in hematopoietic stem cells and leukemia. EMBO J. 2023, 42, e112348. [Google Scholar] [CrossRef] [PubMed]
- Zdzisińska, B.; Żurek, A.; Kandefer-Szerszeń, M. Alpha-ketoglutarate as a molecule with pleiotropic activity: Well-known and novel possibilities of therapeutic use. Arch. Immunol. Ther. Exp. 2017, 65, 21–36. [Google Scholar] [CrossRef]
- Zhong, L.; Mostoslavsky, R. SIRT6: A master epigenetic gatekeeper of glucose metabolism. Transcription. Transcription 2010, 1, 17–21. [Google Scholar] [CrossRef]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.; Vadysirisack, D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1a. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Simithy, J.; Sidoli, S.; Yuan, Z.F.; Coradin, M.; Bhanu, N.V.; Marchione, D.M.; Klein, B.J.; Bazilevsky, G.A.; McCullough, C.E.; Magin, R.S.; et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Viney, I.; Wellen, E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Crispo, F.; Condelli, V.; Lepore, S.; Notarangelo, T.; Sgambato, A.; Esposito, F.; Maddalena, F.; Landriscina, M. Metabolic Dysregulations and Epigenetics: A Bidirectional Interplay that Drives Tumor Progression. Cells 2019, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shah, K.; Khodadadi-Jamayran, A.; Jiang, H. Dpy30 is critical for maintaining the identity and function of adult hematopoietic stem cells. J. Exp. Med. 2016, 213, 2349–2364. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Augustin, J.; Chang, C.; Hu, J.; Shah, K.; Chang, C.W.; Townes, T.; Jiang, H. The DPY30 subunit in SET1/MLL complexes regulates the proliferation and differentiation of hematopoietic progenitor cells. Blood 2014, 124, 2025–2033. [Google Scholar] [CrossRef] [PubMed]
- Welsh, C.; Day, R.; McGurk, C.; Masters, J.R.; Wood, R.D.; Köberle, B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Martínez, J.; Hernández, C.; Pérez-Montiel, D.; Castro, C.; Fabián-Morales, E.; Santibáñez, M.; González-Barrios, R.; Díaz-Chávez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Pan, M.R.; Li, K.; Lin, S.Y.; Hung, W.C. Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci. 2016, 17, 685. [Google Scholar] [CrossRef] [PubMed]
- Biechonski, S.; Yassin, M.; Milyavsky, M. DNA-damage response in hematopoietic stem cells: An evolutionary trade-off between blood regeneration and leukemia suppression. Carcinogenesis 2017, 38, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G.C., Jr. Genetic basis of Fanconi anemia. Curr. Opin. Hematol. 2003, 10, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Kupfer, G.M. Fanconi anemia. Hematol. Oncol. Clin. N. Am. 2009, 23, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; D’Andrea, A.D. The Fanconi Anemia/BRCA pathway: New faces in the crowd. Genes Dev. 2005, 19, 2925–2940. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Hodskinson, M.R.; Silhan, J.; Crossan, G.P.; Garaycoechea, J.I.; Mukherjee, S.; Johnson, C.M.; Schärer, O.D.; Patel, K.J. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell 2014, 54, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kahkonen, M.; Schwartzentruber, J.; Kircher, M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Virts, E.L.; Jankowska, A.; Mackay, C.; Glaas, M.F.; Wiek, C.; Kelich, S.L.; Lottmann, N.; Kennedy, F.M.; Marchal, C.; Lehnert, E.; et al. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum. Mol. Genet. 2015, 24, 5093–5108. [Google Scholar] [CrossRef]
- Dong, H.; Nebert, D.W.; Bruford, E.A.; Thompson, D.C.; Joenje, H.; Vasiliou, V. Update of the human and mouse Fanconi anemia genes. Hum. Genom. 2015, 9, 32. [Google Scholar] [CrossRef]
- Park, J.Y.; Virts, E.L.; Jankowska, A.; Wiek, C.; Othman, M.; Chakraborty, S.C.; Vance, G.H.; Alkuraya, F.S.; Hanenberg, H.; Andreassen, P.R. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J. Med. Genet. 2016, 53, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Knies, K.; Inano, S.; Ramirez, M.J.; Ishiai, M.; Surralles, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Sertorio, M.; Du, W.; Amarachintha, S.; Wilson, A.; Pang, Q. In Vivo RNAi Screen Unveils PPARγ as a Regulator of Hematopoietic Stem Cell Homeostasis. Stem Cell Rep. 2017, 8, 1242–1255. [Google Scholar] [CrossRef]
- Karigane, D.; Takubo, K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int. J. Hematol. 2017, 106, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.S.; Leung, K.S.; Hicks, M.J.; Hastings, P.J.; Youssoufian, H.; Plon, S.E. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 2006, 175, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Rousset, S.; Nocentini, S.; Rouillard, D.; Baroche, C.; Moustacchi, E. Mitochondrial alterations in fanconi anemia fibroblasts following ultraviolet A or psoralen photoactivation. Photochem. Photobiol. 2002, 75, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Talamanca, A.A.; Castello, G.; d’Ischia, M.; Pallardo, F.V.; Petrovic, S.; Porto, B.; Tiano, L.; Zatterale, A. Bone marrow cell transcripts from Fanconi anaemia patients reveal in vivo alterations in mitochondrial, redox and DNA repair pathways. Eur. J. Haematol. 2013, 91, 141–151. [Google Scholar] [CrossRef]
- Usai, C.; Ravera, S.; Cuccarolo, P.; Panfoli, I.; Dufour, C.; Cappelli, E.; Degan, P. Dysregulated Ca2+ homeostasis in Fanconi anemia cells. Sci. Rep. 2015, 5, 8088. [Google Scholar] [CrossRef]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zanier, R.; Briot, D.; Dugas du Villard, J.A.; Sarasin, A.; Rosselli, F. Fanconi anemia C gene product regulates expression of genes involved in differentiation and inflammation. Oncogene 2004, 23, 5004–5013. [Google Scholar] [CrossRef]
- Pagano, G.; Shyamsunder, P.; Verma, R.S.; Lyakhovich, A. Damaged mitochondria in Fanconi anemia—An isolated event or a general phenomenon? Oncoscience 2014, 1, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Columbaro, M.; Capanni, C.; Bartolucci, M.; Panfoli, I.; Morelli, A.; Dufour, C.; Cappelli, E.; et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie 2013, 95, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, E.; Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Bartolucci, M.; Panfoli, I.; Dufour, C.; Degan, P. Mitochondrial respiratory complex I defects in Fanconi anemia. Trends Mol. Med. 2013, 19, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernandez, A.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.S.; Marquez-Loza, L.; Eaton, L.; Duncan, A.W.; Goldman, D.C.; Anur, P.; Watanabe-Smith, K.; Rathbun, R.K.; Fleming, W.H.; Bagby, G.C.; et al. Fancd2−/− mice have hematopoietic defects that can be partially corrected by resveratrol. Blood 2010, 116, 5140–5148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Du, W.; Wilson, A.F.; Namekawa, S.H.; Andreassen, P.R.; Meetei, A.R.; Pang, Q. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci. Rep. 2017, 7, 45626. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, J.; Hu, Q.; Zhou, H.M.; Yi, J.; Yu, Z.; Xu, L.; Wang, X.; Yang, Y.; Loscalzo, J. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab. 2011, 14, 555–566. [Google Scholar] [CrossRef]
- Tao, R.; Zhao, Y.; Chu, H.; Wang, A.; Zhu, J.; Chen, X.; Zou, Y.; Shi, M.; Liu, R.; Su, N.; et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 2017, 14, 720–728. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, F.; Zhang, Y.; Li, X.; Chen, C.; Zhou, M.; Yu, Z.; Liu, Y.; Zhao, Y.; Hao, X.; et al. PPM1K regulates hematopoiesis and leukemogenesis through CDC20-mediated ubiquitination of meis1 and p21. Cell Rep. 2018, 23, 1461–1475. [Google Scholar] [CrossRef]
- Hao, X.; Gu, H.; Chen, C.; Huang, D.; Zhao, Y.; Xie, L.; Zou, Y.; Shu, H.S.; Zhang, Y.; He, X.; et al. Metabolic imaging reveals a unique preference of symmetric cell division and homing of leukemia-initiating cells in an endosteal niche. Cell Metab. 2019, 29, 950–965.e6. [Google Scholar] [CrossRef]
- DeVilbiss, A.W.; Zhao, Z.; Martin-Sandoval, M.S.; Ubellacker, J.M.; Tasdogan, A.; Agathocleous, M.; Mathews, T.P.; Morrison, S.J. Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. eLife 2021, 10, e61980. [Google Scholar] [CrossRef]
- Schönberger, K.; Obier, N.; Romero-Mulero, M.C.; Cauchy, P.; Mess, J.; Pavlovich, P.V.; Zhang, Y.W.; Mitterer, M.; Rettkowski, J.; Lalioti, M.E.; et al. Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell 2022, 29, 131–148.e10. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Sala, B.P.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottgens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef]
- Cao, J.; Yao, Q.J.; Wu, J.; Chen, X.; Huang, L.; Liu, W.; Qian, K.; Wan, J.J.; Zhou, B.O. Deciphering the metabolic heterogeneity of hematopoietic stem cells with single-cell resolution. Cell Metab. 2024, 36, 209–221.e6. [Google Scholar] [CrossRef]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef]
- Che, R.; Zhang, J.; Nepal, M.; Han, B.; Fei, P. Multifaceted Fanconi Anemia Signaling. Trends Genet. 2018, 34, 171–183. [Google Scholar] [CrossRef]
- Zhan, S.; Siu, J.; Wang, Z.; Yu, H.; Bezabeh, T.; Deng, Y.; Du, W.; Fei, P. Focal Point of Fanconi Anemia Signaling. Int. J. Mol. Sci. 2021, 22, 12976. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Fei, P.; Simon, D.W.; Morowitz, M.J.; Mehta, P.A.; Du, W. Crosstalk between DNA Damage Repair and Metabolic Regulation in Hematopoietic Stem Cells. Cells 2024, 13, 733. https://doi.org/10.3390/cells13090733
Xu J, Fei P, Simon DW, Morowitz MJ, Mehta PA, Du W. Crosstalk between DNA Damage Repair and Metabolic Regulation in Hematopoietic Stem Cells. Cells. 2024; 13(9):733. https://doi.org/10.3390/cells13090733
Chicago/Turabian StyleXu, Jian, Peiwen Fei, Dennis W. Simon, Michael J. Morowitz, Parinda A. Mehta, and Wei Du. 2024. "Crosstalk between DNA Damage Repair and Metabolic Regulation in Hematopoietic Stem Cells" Cells 13, no. 9: 733. https://doi.org/10.3390/cells13090733
APA StyleXu, J., Fei, P., Simon, D. W., Morowitz, M. J., Mehta, P. A., & Du, W. (2024). Crosstalk between DNA Damage Repair and Metabolic Regulation in Hematopoietic Stem Cells. Cells, 13(9), 733. https://doi.org/10.3390/cells13090733