

Induced Pluripotent Stem Cells and Organoids in Advancing Neuropathology Research and Therapies

 , , , , and
, , , , and
Abstract
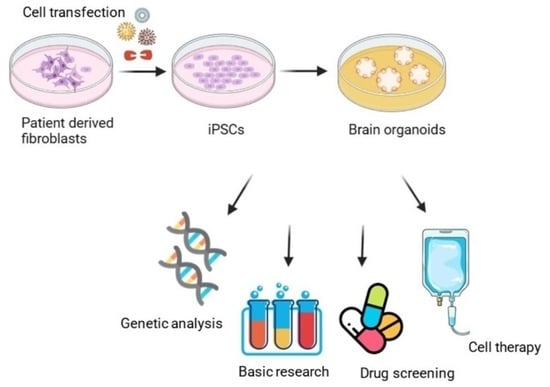
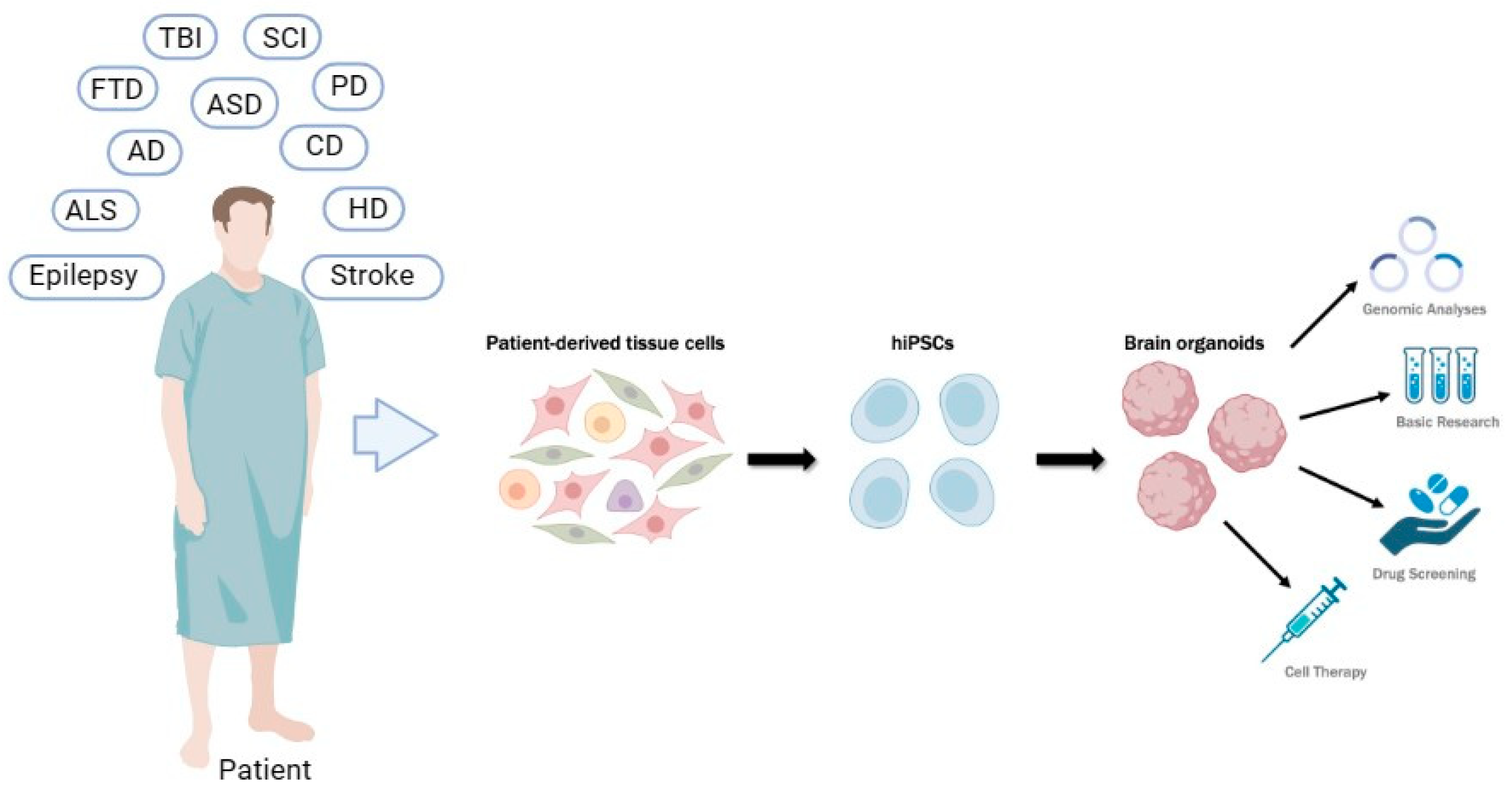
:
1. Introduction
2. Generation and Characterization of iPSCs
2.1. Which Cells Can Be Reprogrammed?
2.2. The Reprogramming Recipe
2.3. Reprogramming Factor Delivery Systems
3. Organoid Models
What They Are, How They Work, and Applications
4. Organoid Models in Neurological Diseases
4.1. Alzheimer’s Disease
4.2. Amyotrophic Lateral Sclerosis
4.3. Attention Deficit Hyperactivity Disorder
4.4. Autism Spectrum Disorder
4.5. Canavan Disease
4.6. Epilepsy
4.7. Frontotemporal Dementia
4.8. Huntington’s Disease
4.9. Multiple Sclerosis
4.10. Parkinson’s Disease
4.11. Spinal Cord Injury
4.12. Stroke
4.13. Traumatic Brain Injury

{kind=link}
{kind=link}
{kind=link}
| Organoid Type | Disease | Cell Type | Result | Reference |
|---|---|---|---|---|
| Cerebral Organoid | AD | iPSC | Modeling sporadic Alzheimer’s disease in human brain organoids under serum exposure | [204] |
| Cerebral Organoid | AD | hiPSC | Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs. isogenic controls | [205] |
| Cerebral Organoid | AD | iPSC | Modeling amyloid beta and tau pathology in human cerebral organoids | [206] |
| Disease Stem Cell | AD | iPSC | Familial Alzheimer’s disease mutations in PSEN1 lead to premature human stem cell neurogenesis | [207] |
| Disease Stem Cell | AD | iPSC and hiPSC | iPSC-derived human microglia-like cells to study neurological diseases | [208] |
| Cerebral Organoid | AD | iPSC | APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patients’ iPSC-derived cerebral organoids | [209] |
| Cerebral Organoid | AD | iPSC | A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids | [210] |
| Cerebral Organoid | AD | iPSC | Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids | [211] |
| Cerebral Organoid | AD | iPSC | Tau pathology epigenetically remodels the neuron-glial cross-talk in Alzheimer’s disease | [212] |
| Disease Stem Cell | AD | iPSC | APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types | [95] |
| Disease Stem Cell | AD | iPSC | Type I interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer’s disease | [213] |
| Cerebral Organoid | AD | iPSC | Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models | [214] |
| Cerebral Organoid | PD | hiPSC | Modeling G2019S-LRRK2 sporadic Parkinson’s disease in 3D midbrain organoids | [215] |
| Cerebral Organoid | PD | hiPSC | Lewy body-like pathology and loss of dopaminergic neurons in midbrain organoids derived from familial Parkinson’s disease patient | [216] |
| Midbrain Organoid | PD | hiPSC | Human iPSC-derived midbrain organoids functionally integrate into striatum circuits and restore motor function in a mouse model of Parkinson’s disease | [217] |
| Neurospheres | PD | hiPSC and iPSC | Patient-derived three-dimensional cortical neurospheres to model Parkinson’s disease | [218] |
| Midbrain Organoid | PD | hiPSC and iPSC | Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease linked DNAJC6 mutations | [219] |
| Midbrain Organoid | PD | iPSC | Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality | [220] |
| Cerebral Organoid | PD | iPSC | Use of 3D organoids as a model to study idiopathic form of Parkinson’s disease | [221] |
| Cerebral Organoid | PD | iPSC | The Parkinson’s disease-associated mutation LRRK2-G2019S alters dopaminergic differentiation dynamics via NR2F1 | [222] |
| Cerebral Organoid | Rett syndrome | hiPSC | Identification of neural oscillations and epileptiform changes in human brain organoids | [223] |
| Cerebral Organoid | TLE | iPSC | Modeling genetic epileptic encephalopathies using brain organoids | [224] |
| Cerebral Organoid | TSC | hiPSC | Amplification of human interneuron progenitors promotes brain tumors and neurological defects | [225] |
| Motor neurons study | ALS | iPSC | Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons | [226] |
| Sensorimotor organoids | ALS | iPSC | Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions | [99] |
| Cerebral Organoid | ALS | iPSC | Spinal cord extracts of amyotrophic lateral sclerosis spread TDP-43 pathology in cerebral organoids | [227] |
| Motor neurons and brain organoids | ALS and FTD | iPSC | CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro | [228] |
| Cerebral organoid slice model | ALS and FTD | iPSC | Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology | [134] |
| Brain organoids | ALS and FTD | iPSC | Granulin loss of function in human mature brain organoids implicates astrocytes in TDP-43 pathology | [229] |
| Motor neurons | ALS | hiPSC | Exploring motor neuron diseases using iPSC platforms | [230] |
| Cerebral organoids | FTD | iPSC | ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids | [133] |
| Molecular study | FTD | iPSC | Pathological progression induced by the frontotemporal dementia-associated R406W tau mutation in patient-derived iPSCs | [231] |
| iPSC-derived astrocytes | MS | iPSC | iPSC-derived reactive astrocytes from patients with multiple sclerosis protect cocultured neurons in inflammatory conditions | [232] |
| Model study | MS | iPSC | Selective PDE4 subtype inhibition provides new opportunities to intervene in neuroinflammatory versus myelin-damaging hallmarks of multiple sclerosis | [233] |
| RRMS and PPMS iPSC cellular models | MS | iPSC | Generation of RRMS- and PPMS-specific iPSCs as a platform for modeling multiple sclerosis | [234] |
| Cerebral organoids | MS | iPSC | Cerebral organoids in primary progressive multiple sclerosis reveal stem cell and oligodendrocyte differentiation defect | [165] |
| Model study | MS | iPSC | Generation and characterization of four multiple sclerosis iPSC lines from a single family | [235] |
| Cerebral organoids | ASD | iPSC | Single-cell brain organoid screening identifies developmental defects in autism | [236] |
| Forebrain organoids/Molecular study | ASD | iPSC | Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder | [237] |
| Organoids/Molecular study | ASD | iPSC | FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders | [113] |
| Brain organoids | ASD | iPSC | Superoxide dismutase isozymes in cerebral organoids from autism spectrum disorder patients | [238] |
| Organoids/Molecular study | ASD | iPSC | CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPSC cells | [239] |
| Cell Therapy | TBI | Rat | Combining enriched environment and induced pluripotent stem cell therapy results in improved cognitive and motor function following traumatic brain injury | [240] |
| Cell Therapy | TBI | Mice | Controlled cortical impact model of mouse brain injury with therapeutic transplantation of human induced pluripotent stem cell-derived neural cells | [241] |
| Cerebral Organoid | TBI | hiPSC | Modeling traumatic brain injury in human cerebral organoids | [198] |
| Cell Therapy | CD | Mice | Cell-based therapy for Canavan disease using human iPSC-derived NPCs and OPCs | [122] |
| Cerebral Organoid | Stroke | hiPSC | Gene expression profiles of human cerebral organoids identify PPAR pathway and PKM2 as key markers for oxygen glucose deprivation and reoxygenation | [242] |
| iPSC derived telencephalon organoids | ADHD | iPSC | Telencephalon organoids derived from an individual with ADHD show altered neurodevelopment of early cortical layer structure | [105] |
| Model study | ASD and ADHD | iPSC | Modeling human cerebellar development in vitro in 2D structure | [243] |
| Molecular study | ADHD | iPSC | Generation of a human induced pluripotent stem cell (iPSC) line from a 51-year-old female with attention-deficit/hyperactivity disorder (ADHD) carrying a duplication of SLC2A3 | [244] |
| Model study | ADHD | iPSC | Generation of four iPSC lines from peripheral blood mononuclear cells (PBMCs) of an attention-deficit/hyperactivity disorder (ADHD) individual and a healthy sibling in a Caucasian family in Australia | [245] |
| Model study | ADHD | iPSCs and NSCs | Growth rates of human induced pluripotent stem cells and neural stem cells from attention-deficit/hyperactivity disorder patients: a preliminary study | [246] |
| Molecular study | HD | iPSC | An alternative splicing modulator decreases mutant HTT and improves the molecular fingerprint in Huntington’s disease patient neurons | [247] |
| Molecular study | HD | iPSC-derived neurons (Mice) | CryoET reveals organelle phenotypes in Huntington’s disease patient iPSC-derived and mouse primary neurons | [248] |
| Model study | HD | iPSC-derived neural cells | Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites | [249] |
| Model study | HD | iPSC-derived neural cells | Extracellular vesicles improve GABAergic transmission in Huntington’s disease iPSC-derived neurons | [250] |
4.14. Limitations in the Use of Organoid Models
5. iPSC-Based Therapies for Neurological Diseases
| Trial Type | Disease | Target | Result | Reference |
|---|---|---|---|---|
| Cell Therapy | AD | Rat | The transplanted rats rescued Alzheimer’s cognition. | [278] |
| Cell Therapy | AD | Mouse | Grafted mice showed improved memory, synaptic plasticity, and reduced AD brain pathology, including a reduction in amyloid and tangle deposits. | [279] |
| Drug Screening | AD | hiPSC | β-secretase inhibitor IV (BSI) and γ-secretase inhibitor XXI/compound E (GSI) showed similar effects as screening in other models. | [280] |
| Drug Screening | AD | hiPSC | Docosahexaenoic acid (DHA) treatment alleviated the stress responses in the AD neural cells. | [270] |
| Drug Screening | AD | hiPSC | The anthelminthic avermectins increase the relative production of short forms of Aβ and reduce the relative production of longer Aβ fragments in human cortical neurons. | [281] |
| Cell Therapy | HD | Mice | iPSCs survived and differentiated into region-specific neurons in both mice groups without tumor formation. | [282] |
| Cell Therapy | HD | Mice | Grafted mice showed a significant increase in lifespan. In iPSC groups, animals showed significant improvement in motor functions and grip strength. | [283] |
| Cell Therapy | HD | Rat | Grafted rats showed significant behavioral improvements for up to 12 weeks. iPSCs enhanced endogenous neurogenesis and reconstituted the damaged neuronal connections. | [166] |
| Cell Therapy | HD | Mice | Improved neuronal dysfunction by SUPT4H1-edited iPSC grafts. | [284] |
| Cell Therapy | MLD | Mice | Transplantation of ARSA-overexpressing precursors into ARSA-deficient mice significantly reduced sulfatide storage up to 300 µm from grafted cells. | [285] |
| Cell Therapy | MLD | Mice | Grafts of iPSCs into neonatal and adult immunodeficient MLD mice stably restored arylsulfatase A (ARSA) activity in the whole CNS and a significant decrease in sulfatide storage when ARSA-overexpressing cells were used. | [165] |
| Cell Therapy | PD | Rat | iPSC graft differentiated into mature mDA neurons that survive over long term and restored motor function. | [286] |
| Cell Therapy | PD | Mice | hiPSCs differentiated into mDA neurons and long-term motor functional recovery was achieved after transplantation. | [287] |
| Cell Therapy | PD | Rat | Grafted iPSCs could survive in Parkinsonian rat brains for at least 150 days, and many of them differentiated into tyrosine hydroxylase (TH)-positive cells. | [288] |
| Cell Therapy | PD | Rat | Intranigral engraftment to the ventral midbrain demonstrated that mDA progenitors cryopreserved on day 17, and cells had a greater capacity than immature mDA neuron cells to innervate over long distances to forebrain structures. | [289] |
| Cell Therapy | PD | Rat | hiPSC-derived dopaminergic progenitor cells integrate better into the striatum of neonates than older rats. | [290] |
| Cell Therapy | PD | Mice | More than 90% of the engrafted cells differentiated into the lineage of mDA neurons, and approximately 15% developed into mature mDA neurons without tumor formation. | [291] |
| Cell Therapy | PD | Rat | There was a neural remodel of basal ganglia circuitry and no tumorigenicity. | [292] |
| Cell Therapy | PD | Mice | iPSCs matured into mDA neurons, reverse motor function, and established bidirectional connections with natural brain target regions without tumor formation. | [217] |
| Cell Therapy | SCI | Rat | Transplanted cells displayed robust integration properties, including synapse formation and myelination by host. | [293] |
| Cell Therapy | SCI | Mice | Due to DREADD expression, it was shown a significant decrease in locomotor dysfunction in SCI-grafted mice, which was exclusively observed following the neurons’ maturation. | [294] |
| Cell Therapy | SCI | Mice | The combination of iPSC graft and rehabilitative training therapy significantly improved motor functions. | [295] |
| Cell Therapy | SCI | Rat | Neuro-pluripotent cells derived from iPSC were able to survive and differentiate into both neurons and astrocytes, which improved forelimb locomotor function. | [296] |
| Cell Therapy | Stroke | Mice | Combination of electroacupuncture and iPSC-derived extracellular vesicle treatment ameliorated neurological impairments and reduced the infarct volume and neuronal apoptosis in MCAO mice. | [297] |
| Cell Therapy | Stroke | Pig | Tanshinone IIA nanoparticles increased iPSC engraftment, enhanced cellular and tissue recovery, and improved neurological function in a translational pig stroke model. | [298] |
| Cell Therapy | Stroke | Rat | Increased glucose metabolism and neurofunctional in iPSC-transplanted rats. | [299] |
| Cell Therapy | Stroke | Rat | Graft of iPSCs inhibited microglial activation and expression of proinflammatory cytokines and suppressed oxidative stress and neuronal death in the cerebral cortex at the ischemic border zone. | [300] |
| Cell Therapy | Stroke | Mice | Graft survived well and primarily differentiated into GABAergic interneurons and significantly restored the sensorimotor deficits of stroke mice for a long time. | [301] |
| Cell Therapy | Stroke | Rat | Generated oligodendrocytes survived and formed myelin-ensheathing human axons in the host tissue after grafting onto adult human cortical organotypic cultures. | [302] |
| Cell Therapy | TLE | Mice | A much-reduced frequency of spontaneous recurrent seizures in grafted animals. | [262] |
6. Conclusions and Future Perspectives
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Specchio, N.; Aronica, E. Advances in the Genetics and Neuropathology of Tuberous Sclerosis Complex: Edging Closer to Targeted Therapy. Lancet Neurol. 2022, 21, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.-L.; Chang, K.-H. Stem Cell Therapy in Treating Epilepsy. Front. Neurosci. 2022, 16, 934507. [Google Scholar] [CrossRef] [PubMed]
- Toman, N.G.; Grande, A.W.; Low, W.C. Neural Repair in Stroke. Cell Transplant. 2019, 28, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, B.F.; da Cruz, B.C.; de Sousa, B.M.; Correia, P.D.; David, N.; Rocha, C.; Almeida, R.D.; Ribeiro da Cunha, M.; Marques Baptista, A.A.; Vieira, S.I. Cell Therapies for Spinal Cord Injury: A Review of the Clinical Trials and Cell-Type Therapeutic Potential. Brain J. Neurol. 2023, 146, 2672–2693. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tan, S. Direct Lineage Conversion of Astrocytes to Induced Neural Stem Cells or Neurons. Neurosci. Bull. 2015, 31, 357–367. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.E. Using Induced Pluripotent Stem Cells Derived Neurons to Model Brain Diseases. Neural Regen. Res. 2017, 12, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Espinosa, M.B.; Paluh, J.L. Human Stem Cell-Derived Neurons and Neural Circuitry Therapeutics: Next Frontier in Spinal Cord Injury Repair. Exp. Biol. Med. 2022, 247, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martello, G.; Smith, A. The Nature of Embryonic Stem Cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 647–675. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Madrid, M.; Sumen, C.; Aivio, S.; Saklayen, N. Autologous Induced Pluripotent Stem Cell-Based Cell Therapies: Promise, Progress, and Challenges. Curr. Protoc. 2021, 1, e88. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.X.; Pek, N.M.Q.; Soh, B.-S. Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 936. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Zhang, M.; Ly, O.T.; Chen, H.; Sridhar, A.; Lambers, E.; Chalazan, B.; Youn, S.-W.; Maienschein-Cline, M.; Feferman, L.; et al. Human Induced Pluripotent Stem Cell-Derived Atrial Cardiomyocytes Carrying an SCN5A Mutation Identify Nitric Oxide Signaling as a Mediator of Atrial Fibrillation. Stem Cell Rep. 2021, 16, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Pasteuning-Vuhman, S.; de Jongh, R.; Timmers, A.; Pasterkamp, R.J. Towards Advanced iPSC-Based Drug Development for Neurodegenerative Disease. Trends Mol. Med. 2021, 27, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Montserrat, N.; Aasen, T.; Gonzalez, F.; Rodríguez-Pizà, I.; Vassena, R.; Raya, A.; Boué, S.; Barrero, M.J.; Corbella, B.A.; et al. Generation of Induced Pluripotent Stem Cells from Human Cord Blood Using OCT4 and SOX2. Cell Stem Cell 2009, 5, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and Rapid Generation of Induced Pluripotent Stem Cells from Human Keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Belmonte, J.C.I. Isolation and Cultivation of Human Keratinocytes from Skin or Plucked Hair for the Generation of Induced Pluripotent Stem Cells. Nat. Protoc. 2010, 5, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of Human Induced Pluripotent Stem Cells from Urine Samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Cai, X.; Wang, L.; Liao, B.; Zhang, H.; Shan, Y.; Chen, Q.; Zhou, T.; Li, X.; Hou, J.; et al. Generating a Non-Integrating Human Induced Pluripotent Stem Cell Bank from Urine-Derived Cells. PLoS ONE 2013, 8, e70573. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-F.; Chen, M.; Zhang, N.-N.; Yang, H.-J.; Rui, Q.; Zhou, Y.-F. In Vitro and in Vivo Differentiation of Induced Pluripotent Stem Cells Generated from Urine-Derived Cells into Cardiomyocytes. Biol. Open 2018, 7, bio029157. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Marino, F.; Salerno, L.; Cianflone, E.; Molinaro, C.; Salerno, N.; De Angelis, A.; Viglietto, G.; Urbanek, K.; Torella, D. From Spheroids to Organoids: The Next Generation of Model Systems of Human Cardiac Regeneration in a Dish. Int. J. Mol. Sci. 2021, 22, 13180. [Google Scholar] [CrossRef] [PubMed]
- Su, R.-J.; Baylink, D.J.; Neises, A.; Kiroyan, J.B.; Meng, X.; Payne, K.J.; Tschudy-Seney, B.; Duan, Y.; Appleby, N.; Kearns-Jonker, M.; et al. Efficient Generation of Integration-Free Ips Cells from Human Adult Peripheral Blood Using BCL-XL Together with Yamanaka Factors. PLoS ONE 2013, 8, e64496. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Markoulaki, S.; Schorderet, P.; Carey, B.W.; Beard, C.; Wernig, M.; Creyghton, M.P.; Steine, E.J.; Cassady, J.P.; Foreman, R.; et al. Direct Reprogramming of Terminally Differentiated Mature B Lymphocytes to Pluripotency. Cell 2008, 133, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Nagano, S.; Maeda, T.; Ichise, H.; Kashima, S.; Ohtaka, M.; Nakanishi, M.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; Masuda, K.; et al. High Frequency Production of T Cell-Derived iPSC Clones Capable of Generating Potent Cytotoxic T Cells. Mol. Ther. Methods Clin. Dev. 2020, 16, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Suwanpitak, S.; Promnakhon, N.; Netsrithong, R.; Wattanapanitch, M. Efficient Generation of iPSC-Derived Hematoendothelial Progenitors and Specification Toward T Cell Lineage. Methods Mol. Biol. 2022, 2454, 423–442. [Google Scholar] [CrossRef] [PubMed]
- Serwold, T.; Hochedlinger, K.; Swindle, J.; Hedgpeth, J.; Jaenisch, R.; Weissman, I.L. T-Cell Receptor-Driven Lymphomagenesis in Mice Derived from a Reprogrammed T Cell. Proc. Natl. Acad. Sci. USA 2010, 107, 18939–18943. [Google Scholar] [CrossRef] [PubMed]
- Dowey, S.N.; Huang, X.; Chou, B.-K.; Ye, Z.; Cheng, L. Generation of Integration-Free Human Induced Pluripotent Stem Cells from Postnatal Blood Mononuclear Cells by Plasmid Vector Expression. Nat. Protoc. 2012, 7, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Martinez, H.; Sun, B.; Li, A.; Zimmer, M.; Katsanis, N.; Davis, E.E.; Kurtzberg, J.; Lipnick, S.; Noggle, S.; et al. Rapid and Efficient Generation of Transgene-Free iPSC from a Small Volume of Cryopreserved Blood. Stem Cell Rev. Rep. 2015, 11, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of Human Peripheral Blood Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Moodley, K.; Sibanda, N.; February, K.; Rossouw, T. “It’s My Blood”: Ethical Complexities in the Use, Storage and Export of Biological Samples: Perspectives from South African Research Participants. BMC Med. Ethics 2014, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Boué, S.; Izpisúa Belmonte, J.C. Methods for Making Induced Pluripotent Stem Cells: Reprogramming à La Carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Eminli, S.; Foudi, A.; Stadtfeld, M.; Maherali, N.; Ahfeldt, T.; Mostoslavsky, G.; Hock, H.; Hochedlinger, K. Differentiation Stage Determines Potential of Hematopoietic Cells for Reprogramming into Induced Pluripotent Stem Cells. Nat. Genet. 2009, 41, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Ng, J.-H.; Heng, J.-C.D.; Ng, H.-H. Molecules That Promote or Enhance Reprogramming of Somatic Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 4, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M.; et al. Pluripotent Stem Cells Induced from Adult Neural Stem Cells by Reprogramming with Two Factors. Nature 2008, 454, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of Pluripotent Stem Cells from Primary Human Fibroblasts with Only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct Reprogramming of Human Neural Stem Cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Araúzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-Induced Pluripotency in Adult Neural Stem Cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Bouwman, B.A.; Ang, Y.S.; Kim, S.J.; Lee, D.F.; Lemischka, I.R.; Rendl, M. Single Transcription Factor Reprogramming of Hair Follicle Dermal Papilla Cells to Induced Pluripotent Stem Cells. Stem Cells 2011, 29, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Saha, K.; Pando, B.; van Zon, J.; Lengner, C.J.; Creyghton, M.P.; van Oudenaarden, A.; Jaenisch, R. Direct Cell Reprogramming Is a Stochastic Process Amenable to Acceleration. Nature 2009, 462, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, X.; Qin, H.; Zhu, F.; Liu, H.; Yang, W.; Zhang, Q.; Xiang, C.; Hou, P.; Song, Z.; et al. Two Supporting Factors Greatly Improve the Efficiency of Human iPSC Generation. Cell Stem Cell 2008, 3, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Tsubooka, N.; Ichisaka, T.; Okita, K.; Takahashi, K.; Nakagawa, M.; Yamanaka, S. Roles of Sall4 in the Generation of Pluripotent Stem Cells from Blastocysts and Fibroblasts. Genes Cells 2009, 14, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of Human Somatic Cells to Pluripotency with Defined Factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, Y.-J.; Jung, H. Protein Kinases and Their Inhibitors in Pluripotent Stem Cell Fate Regulation. Stem Cells Int. 2019, 2019, e1569740. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Long, B.; Han, W.; Yuan, S.; Wang, K. microRNAs: Important Regulators of Stem Cells. Stem Cell Res. Ther. 2017, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple Targets of miR-302 and miR-372 Promote Reprogramming of Human Fibroblasts to Induced Pluripotent Stem Cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Çağlayan, E.S.; Güran, Ş. Importance of Myc-Related microRNAs in Induced Pluripotency. Cell Biol. Int. 2015, 39, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, U.; Davila, J.; Hart, R.P. miRNA in Pluripotent Stem Cells. Regen. Med. 2010, 5, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.M.S.; Costa, I.C.; Santos, J.F.D.; Dourado, P.M.M.; Forni, M.F.; Ferreira, J.C.B. Induced Pluripotent Stem Cells Reprogramming: Epigenetics and Applications in the Regenerative Medicine. Rev. Assoc. Med. Bras. 2017, 63, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Yin, X.; Yang, W.; Du, Y.; Hou, P.; Ge, J.; Liu, C.; Zhang, W.; Zhang, X.; et al. Generation of iPSCs from Mouse Fibroblasts with a Single Gene, Oct4, and Small Molecules. Cell Res. 2011, 21, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y.; et al. Chemical Reprogramming of Human Somatic Cells to Pluripotent Stem Cells. Nature 2022, 605, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond Editing: Repurposing CRISPR-Cas9 for Precision Genome Regulation and Interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Balboa, D.; Katayama, S.; Bespalov, M.; Krjutškov, K.; Jouhilahti, E.-M.; Trokovic, R.; Kere, J.; Otonkoski, T. Human Pluripotent Reprogramming with CRISPR Activators. Nat. Commun. 2018, 9, 2643. [Google Scholar] [CrossRef] [PubMed]
- Sokka, J.; Yoshihara, M.; Kvist, J.; Laiho, L.; Warren, A.; Stadelmann, C.; Jouhilahti, E.-M.; Kilpinen, H.; Balboa, D.; Katayama, S.; et al. CRISPR Activation Enables High-Fidelity Reprogramming into Human Pluripotent Stem Cells. Stem Cell Rep. 2022, 17, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.L.; Halmai, J.A.N.M.; Fink, K.D. The iNs and Outs of Direct Reprogramming to Induced Neurons. Front. Genome Ed. 2020, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; P R Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Brix, J.; Zhou, Y.; Luo, Y. The Epigenetic Reprogramming Roadmap in Generation of iPSCs from Somatic Cells. J. Genet. Genomics Yi Chuan Xue Bao 2015, 42, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Van den Hurk, M.; Kenis, G.; Bardy, C.; van den Hove, D.L.; Gage, F.H.; Steinbusch, H.W.; Rutten, B.P. Transcriptional and Epigenetic Mechanisms of Cellular Reprogramming to Induced Pluripotency. Epigenomics 2016, 8, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Plath, K. Epigenetics of Reprogramming to Induced Pluripotency. Cell 2013, 152, 1324–1343. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of Pluripotent Stem Cells by Defined Factors Is Greatly Improved by Small-Molecule Compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Chou, B.-K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate Greatly Enhances Derivation of Human Induced Pluripotent Stem Cells by Promoting Epigenetic Remodeling and the Expression of Pluripotency-Associated Genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-J.; Park, Y.-I.; So, B.; Kang, H.-G. Sodium Butyrate Efficiently Converts Fully Reprogrammed Induced Pluripotent Stem Cells from Mouse Partially Reprogrammed Cells. Cell. Reprogram. 2014, 16, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Desponts, C.; Do, J.T.; Hahm, H.S.; Schöler, H.R.; Ding, S. Induction of Pluripotent Stem Cells from Mouse Embryonic Fibroblasts by Oct4 and Klf4 with Small-Molecule Compounds. Cell Stem Cell 2008, 3, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Koshino, Y.; Shibata, F.; Oki, T.; Nakajima, H.; Nosaka, T.; Kumagai, H. Retrovirus-Mediated Gene Transfer and Expression Cloning: Powerful Tools in Functional Genomics. Exp. Hematol. 2003, 31, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of Pluripotent Stem Cells from Fibroblast Cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Varas, F.; Stadtfeld, M.; de Andres-Aguayo, L.; Maherali, N.; di Tullio, A.; Pantano, L.; Notredame, C.; Hochedlinger, K.; Graf, T. Fibroblast-Derived Induced Pluripotent Stem Cells Show No Common Retroviral Vector Insertions. Stem Cells 2009, 27, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Sukonnik, T.; Kean, T.; Bharadwaj, R.R.; Pasceri, P.; Ellis, J. Retrovirus Silencing, Variegation, Extinction, and Memory Are Controlled by a Dynamic Interplay of Multiple Epigenetic Modifications. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 10, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Stadtfeld, M.; Murphy, G.J.; Hochedlinger, K.; Kotton, D.N.; Mostoslavsky, G. Induced Pluripotent Stem Cell Generation Using a Single Lentiviral Stem Cell Cassette. Stem Cells 2009, 27, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac Transposition Reprograms Fibroblasts to Induced Pluripotent Stem Cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Bendriem, R.M.; Wu, W.W.; Shen, R.-F. 3D Brain Organoids Derived from Pluripotent Stem Cells: Promising Experimental Models for Brain Development and Neurodegenerative Disorders. J. Biomed. Sci. 2017, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.G.; Kriegstein, A.R. Challenges of Organoid Research. Annu. Rev. Neurosci. 2022, 45, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Garreta, E.; Kamm, R.D.; Chuva De Sousa Lopes, S.M.; Lancaster, M.A.; Weiss, R.; Trepat, X.; Hyun, I.; Montserrat, N. Rethinking Organoid Technology through Bioengineering. Nat. Mater. 2021, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Turhan, A.G.; Hwang, J.W.; Chaker, D.; Tasteyre, A.; Latsis, T.; Griscelli, F.; Desterke, C.; Bennaceur-Griscelli, A. iPSC-Derived Organoids as Therapeutic Models in Regenerative Medicine and Oncology. Front. Med. 2021, 8, 728543. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Parent, J.M. Modeling Genetic Epilepsies in a Dish. Dev. Dyn. 2020, 249, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human Cerebral Organoids Recapitulate Gene Expression Programs of Fetal Neocortex Development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Wray, S. Modelling Neurodegenerative Disease Using Brain Organoids. Semin. Cell Dev. Biol. 2021, 111, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Tanaka, Y.; Shibata, M.; Kimura, Y.; Ishikawa, M.; Higurashi, N.; Yamamoto, T.; Ichise, E.; Chiyonobu, T.; Ishii, A. Application of Induced Pluripotent Stem Cells in Epilepsy. Mol. Cell. Neurosci. 2020, 108, 103535. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized Developmental Patterning and Differentiation in Cerebral Organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J.; Buckley, R.F.; Hohman, T.J. 2022 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63, 168. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The Effect of APOE and Other Common Genetic Variants on the Onset of Alzheimer’s Disease and Dementia: A Community-Based Cohort Study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; Van Der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, V.D.; Qiu, L.; Tan, E.K.; Zeng, L.; Zhang, Y. Modelling Alzheimer’s Disease: Insights from in Vivo to in Vitro Three-Dimensional Culture Platforms. J. Tissue Eng. Regen. Med. 2018, 12, 1944–1958. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.-C.; Yang, X.-H.; Cheng, X.-Y.; Deng, W.-B.; Guo, X.-L.; Yang, H.; Wang, Y.; Li, J.; Yao, Y. Optimization of Cerebral Organoids: A More Qualified Model for Alzheimer’s Disease Research. Transl. Neurodegener. 2021, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, S.; Jarray, R.; Nassor, F.; Guyot, A.-C.; Cottin, S.; Rontard, J.; Mikol, J.; Mabondzo, A.; Deslys, J.-P.; Yates, F. Small-Molecule Induction of Aβ-42 Peptide Production in Human Cerebral Organoids to Model Alzheimer’s Disease Associated Phenotypes. PLoS ONE 2018, 13, e0209150. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modeling Neurodegenerative Microenvironment Using Cortical Organoids Derived from Human Stem Cells. Tissue Eng. Part A 2018, 24, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic Lateral Sclerosis Is a Distal Axonopathy: Evidence in Mice and Man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Brown, R.H. Molecular Biology of Amyotrophic Lateral Sclerosis: Insights from Genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.-C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human Sensorimotor Organoids Derived from Healthy and Amyotrophic Lateral Sclerosis Stem Cells Form Neuromuscular Junctions. Nat. Commun. 2021, 12, 4744. [Google Scholar] [CrossRef] [PubMed]
- Agnew-Blais, J.C.; Polanczyk, G.V.; Danese, A.; Wertz, J.; Moffitt, T.E.; Arseneault, L. Evaluation of the Persistence, Remission, and Emergence of Attention-Deficit/Hyperactivity Disorder in Young Adulthood. JAMA Psychiatry 2016, 73, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, S.P. Attention Deficit Hyperactivity Disorder (ADHD): Controversy, Developmental Mechanisms, and Multiple Levels of Analysis. Annu. Rev. Clin. Psychol. 2018, 14, 291–316. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Rubia, K. Neurobiological Circuits Regulating Attention, Cognitive Control, Motivation, and Emotion: Disruptions in Neurodevelopmental Psychiatric Disorders. J. Am. Acad. Child Adolesc. Psychiatry 2012, 51, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Ball, G.; Malpas, C.B.; Genc, S.; Efron, D.; Sciberras, E.; Anderson, V.; Nicholson, J.M.; Silk, T.J. Multimodal Structural Neuroimaging Markers of Brain Development and ADHD Symptoms. Am. J. Psychiatry 2019, 176, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hartlaub, A.M.; McElroy, C.A.; Maitre, N.L.; Hester, M.E. Modeling Human Brain Circuitry Using Pluripotent Stem Cell Platforms. Front. Pediatr. 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Eguchi, N.; Okazaki, S.; Sora, I.; Hishimoto, A. Telencephalon Organoids Derived from an Individual with ADHD Show Altered Neurodevelopment of Early Cortical Layer Structure. Stem Cell Rev. Rep. 2023, 19, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Kanner, L. Autistic disturbances of aff ective contact. Nerv. Child 1943, 2, 217–250. [Google Scholar]
- Villa, C.; Combi, R.; Conconi, D.; Lavitrano, M. Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges. Pharmaceutics 2021, 13, 280. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; Wu, C.; Wang, J.; Sun, M. Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. Int. J. Mol. Sci. 2023, 24, 1819. [Google Scholar] [CrossRef] [PubMed]
- Baio, J. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Neurodevelopmental disorders. In Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Iles, A. Autism Spectrum Disorders. Prim. Care Clin. Off. Pract. 2021, 48, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Genovese, A.; Butler, M.G. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes 2023, 14, 677. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P.; Christen, Y. (Eds.) A Time for Metabolism and Hormones; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-27068-5. [Google Scholar]
- Forsberg, S.L.; Ilieva, M.; Maria Michel, T. Epigenetics and Cerebral Organoids: Promising Directions in Autism Spectrum Disorders. Transl. Psychiatry 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.d.S.; Araújo, C.d.A.; Rocha, C.A.G.; Costa-Ferro, Z.S.M.; Souza, B.S.d.F. Modeling Autism Spectrum Disorders with Induced Pluripotent Stem Cell-Derived Brain Organoids. Biomolecules 2023, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Russo, F.B.; Brito, A.; de Freitas, A.M.; Castanha, A.; de Freitas, B.C.; Beltrão-Braga, P.C.B. The Use of iPSC Technology for Modeling Autism Spectrum Disorders. Neurobiol. Dis. 2019, 130, 104483. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Bley, A.E.; Eichler, F. Canavan Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lotun, A.; Gessler, D.J.; Gao, G. Canavan Disease as a Model for Gene Therapy-Mediated Myelin Repair. Front. Cell. Neurosci. 2021, 15, 661928. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chao, J.; Ye, P.; Luong, Q.; Sun, G.; Liu, W.; Cui, Q.; Flores, S.; Jackson, N.; Shayento, A.N.H.; et al. Developing Hypoimmunogenic Human iPSC-Derived Oligodendrocyte Progenitor Cells as an Off-The-Shelf Cell Therapy for Myelin Disorders. Adv. Sci. 2023, 10, e2206910. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Feng, L.; Ye, P.; Chen, X.; Cui, Q.; Sun, G.; Zhou, T.; Tian, E.; Li, W.; Hu, W.; et al. Therapeutic Development for Canavan Disease Using Patient iPSCs Introduced with the Wild-Type ASPA Gene. iScience 2022, 25, 104391. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chao, J.; Tian, E.; Li, L.; Ye, P.; Zhang, M.; Chen, X.; Cui, Q.; Sun, G.; Zhou, T.; et al. Cell-Based Therapy for Canavan Disease Using Human iPSC-Derived NPCs and OPCs. Adv. Sci. 2020, 7, 2002155. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chao, J.; Zhang, M.; Pacquing, E.; Hu, W.; Shi, Y. Developing a Human iPSC-Derived Three-Dimensional Myelin Spheroid Platform for Modeling Myelin Diseases. iScience 2023, 26, 108037. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Boas, W.V.E.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Falco-Walter, J. Epilepsy—Definition, Classification, Pathophysiology, and Epidemiology. Semin. Neurol. 2020, 40, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Epilepsy: A Public Health Imperative: Summary; WHO: Geneva, Switzerland, 2019.
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- WHO. Dementia Fact Sheet. March 2023. Available online: https://www.who.int/en/news-room/fact-sheets/detail/dementia (accessed on 1 February 2024).
- Coyle-Gilchrist, I.T.S.; Dick, K.M.; Patterson, K.; Vázquez Rodríquez, P.; Wehmann, E.; Wilcox, A.; Lansdall, C.J.; Dawson, K.E.; Wiggins, J.; Mead, S.; et al. Prevalence, Characteristics, and Survival of Frontotemporal Lobar Degeneration Syndromes. Neurology 2016, 86, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.T.; Caixeta, L.; Machado, S.; Silva, A.C.; Nardi, A.E.; Arias-Carrión, O.; Carta, M.G. Epidemiology of Early-Onset Dementia: A Review of the Literature. Clin. Pract. Epidemiol. Ment. Health CP EMH 2013, 9, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Bowles, K.R.; Silva, M.C.; Whitney, K.; Bertucci, T.; Berlind, J.E.; Lai, J.D.; Garza, J.C.; Boles, N.C.; Mahali, S.; Strang, K.H.; et al. ELAVL4, Splicing, and Glutamatergic Dysfunction Precede Neuron Loss in MAPT Mutation Cerebral Organoids. Cell 2021, 184, 4547–4563.e17. [Google Scholar] [CrossRef] [PubMed]
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD Brain Organoid Slice Cultures Display Distinct Early Astrocyte and Targetable Neuronal Pathology. Nat. Neurosci. 2021, 24, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Finkbeiner, S. Protein Aggregates in Huntington’s Disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Andhale, R.; Shrivastava, D. Huntington’s Disease: A Clinical Review. Cureus 2022, 14, e28484. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington Disease: New Insights into Molecular Pathogenesis and Therapeutic Opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington Disease: Natural History, Biomarkers and Prospects for Therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Pouladi, M.A.; Morton, A.J.; Hayden, M.R. Choosing an Animal Model for the Study of Huntington’s Disease. Nat. Rev. Neurosci. 2013, 14, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; McBride, J.L.; Kordower, J.H. Animal Models of Huntington’s Disease. ILAR J. 2007, 48, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Lerou, P.H.; Zhao, R.; Huo, H.; Daley, G.Q. Generation of Human-Induced Pluripotent Stem Cells. Nat. Protoc. 2008, 3, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; An, M.C.; Montoro, D.; Ellerby, L.M. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010, 2, RRN1193. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.-I.; Kim, D.-W.; Lee, N.; Jeon, Y.-J.; Jeon, I.; Kwon, J.; Kim, J.; Soh, Y.; Lee, D.-S.; Seo, K.S.; et al. Quantitative Proteomic Analysis of Induced Pluripotent Stem Cells Derived from a Human Huntington’s Disease Patient. Biochem. J. 2012, 446, 359–371. [Google Scholar] [CrossRef] [PubMed]
- HD iPSC Consortium. Induced Pluripotent Stem Cells from Patients with Huntington’s Disease Show CAG-Repeat-Expansion-Associated Phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.R.; Tom, C.M.; Wang, Y.; Bresee, C.; Rushton, D.; Mathkar, P.P.; Tang, J.; Mattis, V.B. Human Huntington’s Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018, 25, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD Investigators; REGISTRY Investigators; et al. Identification of Genetic Variants Associated with Huntington’s Disease Progression: A Genome-Wide Association Study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Telenius, H.; Kremer, B.; Goldberg, Y.P.; Theilmann, J.; Andrew, S.E.; Zeisler, J.; Adam, S.; Greenberg, C.; Ives, E.J.; Clarke, L.A. Somatic and Gonadal Mosaicism of the Huntington Disease Gene CAG Repeat in Brain and Sperm. Nat. Genet. 1994, 6, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Świtońska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Marczak, Ł.; Stelmaszczuk, M.; Figlerowicz, M.; Figiel, M. Identification of Altered Developmental Pathways in Human Juvenile HD iPSC With 71Q and 109Q Using Transcriptome Profiling. Front. Cell. Neurosci. 2018, 12, 528. [Google Scholar] [CrossRef] [PubMed]
- Mattis, V.B.; Tom, C.; Akimov, S.; Saeedian, J.; Østergaard, M.E.; Southwell, A.L.; Doty, C.N.; Ornelas, L.; Sahabian, A.; Lenaeus, L.; et al. HD iPSC-Derived Neural Progenitors Accumulate in Culture and Are Susceptible to BDNF Withdrawal Due to Glutamate Toxicity. Hum. Mol. Genet. 2015, 24, 3257–3271. [Google Scholar] [CrossRef] [PubMed]
- Smith-Geater, C.; Hernandez, S.J.; Lim, R.G.; Adam, M.; Wu, J.; Stocksdale, J.T.; Wassie, B.T.; Gold, M.P.; Wang, K.Q.; Miramontes, R.; et al. Aberrant Development Corrected in Adult-Onset Huntington’s Disease iPSC-Derived Neuronal Cultures via WNT Signaling Modulation. Stem Cell Rep. 2020, 14, 406–419. [Google Scholar] [CrossRef]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic Correction of Huntington’s Disease Phenotypes in Induced Pluripotent Stem Cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; An, M.C.; Zhang, N.; O’Brien, R.N.; Ramos, E.M.; Gao, F.; Atwood, R.; Bailus, B.J.; Melov, S.; Mooney, S.D.; et al. Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington’s Disease Neural Stem Cells. Stem Cell Rep. 2015, 5, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.-I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef] [PubMed]
- HD iPSC Consortium. Developmental Alterations in Huntington’s Disease Neural Cells and Pharmacological Rescue in Cells and Mice. Nat. Neurosci. 2017, 20, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between Genetic, Lifestyle and Environmental Risk Factors for Multiple Sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple Sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- McCaughey-Chapman, A.; Connor, B. Cell Reprogramming for Oligodendrocytes: A Review of Protocols and Their Applications to Disease Modeling and Cell-Based Remyelination Therapies. J. Neurosci. Res. 2023, 101, 1000–1028. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights from the Atlas of MS, Third Edition. Mult. Scler. Houndmills Basingstoke Engl. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C. Defining the Clinical Course of Multiple Sclerosis: Results of an International Survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.M.; McCombe, P.A. Role of Gender in Multiple Sclerosis: Clinical Effects and Potential Molecular Mechanisms. J. Neuroimmunol. 2011, 234, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple Sclerosis. Lancet Lond. Engl. 2002, 359, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Daviaud, N.; Chen, E.; Edwards, T.; Sadiq, S.A. Cerebral Organoids in Primary Progressive Multiple Sclerosis Reveal Stem Cell and Oligodendrocyte Differentiation Defect. Biol. Open 2023, 12, bio059845. [Google Scholar] [CrossRef] [PubMed]
- Pouya, A.; Satarian, L.; Kiani, S.; Javan, M.; Baharvand, H. Human Induced Pluripotent Stem Cells Differentiation into Oligodendrocyte Progenitors and Transplantation in a Rat Model of Optic Chiasm Demyelination. PLoS ONE 2011, 6, e27925. [Google Scholar] [CrossRef] [PubMed]
- García-León, J.A.; Kumar, M.; Boon, R.; Chau, D.; One, J.; Wolfs, E.; Eggermont, K.; Berckmans, P.; Gunhanlar, N.; de Vrij, F.; et al. SOX10 Single Transcription Factor-Based Fast and Efficient Generation of Oligodendrocytes from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Petsko, G.A.; Studer, L. Induced Pluripotent Stem Cells: A Tool for Modeling Parkinson’s Disease. Trends Neurosci. 2022, 45, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.; Kim, S.; Krainc, D. Organoid and Pluripotent Stem Cells in Parkinson’s Disease Modeling: An Expert View on Their Value to Drug Discovery. Expert Opin. Drug Discov. 2020, 15, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; Von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-Causing α-Synuclein Missense Mutations Shift Native Tetramers to Monomers as a Mechanism for Disease Initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef] [PubMed]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin Recruitment Is Impaired in Neurons Derived from Mutant PINK1 Induced Pluripotent Stem Cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine Oxidation Mediates Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Eli, I.; Lerner, D.P.; Ghogawala, Z. Acute Traumatic Spinal Cord Injury. Neurol. Clin. 2021, 39, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, C.S.; Nori, S.; Tetreault, L.; Wilson, J.; Kwon, B.; Harrop, J.; Choi, D.; Fehlings, M.G. Traumatic Spinal Cord Injury-Repair and Regeneration. Neurosurgery 2017, 80, S9–S22. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.J.; Martin, M.J. Trauma: Spinal Cord Injury. Surg. Clin. N. Am. 2017, 97, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fang, S.; Deng, S.; Li, H.; Lin, X.; Huang, Y.; Chung, S.; Shu, Y.; Shao, Z. Generation of Neural Organoids for Spinal-Cord Regeneration via the Direct Reprogramming of Human Astrocytes. Nat. Biomed. Eng. 2023, 7, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Shin, H.; Shaker, M.R.; Kim, H.J.; Park, S.-H.; Kim, J.H.; Lee, N.; Kang, M.; Cho, S.; Kwak, T.H.; et al. Production of Human Spinal-Cord Organoids Recapitulating Neural-Tube Morphogenesis. Nat. Biomed. Eng. 2022, 6, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Winanto; Khong, Z.-J.; Hor, J.-H.; Ng, S.-Y. Spinal Cord Organoids Add an Extra Dimension to Traditional Motor Neuron Cultures. Neural Regen. Res. 2019, 14, 1515. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Spellicy, S.E.; Hess, D.C. The Immunomodulatory Capacity of Induced Pluripotent Stem Cells in the Post-Stroke Environment. Front. Cell Dev. Biol. 2021, 9, 647415. [Google Scholar] [CrossRef] [PubMed]
- Juntunen, M.; Hagman, S.; Moisan, A.; Narkilahti, S.; Miettinen, S. In Vitro Oxygen-Glucose Deprivation-Induced Stroke Models with Human Neuroblastoma Cell- and Induced Pluripotent Stem Cell-Derived Neurons. Stem Cells Int. 2020, 2020, e8841026. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Susavila, H.; Bugallo-Casal, A.; Castillo, J.; Campos, F. Adult Stem Cells and Induced Pluripotent Stem Cells for Stroke Treatment. Front. Neurol. 2019, 10, 908. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Susavila, H.; Mora, C.; Aramburu-Núñez, M.; Quintas-Rey, R.; Arias, S.; Collado, M.; López-Arias, E.; Sobrino, T.; Castillo, J.; Dell’Era, P.; et al. Generation and Characterization of the Human iPSC Line IDISi001-A Isolated from Blood Cells of a CADASIL Patient Carrying a NOTCH3 Mutation. Stem Cell Res. 2018, 28, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Liu, Z.; Song, M.; Zhang, W.; Wang, S.; Liu, X.; Ma, S.; Sun, S.; Fu, L.; Chu, Q.; et al. Modeling CADASIL Vascular Pathologies with Patient-Derived Induced Pluripotent Stem Cells. Protein Cell 2019, 10, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-W.; Qian, E.S.; Woodard, L.E.; Bejoy, J. Neural Lineage Differentiation of Human Pluripotent Stem Cells: Advances in Disease Modeling. World J. Stem Cells 2023, 15, 530–547. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Nair, A.L.; Fowke, T.M.; Pontier, M.; Kasi, D.G.; Spijkers, X.M.; Hallard, C.; Rabussier, G.; van Vught, R.; Vulto, P.; et al. Modeling Ischemic Stroke in a Triculture Neurovascular Unit On-a-Chip. Fluids Barriers CNS 2021, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Lacalle-Aurioles, M.; Cassel de Camps, C.; Zorca, C.E.; Beitel, L.K.; Durcan, T.M. Applying hiPSCs and Biomaterials Towards an Understanding and Treatment of Traumatic Brain Injury. Front. Cell. Neurosci. 2020, 14, 594304. [Google Scholar] [CrossRef] [PubMed]
- Imai, R.; Tamura, R.; Yo, M.; Sato, M.; Fukumura, M.; Takahara, K.; Kase, Y.; Okano, H.; Toda, M. Neuroprotective Effects of Genome-Edited Human iPS Cell-Derived Neural Stem/Progenitor Cells on Traumatic Brain Injury. Stem Cells 2023, 41, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of Traumatic Brain Injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, K.; Laskowitz, D.T. Cellular and Molecular Mechanisms of Secondary Neuronal Injury Following Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; Frontiers in Neuroscience; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016; ISBN 978-1-4665-8491-4. [Google Scholar]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. eBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Yang, Z.; Zhu, T.; Shi, Y.; Rubenstein, R.; Tyndall, J.A.; Manley, G.T. An Update on Diagnostic and Prognostic Biomarkers for Traumatic Brain Injury. Expert Rev. Mol. Diagn. 2018, 18, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.; Mukherjee, A.; Sepulveda, S.; Becerra-Calixto, A.; Bravo-Vasquez, N.; Gherardelli, C.; Chavez, M.; Soto, C. Modeling Traumatic Brain Injury in Human Cerebral Organoids. Cells 2021, 10, 2683. [Google Scholar] [CrossRef] [PubMed]
- Snapper, D.M.; Reginauld, B.; Liaudanskaya, V.; Fitzpatrick, V.; Kim, Y.; Georgakoudi, I.; Kaplan, D.L.; Symes, A.J. Development of a Novel Bioengineered 3D Brain-like Tissue for Studying Primary Blast-Induced Traumatic Brain Injury. J. Neurosci. Res. 2023, 101, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Singh, T.; Kayhanian, S.; Tjerkaski, J.; Garcia, N.M.; Carpenter, K.L.H.; Patani, R.; Lindblad, C.; Thelin, E.P.; Syed, Y.A.; et al. Modeling the Inflammatory Response of Traumatic Brain Injury Using Human Induced Pluripotent Stem Cell Derived Microglia. J. Neurotrauma 2023, 40, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.A.; Phillips, J.K.; Costa, J.T.; Cho, F.S.; Oungoulian, S.R.; Finan, J.D. Stretch Injury of Human Induced Pluripotent Stem Cell Derived Neurons in a 96 Well Format. Sci. Rep. 2016, 6, 34097. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.; Mukherjee, A.; Sepulveda, S.E.; Gherardelli, C.; Becerra-Calixto, A.; Bravo-Vasquez, N.; Soto, C. Protocol for Controlled Cortical Impact in Human Cerebral Organoids to Model Traumatic Brain Injury. STAR Protoc. 2021, 2, 100987. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, X.; Xiong, W.; Chen, J. In Vivo Reprogramming Reactive Glia into iPSCs to Produce New Neurons in the Cortex Following Traumatic Brain Injury. Sci. Rep. 2016, 6, 22490. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, G.; Tian, E.; Zhang, M.; Davtyan, H.; Beach, T.G.; Reiman, E.M.; Blurton-Jones, M.; Holtzman, D.M.; Shi, Y. Modeling Sporadic Alzheimer’s Disease in Human Brain Organoids under Serum Exposure. Adv. Sci. 2021, 8, e2101462. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of Hyperexcitability in Alzheimer’s Disease hiPSC-Derived Neurons and Cerebral Organoids vs Isogenic Controls. eLife 2019, 8, e50333. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling Amyloid Beta and Tau Pathology in Human Cerebral Organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Lovejoy, C.; Harris, L.; Willumsen, N.; Alatza, A.; Casey, J.M.; Lines, G.; Kerins, C.; Mueller, A.K.; Zetterberg, H.; et al. Familial Alzheimer’s Disease Mutations in PSEN1 Lead to Premature Human Stem Cell Neurogenesis. Cell Rep. 2021, 34, 108615. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.-C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 Exacerbates Synapse Loss and Neurodegeneration in Alzheimer’s Disease Patient iPSC-Derived Cerebral Organoids. Nat. Commun. 2020, 11, 5540. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-C.; Jang, S.-Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.-H.; Seo, J.; Choi, M.; et al. A Logical Network-Based Drug-Screening Platform for Alzheimer’s Disease Representing Pathological Features of Human Brain Organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of Function of the Mitochondrial Peptidase PITRM1 Induces Proteotoxic Stress and Alzheimer’s Disease-like Pathology in Human Cerebral Organoids. Mol. Psychiatry 2021, 26, 5733–5750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-T.; Liu, D.; Kang, H.-C.; Lu, L.; Huang, H.-Z.; Ai, W.-Q.; Zhou, Y.; Deng, M.-F.; Li, H.; Liu, Z.-Q.; et al. Tau Pathology Epigenetically Remodels the Neuron-Glial Cross-Talk in Alzheimer’s Disease. Sci. Adv. 2023, 9, eabq7105. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Xu, R.; Wang, L.; Alam, M.M.; Ma, Z.; Zhu, S.; Martini, A.C.; Jadali, A.; Bernabucci, M.; Xie, P.; et al. Type-I-Interferon Signaling Drives Microglial Dysfunction and Senescence in Human iPSC Models of Down Syndrome and Alzheimer’s Disease. Cell Stem Cell 2022, 29, 1135–1153. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K.; et al. Acetylation Changes Tau Interactome to Degrade Tau in Alzheimer’s Disease Animal and Organoid Models. Aging Cell 2020, 19, e13081. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Calixto, A.; Mukherjee, A.; Ramirez, S.; Sepulveda, S.; Sinha, T.; Al-Lahham, R.; De Gregorio, N.; Gherardelli, C.; Soto, C. Lewy Body-like Pathology and Loss of Dopaminergic Neurons in Midbrain Organoids Derived from Familial Parkinson’s Disease Patient. Cells 2023, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Han, D.; Liu, W.; Wang, X.; Pan, N.; Wang, Y.; Chen, Z. Human iPSC-Derived Midbrain Organoids Functionally Integrate into Striatum Circuits and Restore Motor Function in a Mouse Model of Parkinson’s Disease. Theranostics 2023, 13, 2673–2692. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Neves, E.; Burke, C.; Jiang, X.; Xu, P.; Rhodes, K.J.; Khurana, V.; Scannevin, R.H.; Chung, C.Y. Patient-Derived Three-Dimensional Cortical Neurospheres to Model Parkinson’s Disease. PLoS ONE 2022, 17, e0277532. [Google Scholar] [CrossRef] [PubMed]
- Wulansari, N.; Darsono, W.H.W.; Woo, H.-J.; Chang, M.-Y.; Kim, J.; Bae, E.-J.; Sun, W.; Lee, J.-H.; Cho, I.-J.; Shin, H.; et al. Neurodevelopmental Defects and Neurodegenerative Phenotypes in Human Brain Organoids Carrying Parkinson’s Disease-Linked DNAJC6 Mutations. Sci. Adv. 2021, 7, eabb1540. [Google Scholar] [CrossRef] [PubMed]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia Integration into Human Midbrain Organoids Leads to Increased Neuronal Maturation and Functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Chlebanowska, P.; Tejchman, A.; Sułkowski, M.; Skrzypek, K.; Majka, M. Use of 3D Organoids as a Model to Study Idiopathic Form of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 694. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Bolognin, S.; Poovathingal, S.K.; Magni, S.; Gérard, D.; Antony, P.M.A.; Nickels, S.L.; Salamanca, L.; Berger, E.; Smits, L.M.; et al. The Parkinson’s-Disease-Associated Mutation LRRK2-G2019S Alters Dopaminergic Differentiation Dynamics via NR2F1. Cell Rep. 2021, 37, 109864. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, R.A.; Miranda, O.A.; Buth, J.E.; Mitchell, S.; Ferando, I.; Watanabe, M.; Allison, T.F.; Kurdian, A.; Fotion, N.N.; Gandal, M.J.; et al. Identification of Neural Oscillations and Epileptiform Changes in Human Brain Organoids. Nat. Neurosci. 2021, 24, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.J.; Repudi, S.; Saleem, A.; Kustanovich, I.; Viukov, S.; Abudiab, B.; Banne, E.; Mahajnah, M.; Hanna, J.H.; Stern, S.; et al. Modeling Genetic Epileptic Encephalopathies Using Brain Organoids. EMBO Mol. Med. 2021, 13, e13610. [Google Scholar] [CrossRef] [PubMed]
- Eichmüller, O.L.; Corsini, N.S.; Vértesy, Á.; Morassut, I.; Scholl, T.; Gruber, V.-E.; Peer, A.M.; Chu, J.; Novatchkova, M.; Hainfellner, J.A.; et al. Amplification of Human Interneuron Progenitors Promotes Brain Tumors and Neurological Defects. Science 2022, 375, eabf5546. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant Axon Branching via Fos-B Dysregulation in FUS-ALS Motor Neurons. eBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Ross, J.P.; Alipour, P.; Castonguay, C.-É.; Li, B.; Catoire, H.; Rochefort, D.; Urushitani, M.; Takahashi, R.; Sonnen, J.A.; et al. Spinal Cord Extracts of Amyotrophic Lateral Sclerosis Spread TDP-43 Pathology in Cerebral Organoids. PLoS Genet. 2023, 19, e1010606. [Google Scholar] [CrossRef] [PubMed]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-Mediated Excision of ALS/FTD-Causing Hexanucleotide Repeat Expansion in C9ORF72 Rescues Major Disease Mechanisms in Vivo and in Vitro. Nat. Commun. 2022, 13, 6286. [Google Scholar] [CrossRef] [PubMed]
- De Majo, M.; Koontz, M.; Marsan, E.; Salinas, N.; Ramsey, A.; Kuo, Y.-M.; Seo, K.; Li, H.; Dräger, N.; Leng, K.; et al. Granulin Loss of Function in Human Mature Brain Organoids Implicates Astrocytes in TDP-43 Pathology. Stem Cell Rep. 2023, 18, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Johns, A.E.; Maragakis, N.J. Exploring Motor Neuron Diseases Using iPSC Platforms. Stem Cells 2022, 40, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Shiozawa, S.; Tsuboi, D.; Amano, M.; Watanabe, H.; Maeda, S.; Kimura, T.; Yoshimatsu, S.; Kisa, F.; Karch, C.M.; et al. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Rep. 2019, 13, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Kerkering, J.; Muinjonov, B.; Rosiewicz, K.S.; Diecke, S.; Biese, C.; Schiweck, J.; Chien, C.; Zocholl, D.; Conrad, T.; Paul, F.; et al. iPSC-Derived Reactive Astrocytes from Patients with Multiple Sclerosis Protect Cocultured Neurons in Inflammatory Conditions. J. Clin. Investig. 2023, 133, e164637. [Google Scholar] [CrossRef] [PubMed]
- Schepers, M.; Paes, D.; Tiane, A.; Rombaut, B.; Piccart, E.; van Veggel, L.; Gervois, P.; Wolfs, E.; Lambrichts, I.; Brullo, C.; et al. Selective PDE4 Subtype Inhibition Provides New Opportunities to Intervene in Neuroinflammatory versus Myelin Damaging Hallmarks of Multiple Sclerosis. Brain Behav. Immun. 2023, 109, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mutukula, N.; Man, Z.; Takahashi, Y.; Iniesta Martinez, F.; Morales, M.; Carreon-Guarnizo, E.; Hernandez Clares, R.; Garcia-Bernal, D.; Martinez Martinez, L.; Lajara, J.; et al. Generation of RRMS and PPMS Specific iPSCs as a Platform for Modeling Multiple Sclerosis. Stem Cell Res. 2021, 53, 102319. [Google Scholar] [CrossRef] [PubMed]
- Fortune, A.J.; Taylor, B.V.; Charlesworth, J.C.; Burdon, K.P.; Blackburn, N.B.; Fletcher, J.L.; Mehta, A.; Young, K.M. Generation and Characterisation of Four Multiple Sclerosis iPSC Lines from a Single Family. Stem Cell Res. 2022, 62, 102828. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fleck, J.S.; Martins-Costa, C.; Burkard, T.R.; Themann, J.; Stuempflen, M.; Peer, A.M.; Vertesy, Á.; Littleboy, J.B.; Esk, C.; et al. Single-Cell Brain Organoid Screening Identifies Developmental Defects in Autism. Nature 2023, 621, 373–380. [Google Scholar] [CrossRef] [PubMed]
- De Jong, J.O.; Llapashtica, C.; Genestine, M.; Strauss, K.; Provenzano, F.; Sun, Y.; Zhu, H.; Cortese, G.P.; Brundu, F.; Brigatti, K.W.; et al. Cortical Overgrowth in a Preclinical Forebrain Organoid Model of CNTNAP2-Associated Autism Spectrum Disorder. Nat. Commun. 2021, 12, 4087. [Google Scholar] [CrossRef] [PubMed]
- Ejlersen, M.; Ilieva, M.; Michel, T.M. Superoxide Dismutase Isozymes in Cerebral Organoids from Autism Spectrum Disorder Patients. J. Neural Transm. 2022, 129, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-Mediated Heterozygous Knockout of the Autism Gene CHD8 and Characterization of Its Transcriptional Networks in Cerebral Organoids Derived from iPS Cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Dunkerson, J.; Moritz, K.E.; Young, J.; Pionk, T.; Fink, K.; Rossignol, J.; Dunbar, G.; Smith, J.S. Combining Enriched Environment and Induced Pluripotent Stem Cell Therapy Results in Improved Cognitive and Motor Function Following Traumatic Brain Injury. Restor. Neurol. Neurosci. 2014, 32, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Furmanski, O.; Nieves, M.D.; Doughty, M.L. Controlled Cortical Impact Model of Mouse Brain Injury with Therapeutic Transplantation of Human Induced Pluripotent Stem Cell-Derived Neural Cells. J. Vis. Exp. 2019, 149, e59561. [Google Scholar] [CrossRef]
- Iwasa, N.; Matsui, T.K.; Iguchi, N.; Kinugawa, K.; Morikawa, N.; Sakaguchi, Y.M.; Shiota, T.; Kobashigawa, S.; Nakanishi, M.; Matsubayashi, M.; et al. Gene Expression Profiles of Human Cerebral Organoids Identify PPAR Pathway and PKM2 as Key Markers for Oxygen-Glucose Deprivation and Reoxygenation. Front. Cell. Neurosci. 2021, 15, 605030. [Google Scholar] [CrossRef] [PubMed]
- Madencioglu, D.A.; Kruth, K.A.; Wassink, T.H.; Magnotta, V.A.; Wemmie, J.A.; Williams, A.J. Modeling Human Cerebellar Development In Vitro in 2D Structure. J. Vis. Exp. 2022, 187, e64462. [Google Scholar] [CrossRef]
- Jansch, C.; Günther, K.; Waider, J.; Ziegler, G.C.; Forero, A.; Kollert, S.; Svirin, E.; Pühringer, D.; Kwok, C.K.; Ullmann, R.; et al. Generation of a Human Induced Pluripotent Stem Cell (iPSC) Line from a 51-Year-Old Female with Attention-Deficit/Hyperactivity Disorder (ADHD) Carrying a Duplication of SLC2A3. Stem Cell Res. 2018, 28, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Lee, K.M.; Liu, X.; Nefzger, C.M.; Vijayakumar, P.; Hawi, Z.; Pang, K.C.; Parish, C.L.; Polo, J.M.; Bellgrove, M.A. Generation of Four iPSC Lines from Peripheral Blood Mononuclear Cells (PBMCs) of an Attention Deficit Hyperactivity Disorder (ADHD) Individual and a Healthy Sibling in an Australia-Caucasian Family. Stem Cell Res. 2019, 34, 101353. [Google Scholar] [CrossRef] [PubMed]
- Yde Ohki, C.M.; Walter, N.M.; Bender, A.; Rickli, M.; Ruhstaller, S.; Walitza, S.; Grünblatt, E. Growth Rates of Human Induced Pluripotent Stem Cells and Neural Stem Cells from Attention-Deficit Hyperactivity Disorder Patients: A Preliminary Study. J. Neural Transm. 2023, 130, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Krach, F.; Stemick, J.; Boerstler, T.; Weiss, A.; Lingos, I.; Reischl, S.; Meixner, H.; Ploetz, S.; Farrell, M.; Hehr, U.; et al. An Alternative Splicing Modulator Decreases Mutant HTT and Improves the Molecular Fingerprint in Huntington’s Disease Patient Neurons. Nat. Commun. 2022, 13, 6797. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-H.; Smith-Geater, C.; Galaz-Montoya, J.G.; Gu, Y.; Gupte, S.R.; Aviner, R.; Mitchell, P.G.; Hsu, J.; Miramontes, R.; Wang, K.Q.; et al. CryoET Reveals Organelle Phenotypes in Huntington Disease Patient iPSC-Derived and Mouse Primary Neurons. Nat. Commun. 2023, 14, 692. [Google Scholar] [CrossRef] [PubMed]
- The HD iPSC Consortium. HD iPSC Consortium Bioenergetic Deficits in Huntington’s Disease iPSC-Derived Neural Cells and Rescue with Glycolytic Metabolites. Hum. Mol. Genet. 2020, 29, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Beatriz, M.; Rodrigues, R.J.; Vilaça, R.; Egas, C.; Pinheiro, P.S.; Daley, G.Q.; Schlaeger, T.M.; Raimundo, N.; Rego, A.C.; Lopes, C. Extracellular Vesicles Improve GABAergic Transmission in Huntington’s Disease iPSC-Derived Neurons. Theranostics 2023, 13, 3707–3724. [Google Scholar] [CrossRef] [PubMed]
- Lewitzky, M.; Yamanaka, S. Reprogramming Somatic Cells towards Pluripotency by Defined Factors. Curr. Opin. Biotechnol. 2007, 18, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling Familial Alzheimer’s Disease with Induced Pluripotent Stem Cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. A Fresh Look at iPS Cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Yamanaka, S. iPS Cell Technologies: Significance and Applications to CNS Regeneration and Disease. Mol. Brain 2014, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Nori, S.; Okada, Y.; Yasuda, A.; Tsuji, O.; Takahashi, Y.; Kobayashi, Y.; Fujiyoshi, K.; Koike, M.; Uchiyama, Y.; Ikeda, E.; et al. Grafted Human-Induced Pluripotent Stem-Cell-Derived Neurospheres Promote Motor Functional Recovery after Spinal Cord Injury in Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16825–16830. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Kwokdinata, C.; Ramanujam, V.; Chen, J.; de Oliveira, P.N.; Nai, M.H.; Chooi, W.H.; Lim, C.T.; Ng, S.Y.; David, L.; Chew, S.Y. Encapsulation of Human Spinal Cord Progenitor Cells in Hyaluronan-Gelatin Hydrogel for Spinal Cord Injury Treatment. ACS Appl. Mater. Interfaces 2023, 15, 50679–50692. [Google Scholar] [CrossRef] [PubMed]
- Sieber-Blum, M. Epidermal Neural Crest Stem Cells and Their Use in Mouse Models of Spinal Cord Injury. Brain Res. Bull. 2010, 83, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Shirai, K.; Hamada, Y.; Arakawa, N.; Yamane, M.; Takaoka, N.; Aki, R.; Hoffman, R.M.; Amoh, Y. Chronic Spinal Cord Injury Functionally Repaired by Direct Implantation of Encapsulated Hair-Follicle-Associated Pluripotent (HAP) Stem Cells in a Mouse Model: Potential for Clinical Regenerative Medicine. PLoS ONE 2022, 17, e0262755. [Google Scholar] [CrossRef] [PubMed]
- Polentes, J.; Jendelova, P.; Cailleret, M.; Braun, H.; Romanyuk, N.; Tropel, P.; Brenot, M.; Itier, V.; Seminatore, C.; Baldauf, K.; et al. Human Induced Pluripotent Stem Cells Improve Stroke Outcome and Reduce Secondary Degeneration in the Recipient Brain. Cell Transplant. 2012, 21, 2587–2602. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Yamashita, T.; Ohta, Y.; Deguchi, K.; Nagotani, S.; Zhang, X.; Ikeda, Y.; Matsuura, T.; Abe, K. Tridermal Tumorigenesis of Induced Pluripotent Stem Cells Transplanted in Ischemic Brain. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, D.; Hattiangady, B.; Castro, O.W.; Shuai, B.; Kodali, M.; Attaluri, S.; Bates, A.; Dong, Y.; Zhang, S.-C.; Prockop, D.J.; et al. Human Induced Pluripotent Stem Cell-Derived MGE Cell Grafting after Status Epilepticus Attenuates Chronic Epilepsy and Comorbidities via Synaptic Integration. Proc. Natl. Acad. Sci. USA 2019, 116, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.; Cho, J.-H.; Leung, A.; Savvidis, G.; Ahn, S.; Moon, M.; Lee, P.K.J.; Han, J.J.; Azimi, N.; Kim, K.-S.; et al. hPSC-Derived Maturing GABAergic Interneurons Ameliorate Seizures and Abnormal Behavior in Epileptic Mice. Cell Stem Cell 2014, 15, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Wagh, V.; Reis, S.A.; Erdin, S.; Beauchamp, R.L.; Shaikh, G.; Talkowski, M.; Thiele, E.; Sheridan, S.D.; Haggarty, S.J.; et al. TSC Patient-Derived Isogenic Neural Progenitor Cells Reveal Altered Early Neurodevelopmental Phenotypes and Rapamycin-Induced MNK-eIF4E Signaling. Mol. Autism 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yang, M.; Poremsky, E.; Kidd, S.; Schneider, J.S.; Iacovitti, L. Dopaminergic Neurons Derived from Human Induced Pluripotent Stem Cells Survive and Integrate into 6-OHDA-Lesioned Rats. Stem Cells Dev. 2010, 19, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, V.; Frati, G.; Sala, D.; De Cicco, S.; Luciani, M.; Cavazzin, C.; Paulis, M.; Mentzen, W.; Morena, F.; Giannelli, S.; et al. Generation of Human Induced Pluripotent Stem Cell-Derived Bona Fide Neural Stem Cells for Ex Vivo Gene Therapy of Metachromatic Leukodystrophy. Stem Cells Transl. Med. 2017, 6, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Kim, H.S.; Hong, C.P.; Li, E.; Jeon, I.; Park, H.J.; Lee, N.; Pei, Z.; Song, J. Neural Transplants From Human Induced Pluripotent Stem Cells Rescue the Pathology and Behavioral Defects in a Rodent Model of Huntington’s Disease. Front. Neurosci. 2020, 14, 558204. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Ting, H.-C.; Liu, C.-A.; Su, H.-L.; Chiou, T.-W.; Lin, S.-Z.; Harn, H.-J.; Ho, T.-J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced Pluripotent Stem Cells (iPSCs)-Roles in Regenerative Therapies, Disease Modelling and Drug Screening. Cells 2021, 10, 2319. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with iPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the Safety of Induced Pluripotent Stem Cell Lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.R.; Patikas, N.; Foskolou, S.; Field, S.F.; Park, J.-E.; Byrne, M.L.; Bassett, A.R.; Metzakopian, E. Single-Cell Transcriptomics of Parkinson’s Disease Human In Vitro Models Reveals Dopamine Neuron-Specific Stress Responses. Cell Rep. 2020, 33, 108263. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Uchimura, K.; Donnelly, E.L.; Kirita, Y.; Morris, S.A.; Humphreys, B.D. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 2018, 23, 869–881.e8. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.S.; Renner, M.; Gennaro, M.D.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of Induced Pluripotent Stem Cells without Myc from Mouse and Human Fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells without Viral Vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Sugai, K.; Sumida, M.; Shofuda, T.; Yamaguchi, R.; Tamura, T.; Kohzuki, T.; Abe, T.; Shibata, R.; Kamata, Y.; Ito, S.; et al. First-in-Human Clinical Trial of Transplantation of iPSC-Derived NS/PCs in Subacute Complete Spinal Cord Injury: Study Protocol. Regen. Ther. 2021, 18, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Hoveizi, E.; Mohammadi, T.; Moazedi, A.A.; Zamani, N.; Eskandary, A. Transplanted Neural-like Cells Improve Memory and Alzheimer-like Pathology in a Rat Model. Cytotherapy 2018, 20, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Armijo, E.; Edwards, G.; Flores, A.; Vera, J.; Shahnawaz, M.; Moda, F.; Gonzalez, C.; Sanhueza, M.; Soto, C. Induced Pluripotent Stem Cell-Derived Neural Precursors Improve Memory, Synaptic and Pathological Abnormalities in a Mouse Model of Alzheimer’s Disease. Cells 2021, 10, 1802. [Google Scholar] [CrossRef] [PubMed]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-Aβ Drug Screening Platform Using Human iPS Cell-Derived Neurons for the Treatment of Alzheimer’s Disease. PLoS ONE 2011, 6, e25788. [Google Scholar] [CrossRef] [PubMed]
- Brownjohn, P.W.; Smith, J.; Portelius, E.; Serneels, L.; Kvartsberg, H.; De Strooper, B.; Blennow, K.; Zetterberg, H.; Livesey, F.J. Phenotypic Screening Identifies Modulators of Amyloid Precursor Protein Processing in Human Stem Cell Models of Alzheimer’s Disease. Stem Cell Rep. 2017, 8, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Al-Gharaibeh, A.; Culver, R.; Stewart, A.N.; Srinageshwar, B.; Spelde, K.; Frollo, L.; Kolli, N.; Story, D.; Paladugu, L.; Anwar, S.; et al. Induced Pluripotent Stem Cell-Derived Neural Stem Cell Transplantations Reduced Behavioral Deficits and Ameliorated Neuropathological Changes in YAC128 Mouse Model of Huntington’s Disease. Front. Neurosci. 2017, 11, 628. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.K.; Hunter, C.E.; Ye, S.; Pongos, A.L.; Chan, A.W.S. Combination of Stem Cell and Gene Therapy Ameliorates Symptoms in Huntington’s Disease Mice. NPJ Regen. Med. 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Han, A.; Kim, J.Y.; Choi, J.; Bae, H.S.; Cho, G.-B.; Shin, H.; Shin, E.J.; Lee, K.-I.; Kim, S.; et al. SUPT4H1-Edited Stem Cell Therapy Rescues Neuronal Dysfunction in a Mouse Model for Huntington’s Disease. NPJ Regen. Med. 2022, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Doerr, J.; Böckenhoff, A.; Ewald, B.; Ladewig, J.; Eckhardt, M.; Gieselmann, V.; Matzner, U.; Brüstle, O.; Koch, P. Arylsulfatase A Overexpressing Human iPSC-Derived Neural Cells Reduce CNS Sulfatide Storage in a Mouse Model of Metachromatic Leukodystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Alekseenko, Z.; Dias, J.M.; Adler, A.F.; Kozhevnikova, M.; van Lunteren, J.A.; Nolbrant, S.; Jeggari, A.; Vasylovska, S.; Yoshitake, T.; Kehr, J.; et al. Robust Derivation of Transplantable Dopamine Neurons from Human Pluripotent Stem Cells by Timed Retinoic Acid Delivery. Nat. Commun. 2022, 13, 3046. [Google Scholar] [CrossRef] [PubMed]
- Brot, S.; Thamrin, N.P.; Bonnet, M.-L.; Francheteau, M.; Patrigeon, M.; Belnoue, L.; Gaillard, A. Long-Term Evaluation of Intranigral Transplantation of Human iPSC-Derived Dopamine Neurons in a Parkinson’s Disease Mouse Model. Cells 2022, 11, 1596. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guan, Y.; Zhu, H.; Sun, T.; Wang, Y.; Huang, Y.; Ma, C.; Emery, R.; Guan, W.; Wang, C.; et al. Therapeutic Function of iPSCs-Derived Primitive Neuroepithelial Cells in a Rat Model of Parkinson’s Disease. Neurochem. Int. 2022, 155, 105324. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.M.; Marmion, D.J.; Thompson, C.A.; Elliott, N.A.; Federoff, H.; Brundin, P.; Mattis, V.B.; McMahon, C.W.; Kordower, J.H. Optimizing Maturity and Dose of iPSC-Derived Dopamine Progenitor Cell Therapy for Parkinson’s Disease. NPJ Regen. Med. 2022, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Grinand, L.; Takahashi, J. Automated Measurement of Fluorescence Signals Reveals a Significant Increase of the Graft-Derived Neurite Extension in Neonates Compared to Aged Rats. Regen. Ther. 2022, 19, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Nonaka, R.; Oyama, G.; Jo, T.; Kamo, H.; Nuermaimaiti, M.; Akamatsu, W.; Ishikawa, K.-I.; Hattori, N. A Defined Method for Differentiating Human iPSCs into Midbrain Dopaminergic Progenitors That Safely Restore Motor Deficits in Parkinson’s Disease. Front. Neurosci. 2023, 17, 1202027. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhu, H.; Wang, Y.; Sun, T.; Xu, J.; Wang, T.; Guan, W.; Wang, C.; Liu, C.; Ma, C. Miniature-Swine iPSC-Derived GABA Progenitor Cells Function in a Rat Parkinson’s Disease Model. Cell Tissue Res. 2023, 391, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, N.S.; Truong, V.; Malone, D.; Pengo, T.; Patil, N.; Dutton, J.R.; Parr, A.M. Human Induced Pluripotent Stem Cells Integrate, Create Synapses and Extend Long Axons after Spinal Cord Injury. J. Cell. Mol. Med. 2022, 26, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, T.; Nagoshi, N.; Kamata, Y.; Kawai, M.; Ago, K.; Kajikawa, K.; Shibata, R.; Sato, Y.; Imaizumi, K.; Shindo, T.; et al. Modulation by DREADD Reveals the Therapeutic Effect of Human iPSC-Derived Neuronal Activity on Functional Recovery after Spinal Cord Injury. Stem Cell Rep. 2022, 17, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Tashiro, S.; Shibata, S.; Shinozaki, M.; Shindo, T.; Hashimoto, S.; Kawai, M.; Kitagawa, T.; Ago, K.; Matsumoto, M.; et al. Rehabilitative Training Enhances Therapeutic Effect of Human iPSC-Derived Neural Stem/Progenitor Cells Transplantation in Chronic Spinal Cord Injury. Stem Cells Transl. Med. 2023, 12, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gallegos, C.M.; Xue, H.; Li, S.; Kim, D.H.; Zhou, H.; Xia, X.; Liu, Y.; Cao, Q. Transplantation of Human Induced Pluripotent Stem Cell-Derived Neural Progenitor Cells Promotes Forelimb Functional Recovery after Cervical Spinal Cord Injury. Cells 2022, 11, 2765. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Wang, L.; Zhang, Q.; Chen, S.; Zhang, Y.; Xu, H.; Chen, H.; Xu, Y.; He, W.; Zhang, J.; et al. Therapeutic Potential of a Combination of Electroacupuncture and Human iPSC-Derived Small Extracellular Vesicles for Ischemic Stroke. Cells 2022, 11, 820. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.E.; Waters, E.S.; Yang, X.; Fagan, M.M.; Scheulin, K.M.; Sneed, S.E.; Cheek, S.R.; Jeon, J.H.; Shin, S.K.; Kinder, H.A.; et al. Tanshinone IIA-Loaded Nanoparticle and Neural Stem Cell Therapy Enhances Recovery in a Pig Ischemic Stroke Model. Stem Cells Transl. Med. 2022, 11, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, F.; Tu, M.; Wu, S.; He, X.; Liu, H.; Xu, C.; Zhang, K.; Zhu, Y.; Zhou, R.; et al. Quantitative Proteomics Revealed Extensive Microenvironmental Changes after Stem Cell Transplantation in Ischemic Stroke. Front. Med. 2022, 16, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Sakamoto, Y.; Miyagawa, Y.; Nito, C.; Takahashi, S.; Nitahara-Kasahara, Y.; Suda, S.; Yamazaki, Y.; Sakai, M.; Kimura, K.; et al. iPSC-Derived Mesenchymal Stem Cells Attenuate Cerebral Ischemia-Reperfusion Injury by Inhibiting Inflammatory Signaling and Oxidative Stress. Mol. Ther. Methods Clin. Dev. 2023, 30, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.-Y.; Tao, M.-D.; Lou, S.-N.; Yang, D.; Lin, Y.-H.; Wu, H.-Y.; Chang, L.; Luo, C.-X.; Xu, Y.; Liu, Y.; et al. Functional Reconstruction of the Impaired Cortex and Motor Function by hMGEOs Transplantation in Stroke. Biochem. Biophys. Res. Commun. 2023, 671, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Curiel, R.; Jansson, L.; Tsupykov, O.; Avaliani, N.; Aretio-Medina, C.; Hidalgo, I.; Monni, E.; Bengzon, J.; Skibo, G.; Lindvall, O.; et al. Oligodendrocytes in Human Induced Pluripotent Stem Cell-Derived Cortical Grafts Remyelinate Adult Rat and Human Cortical Neurons. Stem Cell Rep. 2023, 18, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Hulme, A.J.; Maksour, S.; St-Clair Glover, M.; Miellet, S.; Dottori, M. Making Neurons, Made Easy: The Use of Neurogenin-2 in Neuronal Differentiation. Stem Cell Rep. 2022, 17, 14–34. [Google Scholar] [CrossRef] [PubMed]
- Barak, M.; Fedorova, V.; Pospisilova, V.; Raska, J.; Vochyanova, S.; Sedmik, J.; Hribkova, H.; Klimova, H.; Vanova, T.; Bohaciakova, D. Human iPSC-Derived Neural Models for Studying Alzheimer’s Disease: From Neural Stem Cells to Cerebral Organoids. Stem Cell Rev. Rep. 2022, 18, 792–820. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Cavagnaro, J.; Deans, R.; Feigal, E.; Horowitz, E.; Keating, A.; Rao, M.; Turner, M.; Wilmut, I.; Yamanaka, S. Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat. Biotechnol. 2014, 32, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Nakamura, M.; Yoshida, K.; Okada, Y.; Tsuji, O.; Nori, S.; Ikeda, E.; Yamanaka, S.; Miura, K. Steps toward safe cell therapy using induced pluripotent stem cells. Circ. Res. 2013, 112, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Morimoto, S. iPSC-Based Disease Modeling and Drug Discovery in Cardinal Neurodegenerative Disorders. Cell Stem Cell 2022, 29, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gascón, S.; Masserdotti, G.; Russo, G.L.; Götz, M. Direct Neuronal Reprogramming: Achievements, Hurdles, and New Roads to Success. Cell Stem Cell 2017, 21, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, J.P.; Göritz, C.; Tatarishvili, J.; Dias, D.O.; Smith, E.M.K.; Lindvall, O.; Kokaia, Z.; Frisén, J. A Latent Neurogenic Program in Astrocytes Regulated by Notch Signaling in the Mouse. Science 2014, 346, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Fukaya, M.; Yang, J.K.; Rothstein, J.D.; Bergles, D.E. NG2+ CNS Glial Progenitors Remain Committed to the Oligodendrocyte Lineage in Postnatal Life and Following Neurodegeneration. Neuron 2010, 68, 668–681. [Google Scholar] [CrossRef] [PubMed]
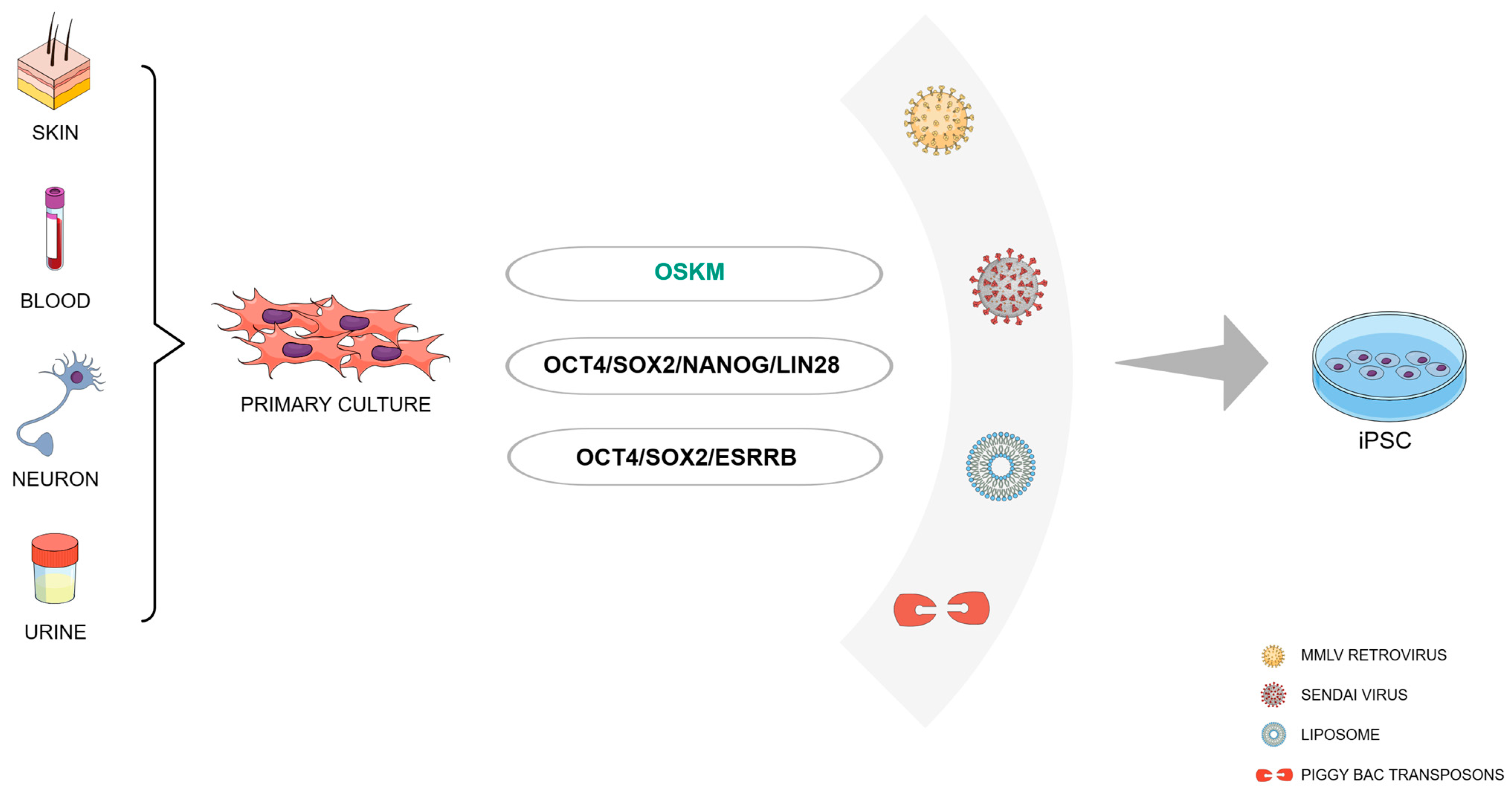

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pazzin, D.B.; Previato, T.T.R.; Budelon Gonçalves, J.I.; Zanirati, G.; Xavier, F.A.C.; da Costa, J.C.; Marinowic, D.R. Induced Pluripotent Stem Cells and Organoids in Advancing Neuropathology Research and Therapies. Cells 2024, 13, 745. https://doi.org/10.3390/cells13090745
Pazzin DB, Previato TTR, Budelon Gonçalves JI, Zanirati G, Xavier FAC, da Costa JC, Marinowic DR. Induced Pluripotent Stem Cells and Organoids in Advancing Neuropathology Research and Therapies. Cells. 2024; 13(9):745. https://doi.org/10.3390/cells13090745
Chicago/Turabian StylePazzin, Douglas Bottega, Thales Thor Ramos Previato, João Ismael Budelon Gonçalves, Gabriele Zanirati, Fernando Antonio Costa Xavier, Jaderson Costa da Costa, and Daniel Rodrigo Marinowic. 2024. "Induced Pluripotent Stem Cells and Organoids in Advancing Neuropathology Research and Therapies" Cells 13, no. 9: 745. https://doi.org/10.3390/cells13090745
APA StylePazzin, D. B., Previato, T. T. R., Budelon Gonçalves, J. I., Zanirati, G., Xavier, F. A. C., da Costa, J. C., & Marinowic, D. R. (2024). Induced Pluripotent Stem Cells and Organoids in Advancing Neuropathology Research and Therapies. Cells, 13(9), 745. https://doi.org/10.3390/cells13090745