
Unveiling Selected Influences on Chronic Kidney Disease Development and Progression

 , , , ,
, , , , 
Abstract
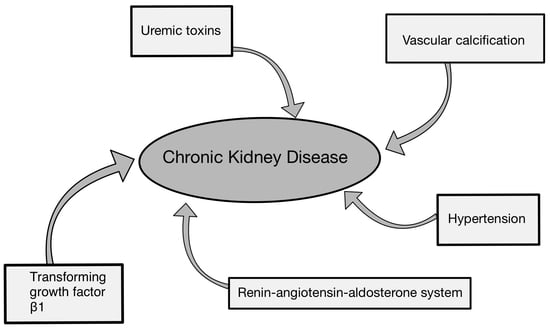
1. Introduction
2. Factors Contributing to the Deterioration of Renal Function and Progression of CKD
2.1. Renin–Angiotensin–Aldosterone System (RAAS) in Chronic Kidney Disease
2.2. Impact of TGF-β on Chronic Kidney Disease
2.3. Vascular Calcification in Chronic Kidney Disease
2.4. Uremic Toxins
2.4.1. Protein-Bound Solutes
2.4.2. Middle Molecules
2.4.3. Free Water-Soluble Low-Molecular Weight Solutes
3. Hypertension and Chronic Kidney Disease
4. Novel Biomarkers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balzer, M.S.; Rohacs, T.; Susztak, K. How Many Cell Types Are in the Kidney and What Do They Do? Annu. Rev. Physiol. 2022, 84, 507–531. [Google Scholar] [CrossRef]
- Ulasi, I.I.; Awobusuyi, O.; Nayak, S.; Ramachandran, R.; Musso, C.G.; Depine, S.A.; Aroca-Martinez, G.; Solarin, A.U.; Onuigbo, M.; Luyckx, V.A.; et al. Chronic Kidney Disease Burden in Low-Resource Settings: Regional Perspectives. Semin. Nephrol. 2022, 42, 151336. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, A.L. Chronic Kidney Disease. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. 1), s03–s09. [Google Scholar] [CrossRef] [PubMed]
- Anger, E.E.; Yu, F.; Li, J. Aristolochic Acid-Induced Nephrotoxicity: Molecular Mechanisms and Potential Protective Approaches. Int. J. Mol. Sci. 2020, 21, 1157. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef] [PubMed]
- Inker, L.A.; Heerspink, H.J.L.; Tighiouart, H.; Levey, A.S.; Coresh, J.; Gansevoort, R.T.; Simon, A.L.; Ying, J.; Beck, G.J.; Wanner, C.; et al. GFR Slope as a Surrogate End Point for Kidney Disease Progression in Clinical Trials: A Meta-Analysis of Treatment Effects of Randomized Controlled Trials. J. Am. Soc. Nephrol. 2019, 30, 1735–1745. [Google Scholar] [CrossRef]
- Rosner, M.H.; Reis, T.; Husain-Syed, F.; Vanholder, R.; Hutchison, C.; Stenvinkel, P.; Blankestijn, P.J.; Cozzolino, M.; Juillard, L.; Kashani, K.; et al. Classification of Uremic Toxins and Their Role in Kidney Failure. Clin. J. Am. Soc. Nephrol. 2021, 16, 1918–1928. [Google Scholar] [CrossRef]
- Lim, Y.J.; Sidor, N.A.; Tonial, N.C.; Che, A.; Urquhart, B.L. Uremic Toxins in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Mechanisms and Therapeutic Targets. Toxins 2021, 13, 142. [Google Scholar] [CrossRef]
- Kim, J.S.; Hwang, H.S. Vascular Calcification in Chronic Kidney Disease: Distinct Features of Pathogenesis and Clinical Implication. Korean Circ. J. 2021, 51, 961–982. [Google Scholar] [CrossRef]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef]
- Chávez-Íñiguez, J.S.; Rifkin, B.S. Dual RAAS Blockade in CKD: Does the Hype have Teeth? Kidney360 2022, 3, 1277–1280. [Google Scholar] [CrossRef]
- Hsu, C.-N.; Tain, Y.-L. Targeting the Renin–Angiotensin–Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.Y.; Cho, H.M.; Kim, M.; Kim, I. Maternal High-Fructose Intake Induces Multigenerational Activation of the Renin-Angiotensin-Aldosterone System. Hypertension 2019, 74, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Dahiya, R.; Singh, Y.; Mishra, A.; Verma, A.; Gothwal, S.K.; Aljabali, A.A.; Dureja, H.; Prasher, P.; Negi, P.; et al. Monotherapy of RAAS blockers and mobilization of aldosterone: A mechanistic perspective study in kidney disease. Chem. Interact. 2020, 317, 108975. [Google Scholar] [CrossRef]
- Schweda, F.; Friis, U.; Wagner, C.; Skott, O.; Kurtz, A.; Rider, S.A.; Mullins, L.J.; Verdon, R.F.; MacRae, C.A.; Mullins, J.J.; et al. Renin Release. Physiology 2007, 22, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. Am. Chin. J. Med. Sci. 2011, 4, 183–190. [Google Scholar] [CrossRef]
- Ma, K.; Gao, W.; Xu, H.; Liang, W.; Ma, G. Role and Mechanism of the Renin-Angiotensin-Aldosterone System in the Onset and Development of Cardiorenal Syndrome. J. Renin-Angiotensin-Aldosterone Syst. 2022, 2022, 3239057. [Google Scholar] [CrossRef] [PubMed]
- Mirabito Colafella, K.M.; Bovée, D.M.; Danser, A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp. Eye Res. 2019, 186, 107680. [Google Scholar] [CrossRef]
- Thomas, M.C. Preventing Progression of Chronic Kidney Disease: Renin–Angiotensin–Aldosterone System Blockade Beyond Blood Pressure. In Management of Chronic Kidney Disease; Arıcı, M., Ed.; Springer: Cham, Switzerland, 2023. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Holappa, M.; Vapaatalo, H.; Vaajanen, A. Local ocular renin–angiotensin–aldosterone system: Any connection with intraocular pressure? A comprehensive review. Ann. Med. 2020, 52, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Alenina, N.; Andrade-Navarro, M.A.; Santos, R.A. MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol. Rev. 2014, 66, 1080–1105. [Google Scholar] [CrossRef] [PubMed]
- Olkowicz, M.; Chlopicki, S.; Smolenski, R.T. Perspectives for angiotensin profiling with liquid chromatography/mass spectrometry to evaluate ACE/ACE2 balance in endothelial dysfunction and vascular pathologies. Pharmacol. Rep. 2015, 67, 778–785. [Google Scholar] [CrossRef]
- Moraes, P.L.; Kangussu, L.M.; da Silva, L.G.; Castro, C.H.; Santos, R.A.; Ferreira, A.J. Cardiovascular effects of small peptides of the renin angiotensin system. Physiol. Rep. 2017, 5, e13505. [Google Scholar] [CrossRef] [PubMed]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the kidney and cardiovascular system. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Leoncini, G.; Viazzi, F.; De Cosmo, S.; Russo, G.; Fioretto, P.; Pontremoli, R. Blood pressure reduction and RAAS inhibition in diabetic kidney disease: Therapeutic potentials and limitations. J. Nephrol. 2020, 33, 949–963. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Veter. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef]
- Ma, T.T.; Meng, X.M. TGF-β/Smad and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 347–364. [Google Scholar] [CrossRef]
- Ruby; Gifford, C.C.; Pandey, R.; Raj, V.S.; Sabbisetti, V.S.; Ajay, A.K. Autophagy as a Therapeutic Target for Chronic Kidney Disease and the Roles of TGF-β1 in Autophagy and Kidney Fibrosis. Cells 2023, 12, 412. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ji, G.; Naeem, H.; Wang, J.; Kantharidis, P.; Powell, D.; Ricardo, S.D. The Use of Targeted Next Generation Sequencing to Explore Candidate Regulators of TGF-β1’s Impact on Kidney Cells. Front. Physiol. 2018, 9, 1755. [Google Scholar] [CrossRef]
- Waasdorp, M.; de Rooij, D.M.; Florquin, S.; Duitman, J.; Spek, C.A. Protease-activated receptor-1 contributes to renal injury and interstitial fibrosis during chronic obstructive nephropathy. J. Cell. Mol. Med. 2019, 23, 1268–1279. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.-K.; Harris, R.C. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.-K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.-C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, N.; Zhuang, S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 2013, 83, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef]
- Bao, J.; Shi, Y.; Tao, M.; Liu, N.; Zhuang, S.; Yuan, W. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clin. Sci. 2018, 132, 2299–2322. [Google Scholar] [CrossRef]
- Tang, C.; Ma, Z.; Zhu, J.; Liu, Z.; Liu, Y.; Liu, Y.; Cai, J.; Dong, Z. P53 in kidney injury and repair: Mechanism and therapeutic potentials. Pharmacol. Ther. 2019, 195, 5–12. [Google Scholar] [CrossRef]
- Ding, Y.; Kim, S.; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef]
- Hu, H.-H.; Chen, D.-Q.; Wang, Y.-N.; Feng, Y.-L.; Cao, G.; Vaziri, N.D.; Zhao, Y.-Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Oxburgh, L.; Chu, G.C.; Michael, S.K.; Robertson, E.J. TGFβ superfamily signals are required for morphogenesis of the kidney mesenchyme progenitor population. Development 2004, 131, 4593–4605. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.-C.; Mao, N.; Yi, S.; Ma, X.; Zou, J.-Q.; Tang, X.; Fan, J.-M. Vascular Calcification in Chronic Kidney Disease: An Update and Perspective. Aging Dis. 2022, 13, 673–697. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Klassen, P.S.; Lazarus, J.M.; Ofsthun, N.; Lowrie, E.G.; Chertow, G.M. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 2004, 15, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Qunibi, W.Y. Reducing the burden of cardiovascular calcification in patients with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16 (Suppl. 2), S95–S102. [Google Scholar] [CrossRef] [PubMed]
- Román-García, P.; Rodríguez-García, M.; Cabezas-Rodríguez, I.; López-Ongil, S.; Díaz-López, B.; Cannata-Andía, J.B. Vascular calcification in patients with chronic kidney disease: types, clinical impact and pathogenesis. Med. Princ. Pract. 2011, 20, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, A.J.; Levine, B.S.; Rodriguez, M. Pathophysiology of Calcium, Phosphorus, and Magnesium Dysregulation in Chronic Kidney Disease. Semin. Dial. 2015, 28, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; van Ballegooijen, A.J. Prevention and treatment of hyperphosphatemia in chronic kidney disease. Kidney Int. 2018, 93, 1060–1072. [Google Scholar] [CrossRef]
- Carrillo-López, N.; Martínez-Arias, L.; Alonso-Montes, C.; Martín-Carro, B.; Martín-Vírgala, J.; Ruiz-Ortega, M.; Fernández-Martín, J.L.; Dusso, A.S.; Rodriguez-García, M.; Naves-Díaz, M.; et al. The receptor activator of nuclear factor κΒ ligand receptor leucine-rich repeat-containing G-protein-coupled receptor 4 contributes to parathyroid hormone-induced vascular calcification. Nephrol. Dial. Transplant. 2021, 36, 618–631. [Google Scholar] [CrossRef]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J.; European Uremic Toxin Work Group. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Devine, E.; Krieter, D.H.; Rüth, M.; Jankovski, J.; Lemke, H.-D. Binding affinity and capacity for the uremic toxin indoxyl sulfate. Toxins 2014, 6, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate—Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Banoglu, E.; King, R.S. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T. Removal of protein-bound uraemic toxins by haemodialysis. Blood Purif. 2013, 35 (Suppl. 2), 20–25. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.; Thomas, M. Angiotensin II and its role in tubular epithelial to mesenchymal transition associated with chronic kidney disease. Cells Tissues Organs 2011, 193, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-beta 1, TIMP-1 and pro-alpha 1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22. [Google Scholar] [PubMed]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-κB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Adelibieke, Y.; Muteliefu, G.; Enomoto, A.; Higashiyama, Y.; Higuchi, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates renal expression of Klotho through production of ROS and activation of nuclear factor-ĸB. Am. J. Nephrol. 2011, 33, 319–324. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S.; Casper, J.; Schmitz, J.; Bräsen, J.H.; Khalifa, A.; Schmidt, B.M.; Einecke, G.; Haller, H.; et al. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.; Caetano-Pinto, P.; Seegers, A.E.; Dankers, A.C.; Broek, P.H.v.D.; Wetzels, J.F.; Brand, J.A.v.D.; Heuvel, L.P.v.D.; Hoenderop, J.G.; Wilmer, M.J.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. Vitr. 2015, 29, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270, Erratum in J. Am. Soc. Nephrol. 2013, 24, 2127–2129. [Google Scholar] [CrossRef]
- Mehaffey, E.; Majid, D.S.A.; Taylor, E.B.; George, E.M.; Ryan, M.J.; Garrett, M.R.; Sasser, J.M.; Duncan, J.W.; Younes, S.T.; Hildebrandt, E.; et al. Tumor necrosis factor-α, kidney function, and hypertension. Am. J. Physiol.-Ren. Physiol. 2017, 313, F1005–F1008. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S3. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Francis, J.; Matrougui, K.; Majid, D.S.A.; Mehaffey, E.; Graham, L.A.; Dominiczak, A.F.; Ferreri, N.R.; Whiting, C.; Castillo, A.; et al. Involvement of tumor necrosis factor-α in natriuretic response to systemic infusion of nitric oxide synthase inhibitor in anesthetized mice. Am. J. Physiol.-Ren. Physiol. 2010, 299, F217–F224. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Bahrami, L.; Castillo, A.; Majid, D.S.A.; Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; et al. TNF-α type 2 receptor mediates renal inflammatory response to chronic angiotensin II administration with high salt intake in mice. Am. J. Physiol.-Ren. Physiol. 2013, 304, F991–F999. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; Zhao, X. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef]
- Ufnal, M.; Zadlo, A.; Ostaszewski, R. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 2015, 6, e02481. [Google Scholar] [CrossRef] [PubMed]
- Cardoza, P.A.; Spillane, M.B.; Marroquin, E.M. Alzheimer’s disease and gut microbiota: Does trimethylamine N-oxide (TMAO) play a role? Nutr. Rev. 2022, 80, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.A.; Faull, R.; Fornasini, G.; Milne, R.W.; Evans, A.M. Accumulation of trimethylamine and trimethylamine-N-oxide in end-stage renal disease patients undergoing haemodialysis. Nephrol. Dial. Transplant. 2006, 21, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Ahmadmehrabi, S.; Tang, W.H.W. Hemodialysis-induced cardiovascular disease. Semin. Dial. 2018, 31, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef]
- Tang, W.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Caglar, K.; Yilmaz, M.; Sonmez, A.; Cakir, E.; Kaya, A.; Acikel, C.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Bilgi, C.; et al. ADMA, proteinuria, and insulin resistance in non-diabetic stage I chronic kidney disease. Kidney Int. 2006, 70, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Anderstam, B.; Katzarski, K.; Bergström, J. Serum levels of NG, NG-dimethyl-L-arginine, a potential endogenous nitric oxide inhibitor in dialysis patients. J. Am. Soc. Nephrol. 1997, 8, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, I.; Sundararajan, S.; Venkatesan, S.; Paadukaana, S.; Balasubramanyam, M.; Mohan, V.; Manickam, N. Asymmetric dimethylarginine (ADMA) accelerates renal cell fibrosis under high glucose condition through NOX4/ROS/ERK signaling pathway. Sci. Rep. 2020, 10, 16005. [Google Scholar] [CrossRef]
- Ku, E.; Lee, B.J.; Wei, J.; Weir, M.R. Hypertension in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Hamrahian, S.M.; Falkner, B. Hypertension in Chronic Kidney Disease. Adv. Exp. Med. Biol. 2017, 956, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F.H.; Williams, B.; Ritz, E. Essential hypertension. Lancet 2007, 370, 591–603. [Google Scholar] [CrossRef]
- Champaneria, M.K.; Patel, R.S.; Oroszi, T.L. When blood pressure refuses to budge: Exploring the complexity of resistant hypertension. Front. Cardiovasc. Med. 2023, 10, 1211199. [Google Scholar] [CrossRef] [PubMed]
- Flack, J.M.; Buhnerkempe, M.G.; Moore, K.T. Resistant Hypertension: Disease Burden and Emerging Treatment Options. Curr. Hypertens. Rep. 2024, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Georgianos, P.I.; Agarwal, R. Resistant Hypertension in Chronic Kidney Disease (CKD): Prevalence, Treatment Particularities, and Research Agenda. Curr. Hypertens. Rep. 2020, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Weldegiorgis, M.; Woodward, M. The impact of hypertension on chronic kidney disease and end-stage renal disease is greater in men than women: A systematic review and meta-analysis. BMC Nephrol. 2020, 21, 506. [Google Scholar] [CrossRef] [PubMed]
- Georgianos, P.I.; Agarwal, R. Hypertension in chronic kidney disease—Treatment standard 2023. Nephrol. Dial. Transplant. 2023, 38, 2694–2703. [Google Scholar] [CrossRef]
- Converse, R.L.; Jacobsen, T.N.; Toto, R.D.; Jost, C.M.; Cosentino, F.; Fouad-Tarazi, F.; Victor, R.G. Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 1992, 327, 1912–1918. [Google Scholar] [CrossRef]
- Greene, E.L.; Kren, S.; Hostetter, T.H. Role of aldosterone in the remnant kidney model in the rat. J. Clin. Investig. 1996, 98, 1063–1068. [Google Scholar] [CrossRef]
- DiBona, G.F. Physiology in perspective: The Wisdom of the Body. Neural control of the kidney. Am. J. Physiol. Integr. Comp. Physiol. 2005, 289, R633–R641. [Google Scholar] [CrossRef] [PubMed]
- Hesse, I.; Johns, E.J. The role of α-adrenoceptors in the regulation of renal tubular sodium reabsorption and renin secretion in the rabbit. Br. J. Pharmacol. 1985, 84, 715–724. [Google Scholar] [CrossRef]
- Terker, A.S.; Yang, C.-L.; McCormick, J.A.; Meermeier, N.P.; Rogers, S.L.; Grossmann, S.; Trompf, K.; Delpire, E.; Loffing, J.; Ellison, D.H.; et al. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 2014, 64, 178–184. [Google Scholar] [CrossRef]
- Davies, M.R.P.; Gleich, K.; Katerelos, M.; Lee, M.; Mount, P.F.; Power, D.A. The Thiazide-Sensitive Co-Transporter Promotes the Development of Sodium Retention in Mice with Diet-Induced Obesity. Kidney Blood Press. Res. 2015, 40, 509–519. [Google Scholar] [CrossRef]
- Lohmeier, T.E.; Iliescu, R.; Liu, B.; Henegar, J.R.; Maric-Bilkan, C.; Irwin, E.D. Systemic and renal-specific sympathoinhibition in obesity hypertension. Hypertension 2012, 59, 331–338. [Google Scholar] [CrossRef]
- Pitzer, A.L.; Van Beusecum, J.P.; Kleyman, T.R.; Kirabo, A. ENaC in Salt-Sensitive Hypertension: Kidney and Beyond. Curr. Hypertens. Rep. 2020, 22, 69. [Google Scholar] [CrossRef]
- Shibata, S.; Mu, S.; Kawarazaki, H.; Muraoka, K.; Ishizawa, K.-I.; Yoshida, S.; Kawarazaki, W.; Takeuchi, M.; Ayuzawa, N.; Miyoshi, J.; et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor–dependent pathway. J. Clin. Investig. 2011, 121, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, A.; Elijovich, F.; Laffer, C.L.; Ertuglu, L.A.; Sahinoz, M.; Saleem, M.; Krishnan, J.; Dola, T.; Aden, L.A.; Sheng, Q.; et al. DC ENaC-Dependent Inflammasome Activation Contributes to Salt-Sensitive Hypertension. Circ. Res. 2022, 131, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart-Bornstein, M.; Lamounier-Zepter, V.; Schraven, A.; Langenbach, J.; Willenberg, H.S.; Barthel, A.; Hauner, H.; McCann, S.M.; Scherbaum, W.A.; Bornstein, S.R. Human adipocytes secrete mineralocorticoid-releasing factors. Proc. Natl. Acad. Sci. USA 2003, 100, 14211–14216. [Google Scholar] [CrossRef]
- El Meouchy, P.; Wahoud, M.; Allam, S.; Chedid, R.; Karam, W.; Karam, S. Hypertension Related to Obesity: Pathogenesis, Characteristics and Factors for Control. Int. J. Mol. Sci. 2022, 23, 12305. [Google Scholar] [CrossRef]
- Huby, A.-C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemèle, E.L. Adipocyte-Derived Hormone Leptin Is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Ball, D.L.; Egan, B.M.; Campbell, W.B.; Nithipatikom, K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension 2004, 43, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Dhaun, N.; Goddard, J.; Webb, D. The endothelin system and its antagonism in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Fonkoue, I.T.; Quyyumi, A.A.; DaCosta, D.; Park, J. Nocturnal blood pressure is associated with sympathetic nerve activity in patients with chronic kidney disease. Physiol. Rep. 2020, 8, e14602. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.R.; Kirkman, D.L.; Edwards, D.G. The Vascular Endothelium in Chronic Kidney Disease: A Novel Target for Aerobic Exercise. Exerc. Sport Sci. Rev. 2016, 44, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Wang, J.; Zhang, L.; Zhao, M.-H.; Chinese Cohort Study of Chronic Kidney Disease (C-STRIDE); Chinese Cohort Study of Chronic Kidney Disease (C-STRIDE) Collaborators. Nocturnal Systolic Hypertension and Adverse Prognosis in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2021, 16, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Uzu, T.; Ishikawa, K.; Fujii, T.; Nakamura, S.; Inenaga, T.; Kimura, G. Sodium restriction shifts circadian rhythm of blood pressure from nondipper to dipper in essential hypertension. Circulation 1997, 96, 1859–1862. [Google Scholar] [CrossRef] [PubMed]
- Stergiou, G.S.; Nasothimiou, E.G.; Destounis, A.; Poulidakis, E.; Evagelou, I.; Tzamouranis, D. Assessment of the diurnal blood pressure profile and detection of non-dippers based on home or ambulatory monitoring. Am. J. Hypertens. 2012, 25, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Kario, K.; Williams, B. Nocturnal Hypertension and Heart Failure: Mechanisms, Evidence, and New Treatments. Hypertension 2021, 78, 564–577. [Google Scholar] [CrossRef]
- Rysz, J.; Gluba-Brzózka, A.; Franczyk, B.; Jabłonowski, Z.; Ciałkowska-Rysz, A. Novel Biomarkers in the Diagnosis of Chronic Kidney Disease and the Prediction of Its Outcome. Int. J. Mol. Sci. 2017, 18, 1702. [Google Scholar] [CrossRef]
- Breznik, B.; Mitrović, A.; Lah, T.T.; Kos, J. Cystatins in cancer progression: More than just cathepsin inhibitors. Biochimie 2019, 166, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.; Olafsson, I.; Palsdottir, A.; Ulvsbäck, M.; Lundwall; Jensson, O.; Grubb, A. Structure and expression of the human cystatin C gene. Biochem. J. 1990, 268, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.; Alvarez-Fernandez, M.; Nathanson, C.-M. Cystatins. Biochem. Soc. Symp. 2003, 70, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Wasung, M.E.; Chawla, L.S.; Madero, M. Biomarkers of renal function, which and when? Clin. Chim. Acta 2015, 438, 350–357. [Google Scholar] [CrossRef]
- Filler, G.; Bökenkamp, A.; Hofmann, W.; Le Bricon, T.; Martínez-Brú, C.; Grubb, A. Cystatin C as a marker of GFR—History, indications, and future research. Clin. Biochem. 2005, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Galteau, M.-M.; Guyon, M.; Gueguen, R.; Siest, G. Determination of serum cystatin C: biological variation and reference values. Clin. Chem. Lab. Med. 2001, 39, 850–857. [Google Scholar] [CrossRef]
- Lamb, E.J.; Levey, A.S.; Stevens, P.E. The Kidney Disease Improving Global Outcomes (KDIGO) guideline update for chronic kidney disease: Evolution not revolution. Clin. Chem. 2013, 59, 462–465. [Google Scholar] [CrossRef]
- Inker, L.A.; Schmid, C.H.; Tighiouart, H.; Eckfeldt, J.H.; Feldman, H.I.; Greene, T.; Kusek, J.W.; Manzi, J.; Van Lente, F.; Zhang, Y.L.; et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N. Engl. J. Med. 2012, 367, 20–29, Erratum in N. Engl. J. Med. 2012, 367, 681Erratum in N. Engl. J. Med. 2012, 367, 2060. [Google Scholar] [CrossRef]
- Banba, N.; Nakamura, T.; Matsumura, M.; Kuroda, H.; Hattori, Y.; Kasai, K. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 2000, 58, 684–690. [Google Scholar] [CrossRef]
- Chow, F.; Nikolic-Paterson, D.; Ozols, E.; Atkins, R.; Rollin, B.; Tesch, G. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, K.; Matsumoto, S.; Aso, Y.; Inukai, T. Aldosterone blockade attenuates urinary monocyte chemoattractant protein-1 and oxidative stress in patients with type 2 diabetes complicated by diabetic nephropathy. J. Clin. Endocrinol. Metab. 2006, 91, 2214–2217. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; O’Kelly, J.; Lu, D.; Leiter, A.; Sohn, J.; Yin, D.; Karlan, B.; Vadgama, J.; Lyons, K.M.; Koeffler, H.P. Expression of connective tissue growth factor (CTGF/CCN2) in breast cancer cells is associated with increased migration and angiogenesis. Int. J. Oncol. 2011, 38, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Dargere, D.; Vidaud, M.; de Gouville, A.-C.; Huet, S.; Martinez, V.; Gauthier, J.-M.; Ba, N.; Sobesky, R.; Ratziu, V.; et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. J. Hepatol. 1999, 30, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.; Tarnow, L.; Andersen, S.; Hovind, P.; Parving, H.H.; Goldschmeding, R.; Van Nieuwenhoven, F.A. Urinary connective tissue growth factor excretion correlates with clinical markers of renal disease in a large population of type 1 diabetic patients with diabetic nephropathy. Diabetes Care 2006, 29, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 Is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-O, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Ju, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; on behalf of the Chronic Renal Insufficiency Cohort (CRIC) Study Group; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutiérrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef]
- Kuncio, G.S.; Neilson, E.G.; Haverty, T. Mechanisms of tubulointerstitial fibrosis. Kidney Int. 1991, 39, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, G.; Rajappa, M. Predictive markers in chronic kidney disease. Clin. Chim. Acta 2022, 535, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Chen, Y.; Lv, J.; Zhang, H.; Tang, J.; Gunaratnam, L.; Li, X.; Yang, L. Kidney injury molecule-1 expression in IgA nephropathy and its correlation with hypoxia and tubulointerstitial inflammation. Am. J. Physiol.-Ren. Physiol. 2014, 306, F885–F895. [Google Scholar] [CrossRef] [PubMed]
- Nowak, N.; Skupien, J.; Niewczas, M.A.; Yamanouchi, M.; Major, M.; Croall, S.; Smiles, A.; Warram, J.H.; Bonventre, J.V.; Krolewski, A.S. Increased plasma kidney injury molecule-1 suggests early progressive renal decline in non-proteinuric patients with type 1 diabetes. Kidney Int. 2016, 89, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Seibert, F.S.; Sitz, M.; Passfall, J.; Haesner, M.; Laschinski, P.; Buhl, M.; Bauer, F.; Babel, N.; Pagonas, N.; Westhoff, T.H. Prognostic Value of Urinary Calprotectin, NGAL and KIM-1 in Chronic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Nigoskar, S.; Thalquotra, M. Comparison of BTP, NGAL, KIM-1, & ADMA biomarkers in CKD and non-CKD subjects. Int. J. Biochem. Mol. Biol. 2023, 14, 32–39. [Google Scholar]
- Buyadaa, O.; Salim, A.; Morton, J.I.; Jandeleit-Dahm, K.; Magliano, D.J.; Shaw, J.E. Examining the factors contributing to the association between non-albuminuric CKD and a low rate of kidney function decline in diabetes. Ther. Adv. Endocrinol. Metab. 2022, 13, 20420188221083518. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Chronic Albuminuria Category | Albumin–Creatinine Ratio in Urine |
|---|---|
| A1 | <3 mg/mmol <30 mg/g |
| A2 | 3–30 mg/mmol 30–300 mg/g |
| A3 | >30 mg/mmol >300 mg/g |
| GFR Category | GFR (mL/min/1.73 m2) |
|---|---|
| G1 | ≥90 |
| G2 | 60–89 |
| G3a | 45–59 |
| G3b | 30–44 |
| G4 | 15–29 |
| G5 | <15 |
| Molecules | Impact on Chronic Kidney Disease |
|---|---|
| TGF-β | Induceskidney fibrosis |
| p53 | Development of acute kidney injury |
| IS | Induceskidney inflammation and fibrosis, nephrotoxic effect |
| pCS | Induceskidney inflammation and fibrosis |
| TNF-α | Leads to reduction in GFR |
| TNFR2 | Glomerulosis and interstitial renal fibrosis |
| TMAO | Accumulation leads to tubulointerstitial renal fibrosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fularski, P.; Czarnik, W.; Frankenstein, H.; Gąsior, M.; Młynarska, E.; Rysz, J.; Franczyk, B. Unveiling Selected Influences on Chronic Kidney Disease Development and Progression. Cells 2024, 13, 751. https://doi.org/10.3390/cells13090751
Fularski P, Czarnik W, Frankenstein H, Gąsior M, Młynarska E, Rysz J, Franczyk B. Unveiling Selected Influences on Chronic Kidney Disease Development and Progression. Cells. 2024; 13(9):751. https://doi.org/10.3390/cells13090751
Chicago/Turabian StyleFularski, Piotr, Witold Czarnik, Hanna Frankenstein, Magdalena Gąsior, Ewelina Młynarska, Jacek Rysz, and Beata Franczyk. 2024. "Unveiling Selected Influences on Chronic Kidney Disease Development and Progression" Cells 13, no. 9: 751. https://doi.org/10.3390/cells13090751
APA StyleFularski, P., Czarnik, W., Frankenstein, H., Gąsior, M., Młynarska, E., Rysz, J., & Franczyk, B. (2024). Unveiling Selected Influences on Chronic Kidney Disease Development and Progression. Cells, 13(9), 751. https://doi.org/10.3390/cells13090751