
Therapeutic Potential of Calcium Channel Blockers in Neuropsychiatric, Endocrine and Pain Disorders





Abstract

{kind=link}
{kind=link}
{kind=link}
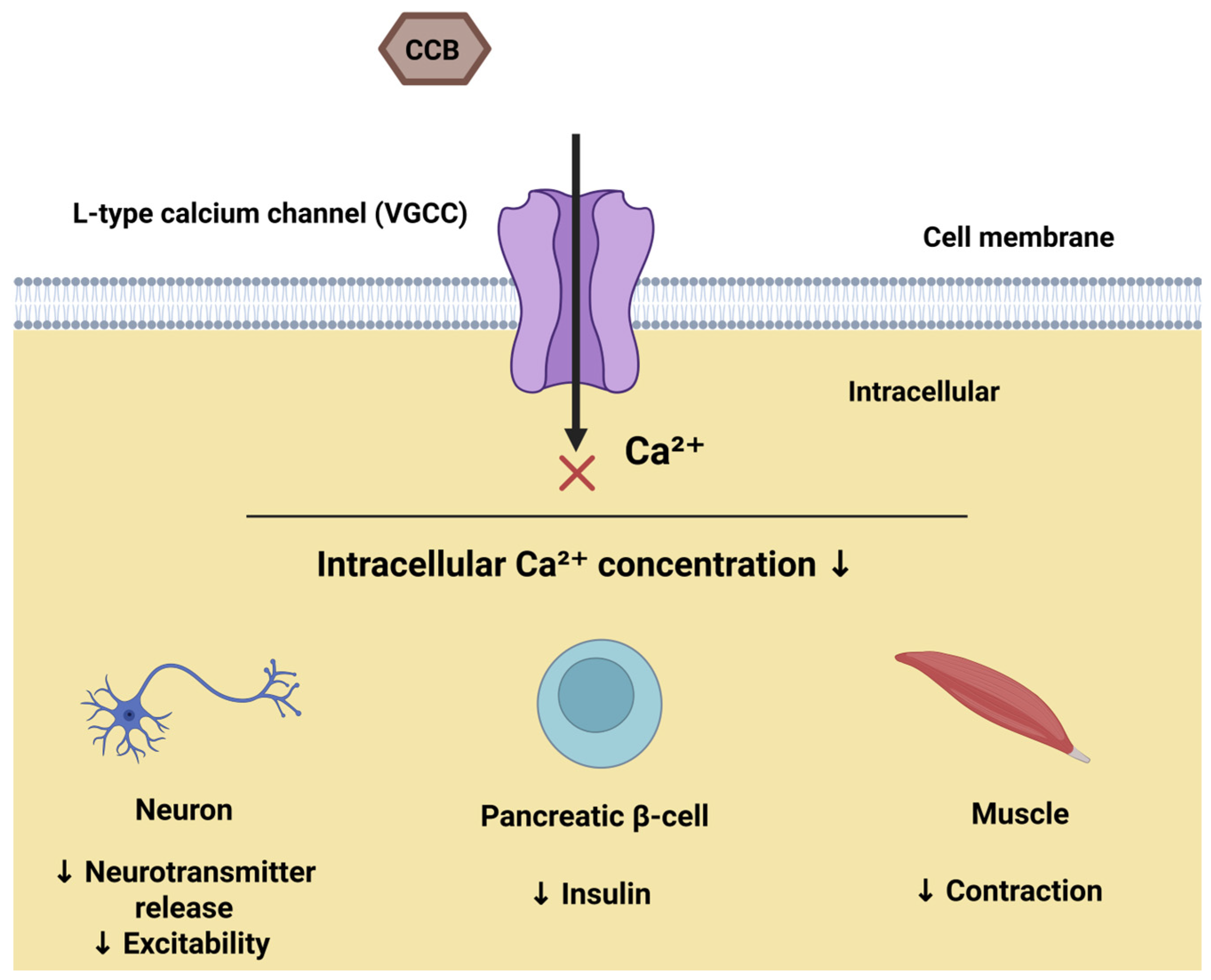
1. Introduction
2. Calcium Channel Blockers in Neuropsychiatric Disorders
2.1. Major Depressive Disorder (MDD)
2.2. Bipolar Disorder and Schizophrenia
2.3. Attention-Deficit Hyperactivity Disorder (ADHD)
2.4. Neurodegenerative Diseases (NDs)
2.5. Epigenetic Considerations in CCB-Mediated Neuropsychiatric Effects
3. Calcium Channel Blockers in Endocrine and Metabolic Disorders
3.1. Diabetes and Insulin Resistance
3.1.1. Effects on Pancreatic β-Cell Function
3.1.2. Clinical Studies on CCBs and Metabolic Outcomes
3.2. Adrenal Disorders
3.2.1. Role in Pheochromocytoma
3.2.2. Role in Primary Aldosteronism
4. CCBs in Pain Disorders
4.1. Calcium Channel Blockers in Neuropathic and Refractory Pain Treatment
4.2. Migraine and Chronic Headache
4.3. Neuropathic, Visceral, and Inflammatory Pain: Therapeutic Potential of Calcium Channel Blockers in Development
4.3.1. N-Type (Cav2.2) Antagonists
4.3.2. T-Type (Cav3.2) Antagonists
4.4. Challenges and Emerging Directions
5. Regulatory and Translational Considerations in CCB Repurposing
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Godfraind, T. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J. Calcium Channel Blockers/Historical Perspectives. In The Evaluation of Beta Blocker and Calcium Antagonist Drugs; Morganroth, J., Moore, E.N., Eds.; Developments in Cardiovascular Medicine; Springer: Dordrecht, The Netherlands, 1982; Volume 18, pp. 289–302. [Google Scholar] [CrossRef]
- McKeever, R.G.; Patel, P.; Hamilton, R.J. Calcium Channel Blockers; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482473/ (accessed on 4 June 2025).
- Elliott, W.J.; Ram, C.V. Calcium Channel Blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.; Lin, L.Y.; Tsai, T.F. Use of Calcium Channel Blockers in Dermatology: A Narrative Review. Expert Rev. Clin. Pharmacol. 2021, 14, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Hollister, L.E.; Trevino, E.S. Calcium Channel Blockers in Psychiatric Disorders: A Review of the Literature. Can. J. Psychiatry 1999, 44, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Nunes, E.J.; Kebede, N.; Rajadhyaksha, A.M.; Addy, N.A. L-Type Calcium Channel Regulation of Depression, Anxiety and Anhedonia-Related Behavioral Phenotypes Following Chronic Stress Exposure. Neuropharmacology 2024, 257, 110031. [Google Scholar] [CrossRef] [PubMed]
- Cativo, E.H.; Lopez, P.D.; Cativo, D.P.; Atlas, S.A.; Rosendorff, C. The Effect of Calcium Channel Blockers on Moderate or Severe Albuminuria in Diabetic, Hypertensive Patients. Am. J. Med. 2021, 134, 104–113.e3. [Google Scholar] [CrossRef] [PubMed]
- Faham, D.E.; Ali, K.; Din, A.S.E.; Bibars, M.; Azmy, O. Can Amlodipine Improve the Pre-Ovulatory Follicle Blood Flow in Women with Polycystic Ovarian Syndrome? J. Reprod. Infertil. 2019, 20, 89–94. [Google Scholar] [PubMed]
- Iimura, O.; Shimamoto, K.; Masuda, A.; Higashiura, K.; Miyazaki, Y.; Hirata, A.; Fukuoka, M.; Murakami, H. Effects of a Calcium Channel Blocker, Manidipine, on Insulin Sensitivity in Essential Hypertensives. J. Diabetes Complicat. 1995, 9, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Effect of Calcium Channel Blockers on Incidence of Diabetes: A Meta-Analysis. Diabetes Metab. Syndr. Obes. 2013, 6, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Muizelaar, J.P.; Kleyer, M.; Hertogs, I.A.; DeLange, D.C. Complex Regional Pain Syndrome (Reflex Sympathetic Dystrophy and Causalgia): Management with the Calcium Channel Blocker Nifedipine and/or the Alpha-Sympathetic Blocker Phenoxybenzamine in 59 Patients. Clin. Neurol. Neurosurg. 1997, 99, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Gelmers, H.J. Calcium-Channel Blockers in the Treatment of Migraine. Am. J. Cardiol. 1985, 55, 139B–143B. [Google Scholar] [CrossRef] [PubMed]
- Tatsushima, Y.; Egashira, N.; Narishige, Y.; Fukui, S.; Kawashiri, T.; Yamauchi, Y.; Oishi, R. Calcium Channel Blockers Reduce Oxaliplatin-Induced Acute Neuropathy: A Retrospective Study of 69 Male Patients Receiving Modified FOLFOX6 Therapy. Biomed. Pharmacother. 2013, 67, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, Y.; Sun, Y.; Alolga, R.N.; Xiao, P.; Ma, G. Antihypertensive Drug Use and the Risk of Depression: A Systematic Review and Network Meta-Analysis. Front. Pharmacol. 2021, 12, 777987. [Google Scholar] [CrossRef] [PubMed]
- Colbourne, L.; Harrison, P.J. Brain-Penetrant Calcium Channel Blockers Are Associated with a Reduced Incidence of Neuropsychiatric Disorders. Mol. Psychiatry 2022, 27, 3904–3912. [Google Scholar] [CrossRef] [PubMed]
- Boal, A.H.; Smith, D.J.; McCallum, L.; Muir, S.; Touyz, R.M.; Dominiczak, A.F.; Padmanabhan, S. Monotherapy with Major Antihypertensive Drug Classes and Risk of Hospital Admissions for Mood Disorders. Hypertension 2016, 68, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz, K.; Sowa-Kućma, M. The Treatment of Depression—Searching for New Ideas. Front. Pharmacol. 2022, 13, 988648. [Google Scholar] [CrossRef] [PubMed]
- Ostacher, M.J.; Iosifescu, D.V.; Hay, A.; Blumenthal, S.R.; Sklar, P.; Perlis, R.H. Pilot Investigation of Isradipine in the Treatment of Bipolar Depression Motivated by Genome-Wide Association. Bipolar Disord. 2014, 16, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Taragano, F.E.; Bagnatti, P.; Allegri, R.F. A Double-Blind, Randomized Clinical Trial to Assess the Augmentation with Nimodipine of Antidepressant Therapy in the Treatment of “Vascular Depression”. Int. Psychogeriatr. 2005, 17, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y.; Lu, Y.; Yin, Y.; Yang, C.; Tang, W.; Song, T.; Tao, X.; Wang, Q. Vascular Depression: A Comprehensive Exploration of the Definition, Mechanisms, and Clinical Challenges. Neurobiol. Dis. 2025, 211, 106946. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, S.L. Applications of Calcium Channel Blockers in Psychiatry: Pharmacokinetic and Pharmacodynamic Aspects of Treatment of Bipolar Disorder. Expert Opin. Drug Metab. Toxicol. 2019, 15, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Lintunen, J.; Lähteenvuo, M.; Tiihonen, J.; Tanskanen, A.; Taipale, H. Adenosine modulators and calcium channel blockers as add-on treatment for schizophrenia. NPJ Schizophr. 2021, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Lintunen, J.; Lähteenvuo, M.; Tanskanen, A.; Tiihonen, J.; Taipale, H. Allopurinol, Dipyridamole and Calcium Channel Blockers in the Treatment of Bipolar Disorder—A Nationwide Cohort Study. J. Affect. Disord. 2022, 313, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yuan, N.; Nurnberger, J.I.; Alliey-Rodriguez, N.; Zhou, J.; Duan, F.; Dai, J.; Chen, Y.; Lu, J.; Xie, L.; et al. A Pilot Pharmacogenetic Study of Calcium Channel Blocker Treatment of Bipolar Mania. Psychiatry Res. 2023, 326, 115281. [Google Scholar] [CrossRef] [PubMed]
- Evaluating Isradipine for Cognitive Enhancement in Schizophrenia and Schizoaffective Disorder. Available online: https://trial.medpath.com/clinical-trial/5eec1264f90cd7d6/evaluating-isradipine-cognitive-enhancement-schizophrenia (accessed on 16 June 2025).
- Þorsteinsson, H.; Baukmann, H.A.; Sveinsdóttir, H.S.; Halldórsdóttir, D.Þ.; Grzymala, B.; Hillman, C.; Rolfe-Tarrant, J.; Parker, M.O.; Cope, J.L.; Ravarani, C.N.J.; et al. Validation of L Type Calcium Channel Blocker Amlodipine as a Novel ADHD Treatment through Cross Species Analysis, Drug Target Mendelian Randomization, and Clinical Evidence from Medical Records. Neuropsychopharmacology 2025, 50, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Holloway, R.; Oakes, D.; Biglan, K.; Lungu, C. A Phase 3 Study of Isradipine as a Disease Modifying Agent in Patients with Early Parkinson’s Disease (STEADY-PD III): Baseline Characteristics and Study Update (P2.039). Neurology 2018, 90 (Suppl. 15), P2-039. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, Y.; Tang, S.; Guo, Y.; Ma, D.; Jiang, X. Mechanism of Nimodipine in Treating Neurodegenerative Diseases: In Silico Target Identification and Molecular Dynamic Simulation. Front. Pharmacol. 2025, 16, 1549953. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Lin, C.L.; Hsu, C.Y.; Wei, C.Y. Flunarizine Induced Parkinsonism in Migraine Group: A Nationwide Population-Based Study. Front. Pharmacol. 2019, 10, 1495. [Google Scholar] [CrossRef] [PubMed]
- Calzetti, S.; Negrotti, A. Permanent Non-Progressive Cinnarizine and Flunarizine-Induced Parkinsonism: An Under-Recognized Tardive Syndrome in the Elderly? J. Neurol. Sci. 2023, 444, 120526. [Google Scholar] [CrossRef] [PubMed]
- Fornarino, S.; Stagnaro, M.; Rinelli, M.; Tiziano, D.; Mancardi, M.M.; Traverso, M.; Veneselli, E.; De Grandis, E. Paroxysmal Features Responding to Flunarizine in a Child with Rapid-Onset Dystonia-Parkinsonism. Neurology 2014, 82, 2037–2038. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Pappalardo, X.G.; Ruggieri, M.; Falsaperla, R.; Parano, E. Alternating Hemiplegia of Childhood: A Distinct Clinical Entity and ATP1A3-Related Disorders: A Narrative Review. Medicine 2022, 101, e29413. [Google Scholar] [CrossRef] [PubMed]
- Serafini, G.; Adavastro, G.; Pardini, M.; Brondino, N.; Amore, M. Early-Life Adversities and Epigenetic Modifications: The Impact on Brain Development, Synaptic Function, and Stress Response. Biol. Psychiatry Glob. Open Sci. 2024, 0, 100560. [Google Scholar] [CrossRef]
- Francis, A.; McKibben, L.A.; Dwivedi, Y. Early-Life Adversity-Induced Epigenetic Reprogramming of Prefrontal Cortex in Rats Subjected to Maternal Separation. Biol. Psychiatry Glob. Open Sci. 2025, 5, 100487. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Wang, T.; Wang, H.; Lian, G.; Xie, L. Causal Relationship between Genetic Proxies for Calcium Channel Blockers and the Risk of Depression: A Drug-Target Mendelian Randomization Study. Front. Psychiatry 2024, 15, 1377705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Gao, H.; Chen, M. Association of Antihypertensive Drugs with COVID-19 Outcomes: A Drug-Target Mendelian Randomization Study. Front. Pharmacol. 2023, 14, 1224737. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Zhao, J.V. Genetic Proxies for Antihypertensive Drugs and Mental Disorders: Mendelian Randomization Study in European and East Asian Populations. BMC Med. 2024, 22, 6. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P. The Pancreatic Beta-Cell as a Fuel Sensor: An Electrophysiologist’s Viewpoint. Diabetologia 1997, 40, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-Gated Ion Channels in Human Pancreatic β-Cells: Electrophysiological Characterization and Role in Insulin Secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Dusonchet, J.; Ashcroft, F. Metabolic Regulation of the Pancreatic Beta-Cell ATP-Sensitive K+ Channel: A Pas de Deux. Diabetes 2004, 53 (Suppl. 3), S113–S122. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C.; Dufrane, D.; Nenquin, M. Nutrient Control of Insulin Secretion in Isolated Normal Human Islets. Diabetes 2006, 55, 3470–3477. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Preventing β-Cell Loss and Diabetes with Calcium Channel Blockers. Diabetes 2012, 61, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, J.; Lu, B.; Sethupathy, P.; Qian, W.-J.; Shalev, A. Verapamil Prevents Decline of IGF-I in Subjects with Type 1 Diabetes and Promotes β-Cell IGF-I Signaling. Diabetes 2023, 72, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sun, P.; Wang, T.; Chen, K.; Zhu, W.; Wang, H. Inhibition of Calcium Influx Reduces Dysfunction and Apoptosis in Lipotoxic Pancreatic β-Cells via Regulation of Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0132411. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Kim, H.E.; Shin, H.C.; Jang, H.J.; Lee, K.W.; Kim, Y.; Kang, S.S.; Chun, J.; Kang, Y. Involvement of Ca2+-Mediated Apoptotic Signals in Palmitate-Induced MIN6N8a Beta Cell Death. Mol. Cell. Endocrinol. 2007, 272, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Gwiazda, K.S.; Yang, T.L.; Lin, Y.; Johnson, J.D. Effects of Palmitate on ER and Cytosolic Ca2+ Homeostasis in Beta-Cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E690–E701. [Google Scholar] [CrossRef] [PubMed]
- Šrámek, J.; Němcová, V.; Kovář, J. Calcium Channel Blockers Do Not Protect against Saturated Fatty Acid-Induced ER Stress and Apoptosis in Human Pancreatic β-Cells. Nutr. Metab. 2021, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- Malayeri, A.; Zakerkish, M.; Ramesh, F.; Galehdari, H.; Hemmati, A.A.; Angali, K.A. The Effect of Verapamil on TXNIP Gene Expression, GLP1R mRNA, FBS, HbA1c, and Lipid Profile in T2DM Patients Receiving Metformin and Sitagliptin. Diabetes Ther. 2021, 12, 2701–2713. [Google Scholar] [CrossRef] [PubMed]
- Ovalle, F.; Grimes, T.; Xu, G.; Patel, A.J.; Grayson, T.B.; Thielen, L.A.; Li, P.; Shalev, A. Verapamil and β-cell Function in Adults with Recent-Onset Type 1 Diabetes. Nat. Med. 2018, 24, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Grimes, T.D.; Grayson, T.B.; Chen, J.; Thielen, L.A.; Tse, H.M.; Li, P.; Kanke, M.; Lin, T.T.; Schepmoes, A.A.; et al. Exploratory Study Reveals Far Reaching Systemic and Cellular Effects of Verapamil Treatment in Subjects with Type 1 Diabetes. Nat. Commun. 2022, 13, 1159. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Kuo, S.-C.; Chang, Y.-Y.; Chen, Y.-T.; Wang, K.-W.K. Verapamil Use Is Associated with Reduction of Newly Diagnosed Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2017, 102, 2604–2610. [Google Scholar] [CrossRef] [PubMed]
- Cooper-DeHoff, R.; Cohen, J.D.; Bakris, G.L.; Messerli, F.H.; Erdine, S.; Hewkin, A.C.; Kupfer, S.; Pepine, C.J.; INVEST Investigators. Predictors of Development of Diabetes Mellitus in Patients with Coronary Artery Disease Taking Antihypertensive Medications (Findings from the INternational VErapamil SR-Trandolapril STudy [INVEST]). Am. J. Cardiol. 2006, 98, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Cooper-DeHoff, R.M.; Aranda, J.M.; Gaxiola, E.; Cangiano, J.L.; Garcia-Barreto, D.; Conti, C.R.; Hewkin, A.; Pepine, C.J. Blood Pressure Control and Cardiovascular Outcomes in High-Risk Hispanic Patients—Findings from the International Verapamil SR/Trandolapril Study (INVEST). Am. Heart J. 2006, 151, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Khodneva, Y.; Shalev, A.; Frank, S.J.; Carson, A.P.; Safford, M.M. Calcium Channel Blocker Use Is Associated with Lower Fasting Serum Glucose among Adults with Diabetes from the REGARDS Study. Diabetes Res. Clin. Pract. 2016, 115, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.H.; Brensinger, C.M.; Bilker, W.B.; Flory, J.H.; Leonard, C.E.; Hennessy, S. Association Between Serious Hypoglycemia and Calcium-Channel Blockers Used Concomitantly with Insulin Secretagogues. JAMA Netw. Open 2021, 4, e2124443. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Huang, K.-C.; Lu, C.-W.; Chu, C.-H.; Huang, C.-N.; Chen, H.-S.; Lee, I.-T.; Chen, J.-F.; Chen, C.-C.; Chen, C.-S.; et al. A Randomized Controlled Trial of R-Form Verapamil Added to Ongoing Metformin Therapy in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2022, 107, e4063–e4071. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, G.P.; McVean, J.; Beck, R.W.; Bauza, C.; Bailey, R.; Buckingham, B.; DiMeglio, L.A.; Sherr, J.L.; Clements, M.; Neyman, A.; et al. Effect of Verapamil on Pancreatic Beta Cell Function in Newly Diagnosed Pediatric Type 1 Diabetes: A Randomized Clinical Trial. JAMA 2023, 329, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Arefanian, H.; Koti, L.; Sindhu, S.; Ahmad, R.; Al Madhoun, A.; Al-Mulla, F. Verapamil Chronicles: Advances from Cardiovascular to Pancreatic β-Cell Protection. Front. Pharmacol. 2023, 14, 1322148. [Google Scholar] [CrossRef] [PubMed]
- Pacak, K. Preoperative Management of the Pheochromocytoma Patient. J. Clin. Endocrinol. Metab. 2007, 92, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Ding, L.; He, Q.; Liu, M. Preoperative Management of Pheochromocytoma and Paraganglioma. Front. Endocrinol. 2020, 11, 586795. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Bravo, E.L. Evolving Concepts in the Pathophysiology, Diagnosis, and Treatment of Pheochromocytoma. Endocr. Rev. 1994, 15, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Ulchaker, J.C.; Goldfarb, D.A.; Bravo, E.L.; Novick, A.C. Successful Outcomes in Pheochromocytoma Surgery in the Modern Era. J. Urol. 1999, 161, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Lebuffe, G.; Dosseh, E.D.; Tek, G.; Tytgat, H.; Moreno, S.; Tavernier, B.; Vallet, B.; Proye, C.A.G. The Effect of Calcium Channel Blockers on Outcome Following the Surgical Treatment of Phaeochromocytomas and Paragangliomas. Anaesthesia 2005, 60, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Yang, H.Y.; Laird, A.M.; Fox, A.C.; Doherty, G.M.; Miller, B.S.; Gauger, P.G. Utility of Oral Nicardipine and Magnesium Sulfate Infusion during Preparation and Resection of Pheochromocytomas. Surgery 2012, 152, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Brunaud, L.; Boutami, M.; Nguyen-Thi, P.L.; Finnerty, B.; Germain, A.; Weryha, G.; Fahey, T.J., 3rd; Mirallie, E.; Bresler, L.; Zarnegar, R. Both Preoperative Alpha and Calcium Channel Blockade Impact Intraoperative Hemodynamic Stability Similarly in the Management of Pheochromocytoma. Surgery 2014, 156, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Nadler, J.L.; Hsueh, W.; Horton, R. Therapeutic Effect of Calcium Channel Blockade in Primary Aldosteronism. J. Clin. Endocrinol. Metab. 1985, 60, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Bravo, E.L.; Fouad, F.M.; Tarazi, R.C. Calcium Channel Blockade with Nifedipine in Primary Aldosteronism. Hypertension 1986, 8 (Suppl. 1), I-191–I-194. [Google Scholar]
- Bursztyn, M.; Grossman, E.; Rosenthal, T. The Absence of Long-Term Therapeutic Effect of Calcium Channel Blockade in the Primary Aldosteronism of Adrenal Adenomas. Am. J. Hypertens. 1988, 1, 88S–90S. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.D.; Du, S.; Bolten, C.W.; Payne, M.A.; Xia, C.; Blinn, J.R.; Funder, J.W.; Hu, X. A Number of Marketed Dihydropyridine Calcium Channel Blockers Have Mineralocorticoid Receptor Antagonist Activity. Hypertension 2008, 51, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Deinum, J.; Riksen, N.P.; Lenders, J.W. Pharmacological Treatment of Aldosterone Excess. Pharmacol. Ther. 2015, 154, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Stölting, G.; Dinh, H.A.; Volkert, M.; Hellmig, N.; Schewe, J.; Hennicke, L.; Seidel, E.; Oberacher, H.; Zhang, J.; Lifton, R.P.; et al. Isradipine Therapy in Cacna1dIle772Met/+ Mice Ameliorates Primary Aldosteronism and Neurologic Abnormalities. JCI Insight 2023, 8, e162468. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.; Penton, D.; Stindl, J.; Humberg, E.; Tegtmeier, I.; Sterner, C.; Beuschlein, F.; Reincke, M.; Barhanin, J.; Bandulik, S.; et al. Pharmacology and Pathophysiology of Mutated KCNJ5 Found in Adrenal Aldosterone-Producing Adenomas. Endocrinology 2014, 155, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, X.; Tong, A.; Zhang, Y.; Wen, J.; Li, Y. The Effects of Different Calcium Channel Blockers on Aldosterone-Producing Adenoma Cells. Front. Endocrinol. 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- De Mingo Alemany, M.C.; Mifsud Grau, L.; Moreno Macián, F.; Ferrer Lorente, B.; León Cariñena, S. A de novo CACNA1D missense mutation in a patient with congenital hyperinsulinism, primary hyperaldosteronism and hypotonia. Channels 2020, 14, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O’Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.S.; Nieto-Rostro, M.; Rahman, W.; Tran-Van-Minh, A.; Ferron, L.; Douglas, L.; Kadurin, I.; Sri Ranjan, Y.; Fernandez-Alacid, L.; Millar, N.S.; et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J. Neurosci. 2009, 29, 4076–4088. [Google Scholar] [CrossRef] [PubMed]
- Freynhagen, R.; Strojek, K.; Griesing, T.; Whalen, E.; Balkenohl, M. Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo-controlled trial of flexible- and fixed-dose regimens. Pain 2005, 115, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.; Javed, S.; Frank, B.; Malik, R.A.; Alam, U. Mirogabalin besylate in the treatment of neuropathic pain. Drugs Today 2020, 56, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Matsui, N.; Kakehi, Y.; Murayama, E.; Ohwada, S.; Sugihara, M. Mirogabalin for the management of postherpetic neuralgia: A randomized, double-blind, placebo-controlled phase 3 study in Asian patients. Pain 2019, 160, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lei, T.; Qin, L.; Li, C.; Lin, X.; Wang, H.; Zhang, G.; Zhang, S.; Shi, K.; Li, L.; et al. Efficacy and Safety of Crisugabalin (HSK16149) in Adults with Postherpetic Neuralgia: A Phase 3 Randomized Clinical Trial. JAMA Dermatol. 2024, 160, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Ma, J.; Li, Y.; Wang, K.; Jiang, C.; Zhang, Y.; Liu, J.; Du, R.; Zhang, W.; Bian, F.; et al. Rapid Onset of Pain Relief with Crisugabalin in Patients with Diabetic Peripheral Neuropathic Pain: Findings from a Multicenter, Randomized, Double-Blind, Controlled Study. Pain Ther. 2025. [Google Scholar] [CrossRef] [PubMed]
- Gou, X.; Liu, Y.; Ye, Q.; He, L.; Chen, Y.; Dong, Y.; Meng, Q.; Shi, Z.; Li, Y.; Lu, Y.; et al. Crisugabalin, a ligand for the α2δ subunit of voltage-gated calcium channels, exhibits no obvious abuse potential in rodents. Pharmacol. Biochem. Behav. 2025, 252, 174015. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, P.; Dou, C.; Shen, J.; Peng, N.; Li, Y.; Wang, J.; Chen, X. Quantification of crisugabalin (HSK16149) in biological matrix by LC-MS/MS method: An application to rat pharmacokinetic and tissue distribution studies. J. Chromatogr. B 2025, 1251, 124396. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chen, S.; Butt, U.D.; Yan, M.; Wu, B. A comprehensive review on ziconotide. Heliyon 2024, 10, e31105. [Google Scholar] [CrossRef] [PubMed]
- Wie, C.S.; Derian, A. Ziconotide; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459151/ (accessed on 29 June 2025).
- Van Zundert, J.; Rauck, R. Intrathecal drug delivery in the management of chronic pain. Best Pract. Res. Clin. Anaesthesiol. 2023, 37, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Gomez, K.; Chen, Y.; Allen, H.N.; Hestehave, S.; Rodríguez-Palma, E.J.; Loya-Lopez, S.; Duran, P.; Nelson, T.S.; Kanumuri, S.R.R.; et al. C2230, a preferential use- and state-dependent CaV2.2 channel blocker, mitigates pain behaviors across multiple pain models. J. Clin. Investig. 2024, 135, e177429. [Google Scholar] [CrossRef] [PubMed]
- Kutzsche, J.; Guzman, G.A.; Willuweit, A.; Kletke, O.; Wollert, E.; Gering, I.; Jürgens, D.; Breitkreutz, J.; Stark, H.; Beck-Sickinger, A.G.; et al. An orally available Cav2.2 calcium channel inhibitor for the treatment of neuropathic pain. Br. J. Pharmacol. 2024, 181, 1734–1756. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kaur, R.; Waziri, A.; Garg, A.; Kadian, R.; Alam, M.S. N-type calcium channel blockers: A new approach towards the treatment of chronic neuropathic pain. Explor. Med. 2023, 4, 85–106. [Google Scholar] [CrossRef]
- Huang, R.; Jiang, L.; Cao, Y.; Liu, H.; Ping, M.; Li, W.; Xu, Y.; Ning, J.; Chen, Y.; Wang, X. Comparative Efficacy of Therapeutics for Chronic Cancer Pain: A Bayesian Network Meta-Analysis. J. Clin. Oncol. 2019, 37, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the trigeminovascular system—40 years and counting. Lancet Neurol. 2019, 18, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y. Migraine Pathophysiology Revisited: Proposal of a New Molecular Theory of Migraine Pathophysiology and Headache Diagnostic Criteria. Int. J. Mol. Sci. 2022, 23, 13002. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Zhong, X.; Zhu, Y.; Li, P.; Zhang, J.; Hou, Y.; Song, L. Ratanasampil is more effective than flunarizine in relieving migraine. Int. J. Neurosci. 2023, 133, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Deligianni, C.I.; Sacco, S.; Ekizoglu, E.; Uluduz, D.; Gil-Gouveia, R.; MaassenVanDenBrink, A.; Ornello, R.; Sanchez-Del-Rio, M.; Reuter, U.; Versijpt, J.; et al. European Headache Federation (EHF) critical re-appraisal and meta-analysis of oral drugs in migraine prevention—Part 2: Flunarizine. J. Headache Pain 2023, 24, 128. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-S.; Choi, J.H. Potential Benefits of Flunarizine in Patients with Sudden Sensorineural Hearing Loss with Incomplete Recovery Following Conventional Steroid Treatment: A Retrospective Analysis. Medicina 2025, 61, 769. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kadian, R. Migraine Prophylaxis; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507873/ (accessed on 29 June 2025).
- Iyengar, S.; Johnson, K.W.; Ossipov, M.H.; Aurora, S.K. CGRP and the Trigeminal System in Migraine. Headache 2019, 59, 659–681. [Google Scholar] [CrossRef] [PubMed]
- Tana, C.; Onan, D.; Messina, R.; Waliszewska-Prosół, M.; Garcia-Azorin, D.; Leal-Vega, L.; Coco-Martin, M.B.; Ornello, R.; Raffaelli, B.; Souza, M.N.P.; et al. From Headache to Heart Health: Investigating the Migraine–Cardiovascular Disease Connection. Neurol. Ther. 2025, 14, 1229–1268. [Google Scholar] [CrossRef] [PubMed]
- Gandini, M.A.; Zamponi, G.W. The N-type calcium channel rises from the ashes. J. Clin. Investig. 2025, 135, e189308. [Google Scholar] [CrossRef] [PubMed]
- Kutzsche, J.; Jürgens, D.; Willuweit, A.; Adermann, K.; Fuchs, C.; Simons, S.; Windisch, M.; Hümpel, M.; Rossberg, W.; Wolzt, M.; et al. Safety and pharmacokinetics of the orally available antiprionic compound PRI-002: A single and multiple ascending dose phase I study. Alzheimers Dement. 2020, 6, e12001. [Google Scholar] [CrossRef] [PubMed]
- Sanner, K. Facilitation of CaV3.2 channel gating in pain pathways reveals a novel mechanism of serum-induced hyperalgesia. bioRxiv 2025. [Google Scholar] [CrossRef]
- Cai, S.; Gomez, K.; Moutal, A.; Khanna, R. Targeting T-type/CaV3.2 channels for chronic pain. Transl. Res. 2021, 234, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Guo, T. Visceral pain from colon and rectum: The mechanotransduction and biomechanics. J. Neural Transm. 2020, 127, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Vergne-Salle, P.; Bertin, P. Chronic pain and neuroinflammation. Jt. Bone Spine 2021, 88, 105222. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Xu, Q.; Shi, Y.; Zhao, R.; Li, H.; Zheng, J.; Liu, F.; Wan, Y.; Wei, B. Pathology of pain and its implications for therapeutic interventions. Signal Transduct. Target. Ther. 2024, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zamponi, G.W. The T-type calcium channelosome. Pflugers Arch. 2024, 476, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Hu, H.; Darido, C.; Vickery, K.; Ranganathan, S. ML218 HCl Is More Efficient Than Capsaicin in Inhibiting Bacterial Antigen-Induced Cal 27 Oral Cancer Cell Proliferation. Int. J. Mol. Sci. 2021, 22, 12559. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Thompson, A.D.; Brogan, J.T.; Schulte, M.L.; Melancon, B.J.; Mi, D.; Lewis, L.M.; Zou, B.; Yang, L.; Morrison, R.; et al. The Discovery and Characterization of ML218: A Novel, Centrally Active T-Type Calcium Channel Inhibitor with Robust Effects in STN Neurons and in a Rodent Model of Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sharma, B.; Ghildiyal, S.; Kharkwal, H. ML218 modulates calcium binding protein, oxidative stress, and inflammation during ischemia-reperfusion brain injury in mice. Eur. J. Pharmacol. 2024, 982, 176919. [Google Scholar] [CrossRef] [PubMed]
- Harding, E.K.; Dedek, A.; Bonin, R.P.; Salter, M.W.; Snutch, T.P.; Hildebrand, M.E. The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. Br. J. Pharmacol. 2021, 178, 3517–3532. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.F.; Scott, V.E.; McGaraughty, S.; Chu, K.L.; Xu, J.; Niforatos, W.; Milicic, I.; Joshi, S.; Zhang, Q.; Xia, Z. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem. Pharmacol. 2014, 89, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Duan, W.R.; An, G.; Thomas, J.W.; Nothaft, W. A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain 2015, 156, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.T.T.; Huang, S.; Chen, L.; Zamponi, G.W. Effect of ABT-639 on Cav3.2 channel activity and its analgesic actions in mouse models of inflammatory and neuropathic pain. Eur. J. Pharmacol. 2024, 967, 176416. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. Z944: A first in class T-type calcium channel modulator for the treatment of pain. J. Peripher. Nerv. Syst. 2014, 19, S11–S12. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Montagut-Bordas, C.; Dickenson, A.H. Calcium channel modulation as a target in chronic pain control. Br. J. Pharmacol. 2018, 175, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.C.; Marliac, M.A.; Geoffroy, C.; Schmit, M.; Bispat, A.; Lewis, R.J.; Tuck, K.L.; Duggan, P.J. The neuronal calcium ion channel activity of constrained analogues of MONIRO-1. Bioorg. Med. Chem. 2020, 28, 115655. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Taketani, Y.; Musayeva, A.; Wang, S.; Yung, A.; Bean, B.; Chen, Y.; Woolf, C.; Dana, R. A Dual Sodium Channel and Calcium Channel inhibitor Ameliorates Ocular Pain in a Murine Dry Eye Disease Model. Invest. Ophthalmol. Vis. Sci. 2023, 64, 3287. [Google Scholar]
- Lee, S.; Jo, S.; Talbot, S.; Zhang, H.B.; Kotoda, M.; Andrews, N.A.; Puopolo, M.; Liu, P.W.; Jacquemont, T.; Pascal, M.; et al. Novel charged sodium and calcium channel inhibitor active against neurogenic inflammation. eLife 2019, 8, e48118. [Google Scholar] [CrossRef] [PubMed]
- Palhares, M.R.; Silva, J.F.; Rezende, M.J.S.; Santos, D.C.; Silva-Junior, C.A.; Borges, M.H.; Ferreira, J.; Gomez, M.V.; Castro-Junior, C.J. Synergistic antinociceptive effect of a calcium channel blocker and a TRPV1 blocker in an acute pain model in mice. Life Sci. 2017, 182, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Li, M.; Wang, W.; Liu, Z.; Guo, Y. The dual role of TRPV1 in peripheral neuropathic pain: Pain switches caused by its sensitization or desensitization. Front. Mol. Neurosci. 2024, 17, 1400118. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Capistrano, R.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.M.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The Repurposing Drugs in Oncology Database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Off-Label Use vs Off-Label Marketing: Part 2: Off-Label Marketing—Consequences for Patients, Clinicians, and Researchers. JACC Basic Transl. Sci. 2023, 8, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Nosengo, N. Can You Teach Old Drugs New Tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, Y.O.; Husain, M.; Patil, K.D.; Sodgir, V.; Patil, T.S.; Agnihotri, V.V.; Mahajan, H.S.; Sharma, C.; Ojha, S.; Goyal, S.N. Verapamil Hydrochloride Loaded Solid Lipid Nanoparticles: Preparation, Optimization, Characterisation, and Assessment of Cardioprotective Effect in Experimental Model of Myocardial Infarcted Rats. Biomed. Pharmacother. 2022, 154, 113429. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Hao, Y.; Tan, X.; Huang, H.; Wang, L.; Zheng, W. Microneedle-Mediated Delivery of Ziconotide-Loaded Liposomes Fused with Exosomes for Analgesia. J. Control. Release 2023, 356, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K.; Green, A.R. Me-Too Pharmaceutical Products: History, Definitions, Examples, and Relevance to Drug Shortages and Essential Medicines Lists. Br. J. Clin. Pharmacol. 2020, 86, 2114–2122. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, Z.; Yang, X.; Shi, S.; Zeng, X.; Cao, D. The Present State and Challenges of Active Learning in Drug Discovery. Drug Discov. Today 2024, 29, 103985. [Google Scholar] [CrossRef] [PubMed]
- Ekhlaspour, L.; Buckingham, B.; Bauza, C.; Clements, M.; Forlenza, G.P.; Neyman, A.; Norlander, L.; Schamberger, M.; Sherr, J.L.; Bailey, R.; et al. Safety and Prescribing Recommendations for Verapamil in Newly Diagnosed Pediatric Type 1 Diabetes (T1D): The CLVer Experience. J. Clin. Transl. Endocrinol. 2024, 36, 100352. [Google Scholar] [CrossRef] [PubMed]
- Saranraj, K.; Kiran, P.U. Drug Repurposing: Clinical Practices and Regulatory Pathways. Perspect. Clin. Res. 2025, 16, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Off-Label Use vs Off-Label Marketing of Drugs: Part 1: Off-Label Use—Patient Harms and Prescriber Responsibilities. JACC Basic Transl. Sci. 2023, 8, 224–233. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzar, A.; Sic, A.; Banh, C.; Knezevic, N.N. Therapeutic Potential of Calcium Channel Blockers in Neuropsychiatric, Endocrine and Pain Disorders. Cells 2025, 14, 1114. https://doi.org/10.3390/cells14141114
Manzar A, Sic A, Banh C, Knezevic NN. Therapeutic Potential of Calcium Channel Blockers in Neuropsychiatric, Endocrine and Pain Disorders. Cells. 2025; 14(14):1114. https://doi.org/10.3390/cells14141114
Chicago/Turabian StyleManzar, Aarish, Aleksandar Sic, Crystal Banh, and Nebojsa Nick Knezevic. 2025. "Therapeutic Potential of Calcium Channel Blockers in Neuropsychiatric, Endocrine and Pain Disorders" Cells 14, no. 14: 1114. https://doi.org/10.3390/cells14141114
APA StyleManzar, A., Sic, A., Banh, C., & Knezevic, N. N. (2025). Therapeutic Potential of Calcium Channel Blockers in Neuropsychiatric, Endocrine and Pain Disorders. Cells, 14(14), 1114. https://doi.org/10.3390/cells14141114