
Role of Thyroid Hormone in Neurodegenerative Disorders of Older People


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of THs in Dementia
3. Role of TH in PD
4. Pathophysiology of TH-Related Neurodegeneration
4.1. Cerebral Vasculature and TH
4.2. Role of TH in Neuronal Cell Viability
4.3. Role of TH in Cellular Stress
4.4. TH Effect on β-Amyloid Precursor Protein
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jurado-Flores, M.; Warda, F.; Mooradian, A.D. Pathophysiology and Clinical Features of Neuropsychiatric Manifestations of Thyroid Disease. J. Endocr. Soc. 2022, 6, bvab194. [Google Scholar] [CrossRef] [PubMed]
- Joffe, R.T.; Sokolov, S.T. Thyroid hormones, the brain, and affective disorders. Crit. Rev. Neurobiol. 1994, 8, 45–63. [Google Scholar] [PubMed]
- Bauer, M.; Goetz, T.; Glenn, T.; Whybrow, P.C. The thyroid-brain interaction in thyroid disorders and mood disorders. J. Neuroendocrinol. 2008, 20, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pei, Y.; Wang, F.; Xu, D.; Cui, W. Higher FT4 or TSH below the normal range are associated with increased risk of dementia: A meta-analysis of 11 studies. Sci. Rep. 2016, 6, 31975. [Google Scholar] [CrossRef]
- Winkler, A.; Weimar, C.; Jöckel, K.-H.; Erbel, R.; Dragano, N.; Broecker-Preuss, M.; Moebus, S.; Führer-Sakel, D.; Dlugaj, M. Thyroid-stimulating hormone and mild cognitive impairment: Results of the Heinz Nixdorf Recall study. J. Alzheimers Dis. 2016, 49, 797–807. [Google Scholar] [CrossRef]
- Correia, N.; Mullally, S.; Cooke, G.; Tun, T.K.; Phelan, N.; Feeney, J.; Fitzgibbon, M.; Boran, G.; O’Mara, S.; Gibney, J. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 2009, 94, 3789–3797. [Google Scholar] [CrossRef] [PubMed]
- Rieben, C.; Segna, D.; da Costa, B.R.; Collet, T.-H.; Chaker, L.; Aubert, C.E.; Baumgartner, C.; Almeida, O.P.; Hogervorst, E.; Trompet, S.; et al. Subclinical thyroid dysfunction and the risk of cognitive decline: A meta-analysis of prospective cohort studies. J. Clin. Endocrinol. Metab. 2016, 101, 4945–4954. [Google Scholar] [CrossRef] [PubMed]
- Forti, P.; Olivelli, V.; Rietti, E.; Maltoni, B.; Pirazzoli, G.; Gatti, R.; Gioia, M.G.; Ravaglia, G. Serum thyroid-stimulating hormone as a predictor of cognitive impairment in an elderly cohort. Gerontology 2011, 58, 41–49. [Google Scholar] [CrossRef]
- Pasqualetti, G.; Pagano, G.; Rengo, G.; Ferrara, N.; Monzani, F. Subclinical hypothyroidism and cognitive impairment: Systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2015, 100, 4240–4248. [Google Scholar] [CrossRef] [PubMed]
- Ojala, A.K.; Schalin-Jäntti, C.; Pitkälä, K.H.; Tilvis, R.S.; Strandberg, T.E. Serum thyroid-stimulating hormone and cognition in older people. Age Ageing 2016, 45, 155–157. [Google Scholar] [CrossRef]
- Gussekloo, J.; van Exel, E.; de Craen, A.J.M.; Meinders, A.E.; Frölich, M.; Westendorp, R.G.J. Thyroid status, disability and cognitive function, and survival in old age. JAMA 2004, 292, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Cappola, A.R.; Fried, L.P.; Arnold, A.M.; Danese, M.D.; Kuller, L.H.; Burke, G.L.; Tracy, R.P.; Ladenson, P.W. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA 2006, 295, 1033–1041. [Google Scholar] [CrossRef]
- Chueire, V.B.; Romaldini, J.H.; Ward, L.S. Subclinical hypothyroidism increases the risk for depression in the elderly. Arch. Gerontol. Geriatr. 2007, 44, 21–28. [Google Scholar] [CrossRef] [PubMed]
- St John, J.A.; Henderson, V.W.; Gatto, N.M.; McCleary, C.A.; Spencer, C.A.; Hodis, H.N.; Mack, W.J. Mildly elevated TSH and cognition in middle-aged and older adults. Thyroid 2009, 19, 111–117. [Google Scholar] [CrossRef]
- Jorde, R.; Waterloo, K.; Storhaug, H.; Nyrnes, A.; Sundsfjord, J.; Jenssen, T.G. Neuropsychological function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. J. Clin. Endocrinol. Metab. 2006, 91, 145–153. [Google Scholar] [CrossRef]
- Roberts, L.M.; Pattison, H.; Roalfe, A.; Franklyn, J.; Wilson, S.; Hobbs, F.R.; Parle, J.V. Is subclinical thyroid dysfunction in the elderly associated with depression or cognitive dysfunction? Ann. Intern. Med. 2006, 145, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.H.; Pearce, S.H.S. Clinical review: The thyroid in mind: Cognitive function and low thyrotropin in older people. J. Clin. Endocrinol. Metab. 2012, 97, 3438–3449. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Park, Y.J.; Kim, T.H.; Han, J.W.; Choi, S.H.; Lim, S.; Park, D.J.; Kim, K.W.; Jang, H.C. Lower-but-normal serum TSH level is associated with the development or progression of cognitive impairment in elderly: Korean Longitudinal Study on Health and Aging (KLoSHA). J. Clin. Endocrinol. Metab. 2014, 99, 424–432. [Google Scholar] [CrossRef]
- Jaracz, J.; Kucharska, A.; Rajewska-Rager, A.; Lacka, K. Cognitive functions and mood during chronic thyrotropin-suppressive therapy with L-thyroxine in patients with differentiated thyroid carcinoma. J. Endocrinol. Investig. 2012, 35, 760–765. [Google Scholar]
- Moon, J.H.; Ahn, S.; Seo, J.; Han, J.W.; Kim, K.M.; Choi, S.H.; Lim, S.; Park, Y.J.; Park, D.J.; Kim, K.W.; et al. The effect of long-term thyroid-stimulating hormone suppressive therapy on the cognitive function of elderly patients with differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, 3782–3789. [Google Scholar] [CrossRef]
- de Oliveira Chachamovitz, D.S.; dos Santos Vigário, P.; e Silva, S.O.; de Mello Teixeira, L.B.; Fagundes, M.L.; Vaisman, M.; Teixeira, P.D.F.D.S. Does low-normal serum TSH level adversely impact cognition in elderly adults and might methimazole therapy improve outcomes? Endocr. J. 2016, 63, 495–505. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, C.; Liu, Y.; Xu, C.; Qin, B.; Liang, H. Genetic correlation between thyroid hormones and Parkinson’s disease. Clin. Exp. Immunol. 2022, 208, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-F.; Yang, Y.-C.; Hsu, C.-Y.; Shen, Y.-C. Risk of Parkinson’s disease in patients with hypothyroidism: A nationwide population-based cohort study. Park. Relat. Disord. 2020, 74, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Charoenngam, N.; Rittiphairoj, T.; Ponvilawan, B.; Prasongdee, K. Thyroid dysfunction and risk of Parkinson’s disease: A systematic review and meta-analysis. Front. Endocrinol. 2022, 13, 863281. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Dolatshahi, M.; Rahmani, F. Shedding light on thyroid hormone disorders and Parkinson disease pathology: Mechanisms and risk factors. J. Endocrinol. Investig. 2021, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhu, L.; Bai, Q.; Wang, L.; Li, Q. Associations between thyroid hormones and cognitive impairment in patients with Parkinson’s disease. eNeuro 2024, 11, ENEURO.0239-24.2024. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Pinna, A.; Costa, G.; Usiello, A.; Pasqualetti, M.; Avallone, L.; Morelli, M.; Napolitano, F. Involvement of the protein Ras homolog enriched in the striatum, Rhes, in dopaminergic neurons’ degeneration: Link to Parkinson’s disease. Int. J. Mol. Sci. 2021, 22, 5326. [Google Scholar] [CrossRef]
- Lee, E.-H.; Kim, S.-M.; Kim, C.-H.; Pagire, S.H.; Pagire, H.S.; Chung, H.Y.; Ahn, J.H.; Park, C.-H. Dopamine neuron induction and the neuroprotective effects of thyroid hormone derivatives. Sci. Rep. 2019, 9, 13659. [Google Scholar] [CrossRef]
- Hochbaum, D.R.; Hulshof, L.; Urke, A.; Wang, W.; Dubinsky, A.C.; Farnsworthe, H.C.; Hakim, R.; Lin, S.; Kleinberg, G.; Robertson, K.; et al. Thyroid hormone remodels cortex to coordinate body-wide metabolism and exploration. Cell 2024, 187, 5679–5697.e23. [Google Scholar] [CrossRef] [PubMed]
- Valcárcel-Hernández, V.; Mayerl, S.; Guadaño-Ferraz, A.; Remaud, S. Thyroid hormone action in adult neurogliogenic niches: The known and unknown. Front. Endocrinol. 2024, 15, 1347802. [Google Scholar] [CrossRef]
- Vatine, G.D.; Al-Ahmad, A.; Barriga, B.K.; Svendsen, S.; Salim, A.; Garcia, L.; Svendsen, C.N. Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood-brain barrier. Cell Stem Cell 2017, 20, 831–843.E5. [Google Scholar] [CrossRef] [PubMed]
- Di Cosmo, C.; Liao, X.H.; Dumitrescu, A.M.; Weiss, R.E.; Refetoff, S. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology 2009, 150, 4450–4458. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Kersseboom, S.; Mayerl, S.; Muller, J.; Groba, C.; Trajkovic-Arsic, M.; Ackermann, T.; Visser, T.J.; Heuer, H. Tetrac can replace thyroid hormone during brain development in mouse mutants deficient in the thyroid hormone transporter mct8. Endocrinology 2013, 154, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Hartley, M.D.; Kirkemo, L.L.; Banerji, T.; Scanlan, T.S. A thyroid hormone—Based strategy for correcting the biochemical abnormality in X-linked adrenoleukodystrophy (X-ALD). Endocrinology 2017, 158, 1328–1338. [Google Scholar] [CrossRef]
- Mooradian, A.D. Blood-brain transport of triiodothyronine is reduced in aged rats. Mech. Ageing Dev. 1990, 52, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Metabolic fuel and amino acid transport into the brain in experimental hypothyroidism. Acta Endocrinol. 1990, 122, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Squizzato, A.; Gerdes, V.E.A.; Brandjes, D.P.M.; Büller, H.R.; Stam, J. Thyroid diseases and cerebrovascular disease. Stroke 2005, 36, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Chaker, L.; Baumgartner, C.; den Elzen, W.P.J.; Ikram, M.A.; Blum, M.R.; Collet, T.-H.; Bakker, S.J.L.; Dehghan, A.; Drechsler, C.; Luben, R.N.; et al. Subclinical hypothyroidism and the risk of stroke events and fatal stroke: An individual participant data analysis. J. Clin. Endocrinol. Metab. 2015, 100, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Brent, G.A. Thyroid hormone and the brain: Mechanisms of action in development and role in protection and promotion of recovery after brain injury. Pharmacol. Ther. 2018, 186, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Donangelo, I.; Abe, K.; Scremin, O.; Ke, S.; Li, F.; Milanesi, A.; Liu, Y.Y.; Brent, G.A. Thyroid hormone treatment activates protective pathways in both in vivo and in vitro models of neuronal injury. Mol. Cell. Endocrinol. 2017, 452, 120–130. [Google Scholar] [CrossRef]
- Crupi, R.; Paterniti, I.; Campolo, M.; Di Paola, R.; Cuzzocrea, S.; Esposito, E. Exogenous T3 administration provides neuroprotection in a murine model of traumatic brain injury. Pharmacol. Res. 2013, 70, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abe, K.; Milanesi, A.; Liu, Y.-Y.; Brent, G.A. Thyroid hormone protects primary cortical neurons exposed to hypoxia by reducing DNA methylation and apoptosis. Endocrinology 2019, 160, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Li, J.; Liu, Y.-Y.; Brent, G.A. Thyroid hormone–mediated histone modification protects cortical neurons from the toxic effects of hypoxic injury. J. Endocr. Soc. 2022, 6, bvac139. [Google Scholar] [CrossRef] [PubMed]
- Sadana, P.; Coughlin, L.; Burke, J.; Woods, R.; Mdzinarishvili, A. Antiedema action of thyroid hormone in MCAO model of ischemic brain stroke: Possible association with AQP4 modulation. J. Neurol. Sci. 2015, 354, 37–45. [Google Scholar] [CrossRef] [PubMed]
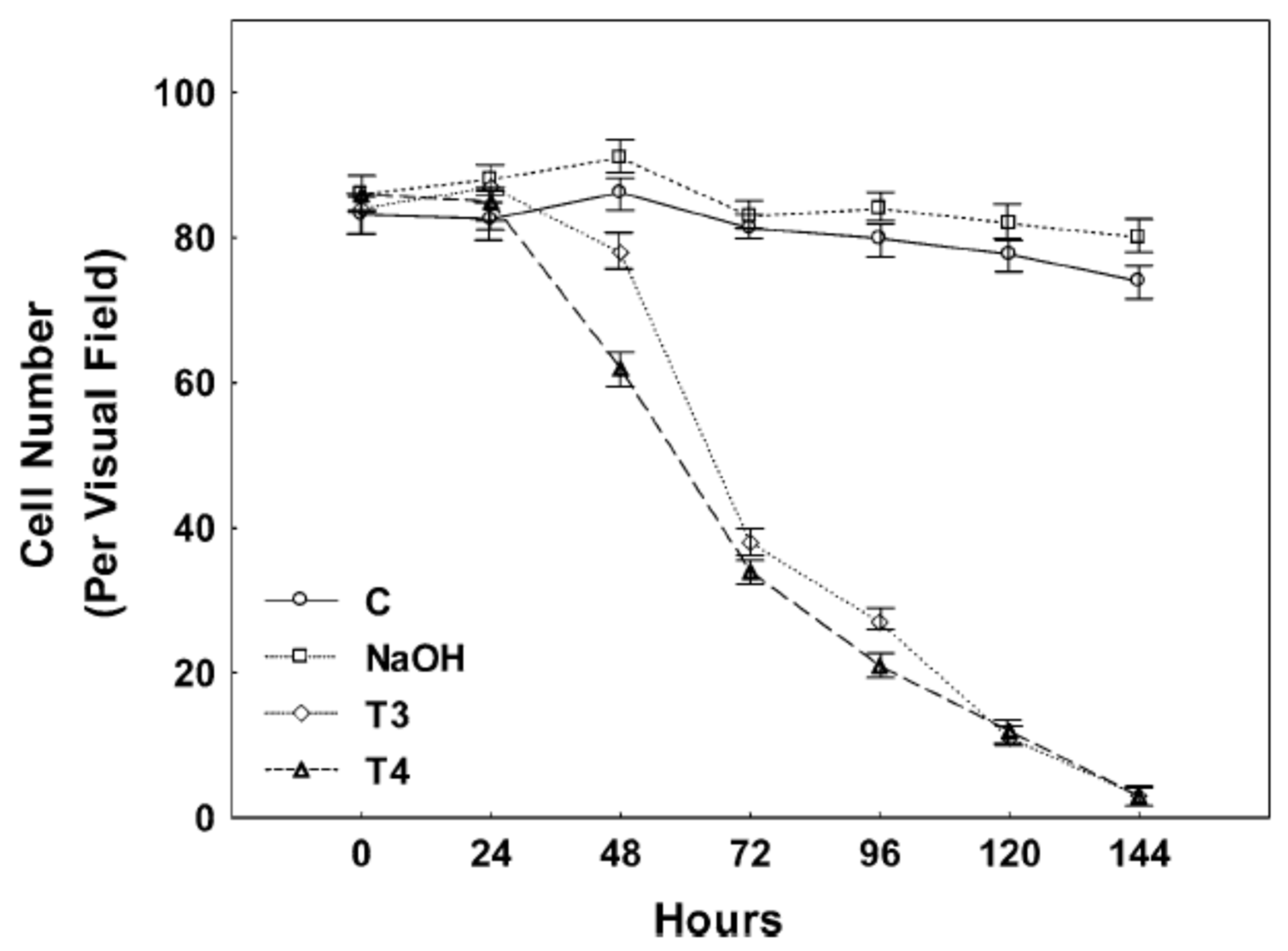
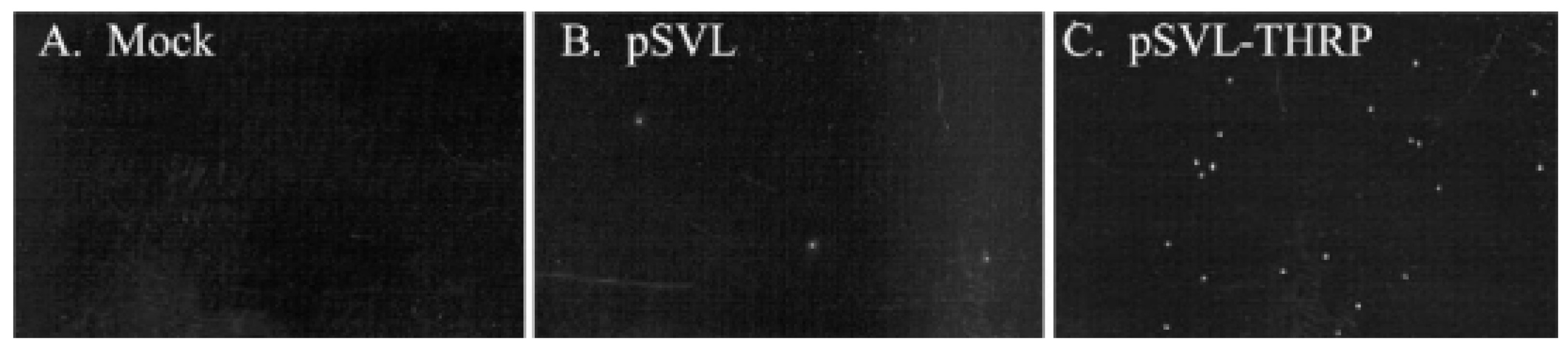
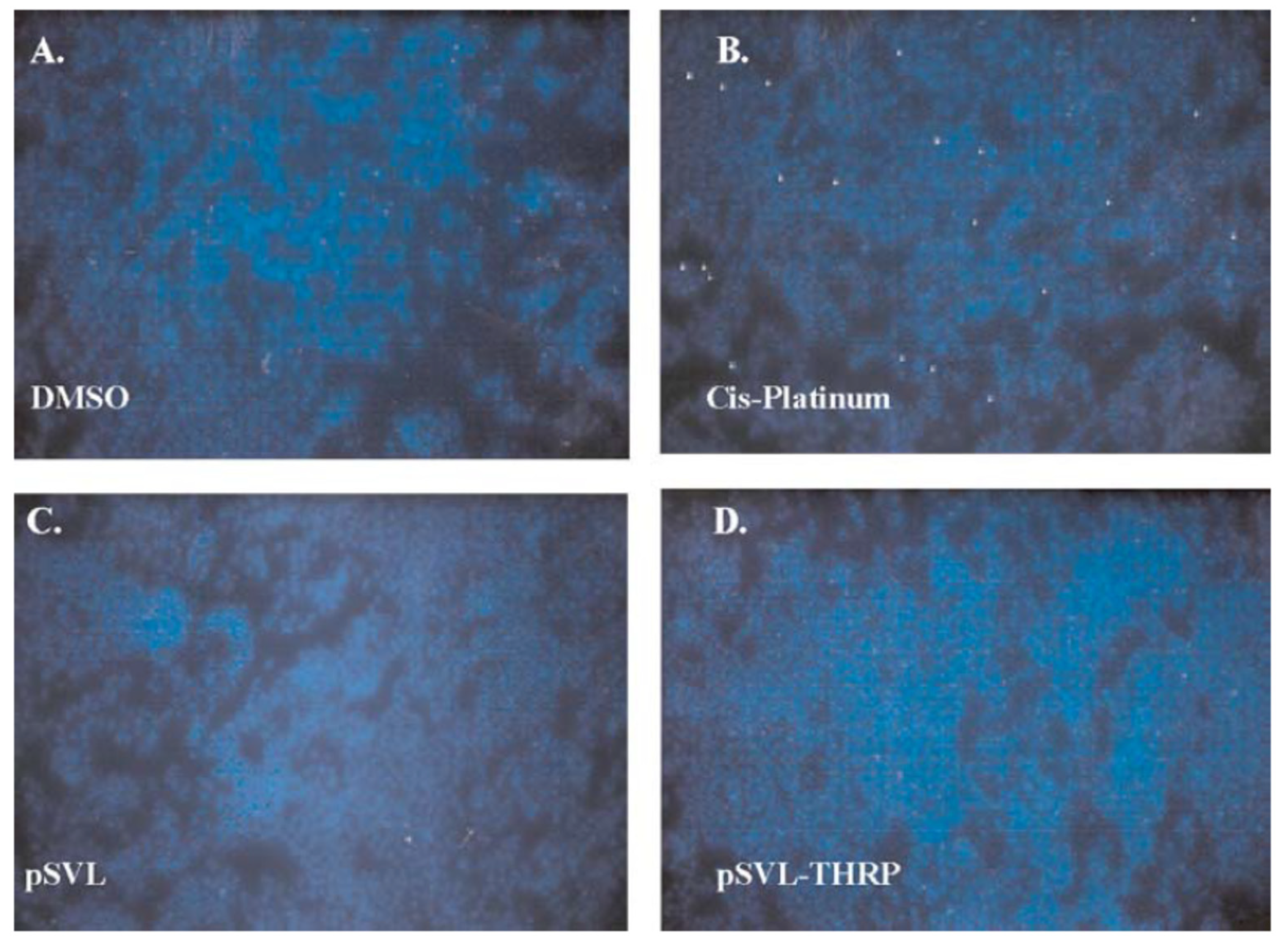
- Haas, M.J.; Fishman, M.; Mreyoud, A.; Mooradian, A.D. Thyroid hormone responsive protein (THRP) mediates thyroid hormone-induced cytotoxicity in primary neuronal cultures. Exp. Brain Res. 2005, 160, 424–432. [Google Scholar] [CrossRef]
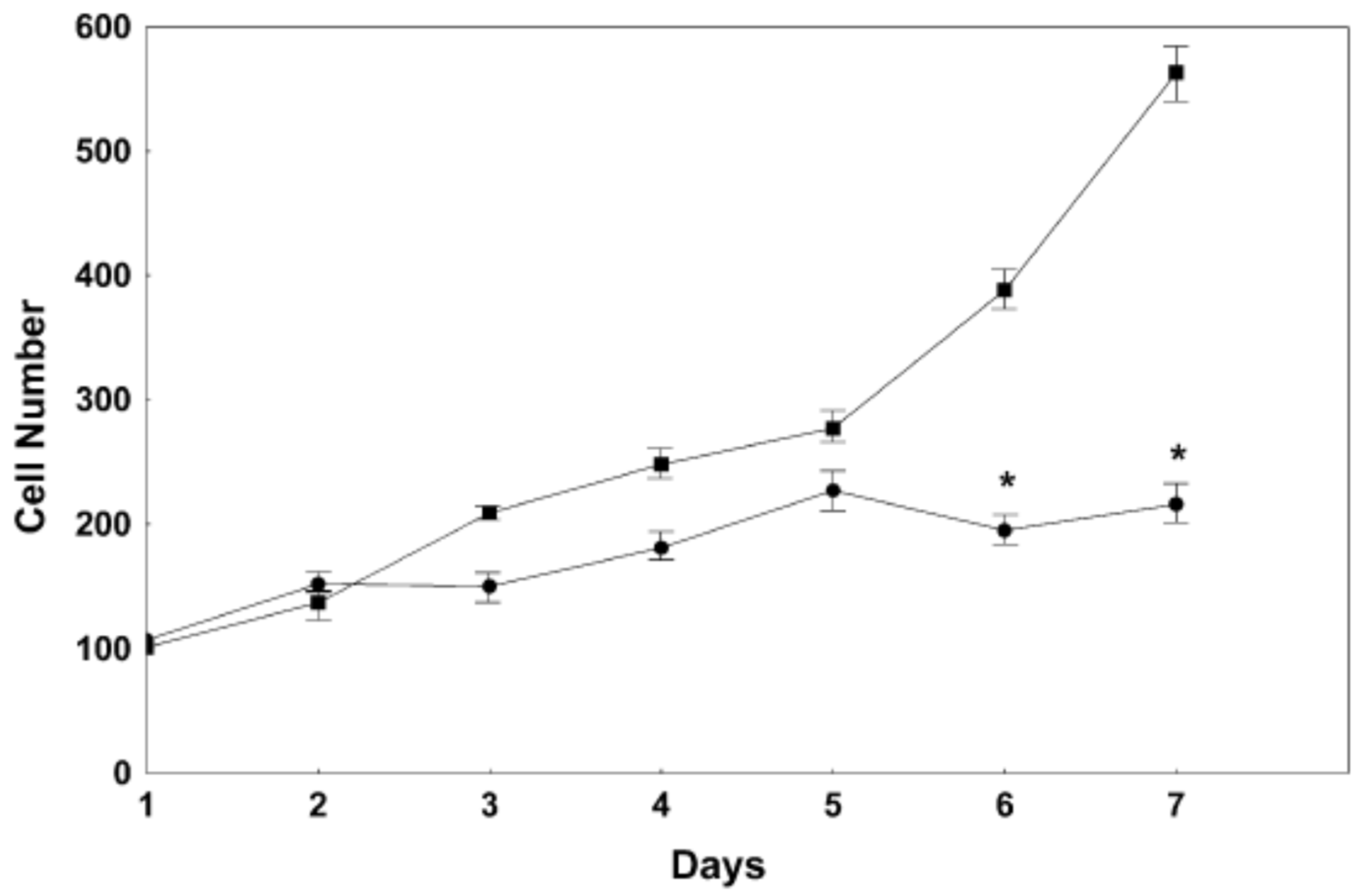
- Haas, M.J.; Parseghian, S.A.; Sajid, R.M.; Mooradian, A.D. Effect of thyroid hormone responsive protein (THRP) expression on PC12 cell survival. Exp. Brain Res. 2003, 150, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.G.; Li, J.; Schneiderjohn, P.; Mooradian, D.A. Cloning and characterization of a complementary DNA for a thyroid hormone-responsive protein in mature rat cerebral tissue. Biochem. J. 1997, 327, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.J.; Li, J.-P.; Pun, K.; Mooradian, A.D. Partial characterization of a cerebral thyroid hormone-responsive protein. Arch. Biochem. Biophys. 2002, 399, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Grove, M.; Vandongen, H.; Vandongen, A.; LaMantia, A.-S.; Penergast, A.M. Localization and phosphorylation of Abl-interactor proteins, Abi-1 and Abi-2, in the developing nervous system. Mol. Cell. Neurosci. 2000, 16, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Grove, M.; Demyanenko, G.; Echarri, A.; Zipfel, P.A.; Quiroz, M.E.; Rodriguiz, R.M.; Playford, M.; Martensen, S.A.; Robinson, M.R.; Wetsel, W.C.; et al. Abi2-deficient mice exhibit defective cell migration, aberrant dendritic spine morphogenesis, and deficits in learning and memory. Mol. Cell. Biol. 2004, 24, 10905–10922. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Haas, M.J.; Mooradian, A.D. The thyroid hormone responsive protein (THRP) has a critical role in the embryogenesis of Xenopus laevis. Neurosci. Lett. 2011, 488, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.N.; Li, J.; Mooradian, A.D. Novel translational repressor (NAT-1) expression is modified by thyroid state and age in brain and liver. Eur. J. Endocrinol. 1998, 139, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Li, J.; Shah, G.N. Age-related changes in thyroid hormone responsive protein (THRP) expression in cerebral tissue of rats. Brain Res. 1998, 793, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Age-related thyroid hormone resistance: A friend or foe. Endocr. Metab. Sci. 2023, 11, 100132. [Google Scholar] [CrossRef]
- Sharlin, D.S.; Visser, T.J.; Forrest, D. Developmental and cell-specific expression of thyroid hormone transporters in the mouse cochlea. Endocrinology 2011, 152, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Visser, W.E.; Friesema, E.C.; Visser, T.J. Minireview: Thyroid hormone transporters: The knowns and the unknowns. Mol. Endocrinol. 2011, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gereben, B.; Zeold, A.; Dentice, M.; Salvatore, D.; Bianco, A.C. Activation and inactivation of thyroid hormone by deiodinases: Local action with general consequences. Cell. Mol. Life Sci. 2008, 65, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeold, A.; Bianco, A.C. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endodrc. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Martinez, M.E.; Liao, X.H.; Van Sande, J.; Refetoff, S.; Galton, V.A.; St Germain, D.L. Type 3 deiodinase deficiency results in functional abnormalities at multiple levels of the thyroid axis. Endocrinology 2007, 148, 5680–5687. [Google Scholar] [CrossRef] [PubMed]
- Desouza, L.A.; Sathanoori, M.; Kapoor, R.; Rajadhyaksha, N.; Gonzalez, L.E.; Kottmann, A.H.; Tole, S.; Vaidya, V.A. Thyroid hormone regulates the expression of the sonic hedgehog signaling pathway in the embryonic and adult mammalian brain. Endocrinology 2011, 152, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; Barrett, T.G.; E Mancilla, E.; Svensson, J.; Kester, M.H.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- Di Cosmo, C.; Liao, X.H.; Dumitrescu, A.M.; Philp, N.J.; Weiss, R.E.; Refetoff, S. Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J. Clin. Investig. 2010, 120, 3377–3388. [Google Scholar] [CrossRef] [PubMed]
- Farwell, A.P. Nonthyroidal illness syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Woolf, P.D.; Lee, L.A.; Hamill, R.W.; McDonald, J.V. Thyroid test abnormalities in traumatic brain injury: Correlation with neurologic impairment and sympathetic nervous system activation. Am. J. Med. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Romine, J.; Gao, X.; Chen, J. Controlled cortical impact model for traumatic brain injury. J. Vis. Exp. 2014, 90, e51781. [Google Scholar] [CrossRef]
- McAllister, R.M.; Albarracin, I.; Price, E.M.; Smith, T.K.; Turk, J.R.; Wyatt, K.D. Thyroid status and nitric oxide in rat arterial vessels. J. Endocrinol. 2005, 185, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kahaly, G.J.; Dillmann, W.H. Thyroid hormone action in the heart. Endocr. Rev. 2005, 26, 704–728. [Google Scholar] [CrossRef]
- Shulga, A.; Blaesse, A.; Kysenius, K.; Huttunen, H.J.; Tanhuanpaa, K.; Saarma, M.; Rivera, C. Thyroxin regulates BDNF expression to promote survival of injured neurons. Mol. Cell. Neurosci. 2009, 42, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Scarpace, P.J. β-Adrenergic receptor activity of cerebral microvessels is reduced in aged rats. Neurochem. Res. 1991, 16, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Scarpace, P.J. 3,5,3′-l-Triiodothyronine regulation of β-adrenergic receptor density and adenylyl cyclase activity in synaptosomal membranes of aged rats. Neurosci. Lett. 1993, 161, 101–104. [Google Scholar] [CrossRef]
- Figueroa, P.B.S.; Ferreira, A.F.F.; Britto, L.R.; Doussoulin, A.P.; Torrão, A.d.S. Association between thyroid function and Alzheimer’s disease: A systematic review. Metab. Brain Dis. 2021, 36, 1523–1543. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Di Segni, C.; Raimondo, S.; Olivieri, G.; Silvestrini, A.; Meucci, E.; Currò, D. Thyroid hormones, oxidative stress, and inflammation. Mediat. Inflamm. 2016, 2016, 6757154. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Habib, M.P.; Dickerson, F.; Yetskievych, T. Effect of age on L-3,5,3’-triiodothyronine-induced ethane exhalation. J. Appl. Physiol. 1994, 77, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Chehade, J.; Kim, J.; Pinnas, J.L.; Mooradian, A.D. Malondialdehyde binding of rat cerebral proteins is reduced in experimental hypothyroidism. Brain Res. 1999, 829, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Raghupathi, R.; Conti, A.C.; Graham, D.I.; Krajewski, S.; Reed, J.C.; Grady, M.S.; Trojanowski, J.; McIntosh, T.K. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in cellular Bcl-2 immunoreactivity. Neuroscience 2002, 110, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W. Cardiac hypertrophy and thyroid hormone signaling. Heart Fail. Rev. 2010, 15, 125–132. [Google Scholar] [CrossRef]
- Latasa, M.J.; Belandia, B.; Pascual, A. Thyroid hormones regulate beta-amyloid gene splicing and protein secretion in neuroblastoma cells. Endocrinology 1998, 139, 2692–2698. [Google Scholar] [CrossRef] [PubMed]
- Dolatshahi, M.; Salehipour, A.; Saghazadeh, A.; Moghaddam, H.S.; Aghamollaii, V.; Fotouhi, A.; Tafakhori, A. Thyroid hormone levels in Alzheimer disease: A systematic review and meta-analysis. Endocrine 2023, 79, 252–272. [Google Scholar] [CrossRef] [PubMed]
- Sawicka-Gutaj, N.; Zawalna, N.; Gut, P.; Ruchała, M. Relationship between thyroid hormones and central nervous system metabolism in physiological and pathological conditions. Pharmacol. Rep. 2022, 74, 847–858. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mooradian, A.D.; Haas, M.J. Role of Thyroid Hormone in Neurodegenerative Disorders of Older People. Cells 2025, 14, 140. https://doi.org/10.3390/cells14020140
Mooradian AD, Haas MJ. Role of Thyroid Hormone in Neurodegenerative Disorders of Older People. Cells. 2025; 14(2):140. https://doi.org/10.3390/cells14020140
Chicago/Turabian StyleMooradian, Arshag D., and Michael J. Haas. 2025. "Role of Thyroid Hormone in Neurodegenerative Disorders of Older People" Cells 14, no. 2: 140. https://doi.org/10.3390/cells14020140
APA StyleMooradian, A. D., & Haas, M. J. (2025). Role of Thyroid Hormone in Neurodegenerative Disorders of Older People. Cells, 14(2), 140. https://doi.org/10.3390/cells14020140