
810-nm Photobiomodulation Evokes Glutamate Release in Normal and Rotenone-Dysfunctional Cortical Nerve Terminals by Modulating Mitochondrial Energy Metabolism

 ,
,  ,
,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Preparation of Synaptosomes
2.4. Technical Characteristics of the Equipment Used and the Irradiation Setup
2.5. Evaluation of OxPhos Activity and Efficiency
2.6. Evaluation of Lipid Peroxidation
2.7. Superfusion Experiments and Endogenous Glutamate Assessment
2.8. Statistical Analysis
3. Results
3.1. PBM Increases OxPhos Activity Without Affecting Its Efficiency
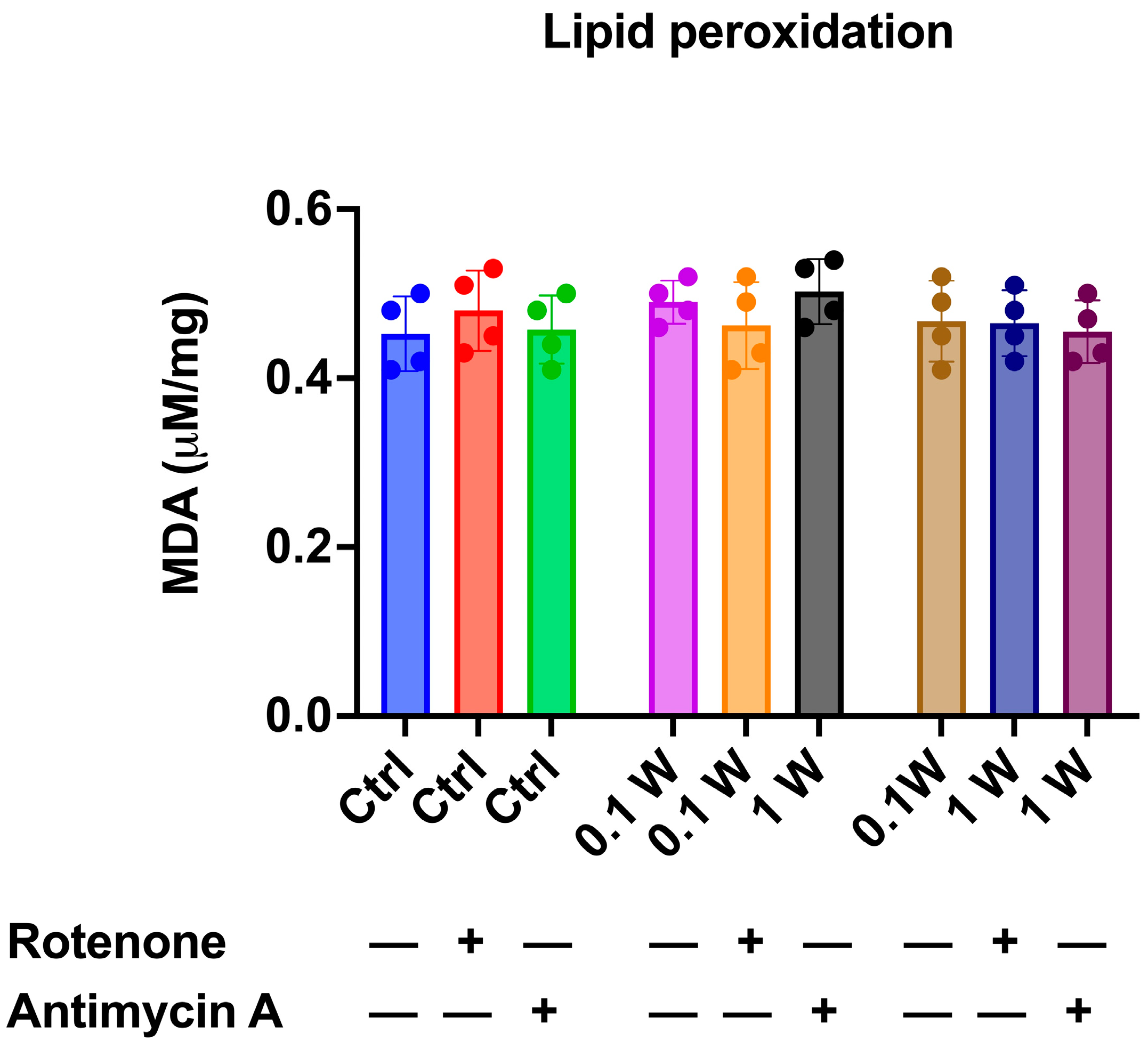
3.2. OxPhos Activity Increment Was Not Associated with Oxidative Damage Accumulation
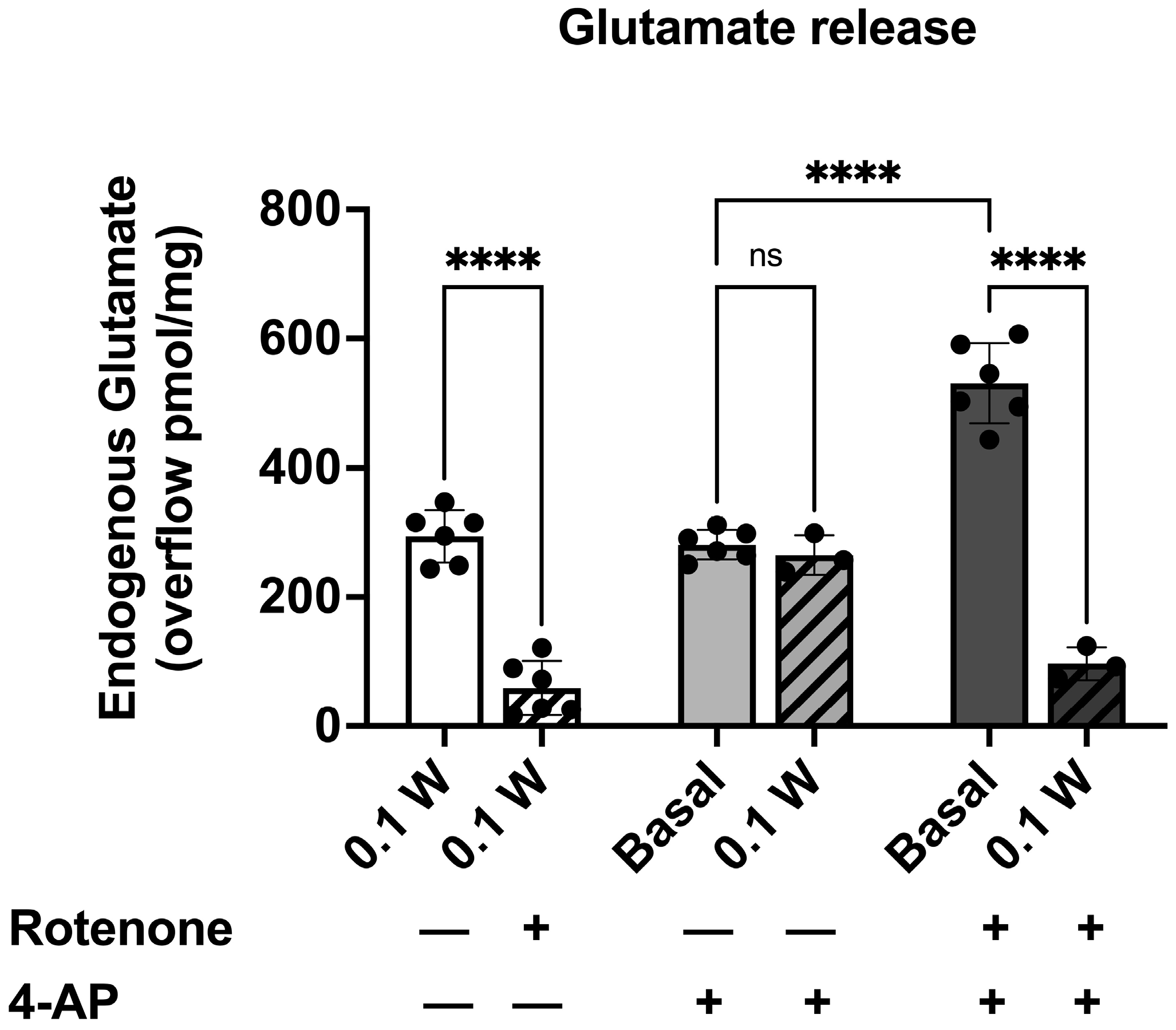
3.3. PBM Induced Endogenous Glutamate Release in Cortical Nerve Terminals and Controlled the Depolarization-Evoked Excessive Efflux in Rotenone-Dependent Mitochondrial Dysfunction
3.3.1. PBM-Induced Endogenous Glutamate Release in Cortical Nerve Terminals in the Presence of Rotenone
3.3.2. PBM Did Not Affect 4-AP-Induced Endogenous Glutamate Release
3.3.3. PBM Affected the 4-AP-Induced Endogenous Glutamate Release in Cortical Nerve Terminals Exposed to Rotenone
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baskerville, R.; Krijgsveld, N.; Esser, P.; Jeffery, G.; Poulton, J. The Effect of Photobiomodulation on the Treatment of Hereditary Mitochondrial Diseases. J. Lasers Med. Sci. 2023, 14, e41. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Ravera, S.; Zekiy, A.; Benedicenti, S.; Pasquale, C. A Narrative Review on Oral and Periodontal Bacteria Microbiota Photobiomodulation, through Visible and Near-Infrared Light: From the Origins to Modern Therapies. Int. J. Mol. Sci. 2022, 23, 1372. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Mechanisms and Mitochondrial Redox Signaling in Photobiomodulation. Photochem. Photobiol. 2018, 94, 199–212. [Google Scholar] [CrossRef]
- Karu, T.I. Special Issue Papers. Photobiological Fundamentals of Low-Power Laser Therapy. IEEE J. Quantum Electron. 1987, 23, 1703–1717. [Google Scholar] [CrossRef]
- Karu, T.I.; Letokhov, V.S.; Lobko, V.V. Biostimulation of HeLa Cells by Low-Intensity Visible Light–IV.-Dichromatic Irradiation. Il Nuovo C. D 1985, 5, 483–496. [Google Scholar] [CrossRef]
- Karu, T.I. Molecular Mechanisms of Therapeutic Effects of Low Intensity Laser Radiation. Laser Life Sci. 1988, 2, 53. [Google Scholar]
- Passarella, S.; Ostuni, A.; Atlante, A.; Quagliariello, E. Increase in the Adp/Atp Exchange in Rat Liver Mitochondria Irradiated in Vitro by Helium-Neon Laser. Biochem. Biophys. Res. Commun. 1988, 156, 978–986. [Google Scholar] [CrossRef]
- Passarella, S.; Casamassima, E.; Molinari, S.; Pastore, D.; Quagliariello, E.; Catalano, I.M.; Cingolani, A. Increase of Proton Electrochemical Potential and ATP Synthesis in Rat Liver Mitochondria Irradiated in Vitro by Helium-Neon Laser. FEBS Lett. 1984, 175, 95–99. [Google Scholar] [CrossRef]
- Amaroli, A.; Ferrando, S.; Benedicenti, S. Photobiomodulation Affects Key Cellular Pathways of All Life-Forms: Considerations on Old and New Laser Light Targets and the Calcium Issue. Photochem. Photobiol. 2019, 95, 455–459. [Google Scholar] [CrossRef]
- Amaroli, A.; Ravera, S.; Parker, S.; Panfoli, I.; Benedicenti, A.; Benedicenti, S. The Protozoan, Paramecium Primaurelia, as a Non-Sentient Model to Test Laser Light Irradiation: The Effects of an 808nm Infrared Laser Diode on Cellular Respiration. Altern. Lab. Anim. 2015, 43, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Ravera, S.; Parker, S.; Panfoli, I.; Benedicenti, A.; Benedicenti, S. An 808-Nm Diode Laser with a Flat-Top Handpiece Positively Photobiomodulates Mitochondria Activities. Photomed. Laser Surg. 2016, 34, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Pasquale, C.; Zekiy, A.; Utyuzh, A.; Benedicenti, S.; Signore, A.; Ravera, S. Photobiomodulation and Oxidative Stress: 980 Nm Diode Laser Light Regulates Mitochondrial Activity and Reactive Oxygen Species Production. Oxid. Med. Cell. Longev. 2021, 2021, 6626286. [Google Scholar] [CrossRef]
- Ravera, S.; Ferrando, S.; Agas, D.; De Angelis, N.; Raffetto, M.; Sabbieti, M.G.; Signore, A.; Benedicenti, S.; Amaroli, A. 1064 Nm Nd:YAG Laser Light Affects Transmembrane Mitochondria Respiratory Chain Complexes. J. Biophotonics 2019, 12, e201900101. [Google Scholar] [CrossRef]
- Ravera, S.; Colombo, E.; Pasquale, C.; Benedicenti, S.; Solimei, L.; Signore, A.; Amaroli, A. Mitochondrial Bioenergetic, Photobiomodulation and Trigeminal Branches Nerve Damage, What’s the Connection? A Review. Int. J. Mol. Sci. 2021, 22, 4347. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Majdi, A.; Pazhuhi, M.; Ghasemi, F.; Khademi, M.; Pashazadeh, F.; Hamblin, M.R.; Cassano, P. Transcranial Photobiomodulation Improves Cognitive Performance in Young Healthy Adults: A Systematic Review and Meta-Analysis. Photobiomodul. Photomed. Laser Surg. 2019, 37, 635–643. [Google Scholar] [CrossRef]
- Cassano, P.; Petrie, S.R.; Mischoulon, D.; Cusin, C.; Katnani, H.; Yeung, A.; De Taboada, L.; Archibald, A.; Bui, E.; Baer, L.; et al. Transcranial Photobiomodulation for the Treatment of Major Depressive Disorder. The ELATED-2 Pilot Trial. Photomed. Laser Surg. 2018, 36, 634–646. [Google Scholar] [CrossRef]
- Eghbaldoost, A.; Mashhadsari, S.P.S.; Ghadirzadeh, E.; Ghoreifi, A.; Allameh, F. Therapeutic Effects of Low-Level Laser on Male Infertility: A Systematic Review. J. Lasers Med. Sci. 2023, 14, e36. [Google Scholar] [CrossRef]
- Abijo, A.; Lee, C.Y.; Huang, C.Y.; Ho, P.C.; Tsai, K.J. The Beneficial Role of Photobiomodulation in Neurodegenerative Diseases. Biomedicines 2023, 11, 1828. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Sadigh-Eteghad, S.; Mahmoudi, J.; Kamari, F.; Cassano, P.; Hamblin, M.R. Light Delivery Approaches for Brain Photobiomodulation. Synth. Lect. Biomed. Eng. 2023, 103–136. [Google Scholar]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial Dysfunction and Molecular Pathways of Disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef]
- Finsterer, J. Mitochondrial Disorders, Cognitive Impairment and Dementia. J. Neurol. Sci. 2009, 283, 143–148. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain Mitochondrial Dysfunction in Aging, Neurodegeneration, and Parkinson’s Disease. Front. Aging Neurosci. 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.M.; Trojanowski, J.Q. Increase of Brain Oxidative Stress in Mild Cognitive Impairment: A Possible Predictor of Alzheimer Disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial Dysfunction in Parkinson’s Disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial Biology and Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Perier, C.; Bové, J.; Dehay, B.; Jackson-Lewis, V.; Rabinovitch, P.S.; Przedborski, S.; Vila, M. Apoptosis-Inducing Factor Deficiency Sensitizes Dopaminergic Neurons to Parkinsonian Neurotoxins. Ann. Neurol. 2010, 68, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Et Biophys. Acta (BBA)–Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef]
- Balázs, R.; Bridges, R.J.; Cotman, C.W. Glutamate and Glutamate Receptors in Neurological Diseases. In Excitatory Amino Acid Transmission in Health and Disease; Oxford University Press: Oxford, UK, 2005; pp. 1–46. [Google Scholar]
- Riedel, G.; Platt, B.; Micheau, J. Glutamate Receptor Function in Learning and Memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [PubMed]
- Koutsilieri, E.; Riederer, P. Excitotoxicity and New Antiglutamatergic Strategies in Parkinson’s Disease and Alzheimer’s Disease. Park. Relat. Disord. 2007, 13 (Suppl. 3), S329–S331. [Google Scholar] [CrossRef] [PubMed]
- de Ceglia, R.; Ledonne, A.; Litvin, D.G.; Lind, B.L.; Carriero, G.; Latagliata, E.C.; Bindocci, E.; Di Castro, M.A.; Savtchouk, I.; Vitali, I.; et al. Specialized Astrocytes Mediate Glutamatergic Gliotransmission in the CNS. Nature 2023, 622, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, S.S.; Buskila, Y. Astrocytic Modulation of Neuronal Signalling. Front. Netw. Physiol. 2023, 3, 1205544. [Google Scholar] [CrossRef]
- Bazzari, A.H.; Parri, H.R. Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective. Brain Sci. 2019, 9, 300. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yang, X.; Zhang, M.; Wang, C.; Chen, L. Glutamate Metabolism in Mitochondria Is Closely Related to Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 557–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Pace, L.; Bedse, G.; Lavecchia, A.M.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and Mitochondria: Two Prominent Players in the Oxidative Stress-Induced Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-Induced Excitotoxicity in Parkinson’s Disease: The Role of Glial Cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.H.T.P.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Siddoway, B.; Hou, H.; Xia, H. Glutamatergic Synapses: Molecular Organisation. eLS 2011. [Google Scholar]
- Langlais, P.J.; Mair, R.G. Protective Effects of the Glutamate Antagonist MK-801 on Pyrithiamine-Induced Lesions and Amino Acid Changes in Rat Brain. J. Neurosci. 1990, 10, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Zott, B.; Konnerth, A. Impairments of Glutamatergic Synaptic Transmission in Alzheimer’s Disease. Semin. Cell Dev. Biol. 2023, 139, 24–34. [Google Scholar] [CrossRef]
- Pagonabarraga, J.; Tinazzi, M.; Caccia, C.; Jost, W.H. The Role of Glutamatergic Neurotransmission in the Motor and Non-Motor Symptoms in Parkinson’s Disease: Clinical Cases and a Review of the Literature. J. Clin. Neurosci. 2021, 90, 178–183. [Google Scholar] [CrossRef]
- Chamkouri, H.; Liu, Q.; Zhang, Y.; Chen, C.; Chen, L. Brain Photobiomodulation Therapy on Neurological and Psychological Diseases. J. Biophotonics 2023, 17, e202300145. [Google Scholar] [CrossRef]
- Hamblin, M.R. Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned? Photonics 2019, 6, 77. [Google Scholar] [CrossRef]
- Salehpour, F.; Rasta, S.H. The Potential of Transcranial Photobiomodulation Therapy for Treatment of Major Depressive Disorder. Rev. Neurosci. 2017, 28, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Spera, V.; Sitnikova, T.; Ward, M.J.; Farzam, P.; Hughes, J.; Gazecki, S.; Bui, E.; Maiello, M.; De Taboada, L.; Hamblin, M.R.; et al. Pilot Study on Dose-Dependent Effects of Transcranial Photobiomodulation on Brain Electrical Oscillations: A Potential Therapeutic Target in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 1481–1498. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Marcoli, M.; Venturini, A.; Passalacqua, M.; Agnati, L.F.; Signore, A.; Raffetto, M.; Maura, G.; Benedicenti, S.; Cervetto, C. Near-Infrared Laser Photons Induce Glutamate Release from Cerebrocortical Nerve Terminals. J. Biophotonics 2018, 11, e201800102. [Google Scholar] [CrossRef]
- Cervetto, C.; Amaroli, A.; Amato, S.; Gatta, E.; Diaspro, A.; Maura, G.; Signore, A.; Benedicenti, S.; Marcoli, M. Photons Induce Vesicular Exocytotic Release of Glutamate in a Power-Dependent Way. Int. J. Mol. Sci. 2023, 24, 10977. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, C.; Vergani, L.; Passalacqua, M.; Ragazzoni, M.; Venturini, A.; Cecconi, F.; Berretta, N.; Mercuri, N.; D’Amelio, M.; Maura, G.; et al. Astrocyte-Dependent Vulnerability to Excitotoxicity in Spermine Oxidase-Overexpressing Mouse. Neuromolecular Med. 2016, 18, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Arany, P.; Pasquale, C.; Benedicenti, S.; Bosco, A.; Ravera, S. Improving Consistency of Photobiomodulation Therapy: A Novel Flat-Top Beam Hand-Piece versus Standard Gaussian Probes on Mitochondrial Activity. Int. J. Mol. Sci. 2021, 22, 7788. [Google Scholar] [CrossRef]
- Hanna, R.; Agas, D.; Benedicenti, S.; Ferrando, S.; Laus, F.; Cuteri, V.; Lacava, G.; Sabbieti, M.G.; Amaroli, A. A Comparative Study between the Effectiveness of 980 Nm Photobiomodulation Delivered by Hand-Piece with Gaussian vs. Flat-Top Profiles on Osteoblasts Maturation. Front. Endocrinol. (Lausanne) 2019, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Clemente Vargas, M.R.; Pasquale, C.; Raffetto, M.; Ravera, S. Photobiomodulation on Isolated Mitochondria at 810 Nm: First Results on the Efficiency of the Energy Conversion Process. Sci. Rep. 2024, 14, 11060. [Google Scholar]
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Characterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, P.C. P/O Ratios of Mitochondrial Oxidative Phosphorylation. Biochim. Et Biophys. Acta (BBA)–Bioenerg. 2005, 1706, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Podestà, M.; Sabatini, F.; Dagnino, M.; Cilloni, D.; Fiorini, S.; Barla, A.; Frassoni, F. Discrete Changes in Glucose Metabolism Define Aging. Sci. Rep. 2019, 9, 10347. [Google Scholar] [CrossRef]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93A Mouse Spinal Cord and Motor Cortex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. J. Neurochem. 2019, 151, 336–350. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Galván, E.; Sitges, M. Characterization of the Participation of Sodium Channels on the Rise in Na+ Induced by 4-Aminopyridine (4-AP) in Synaptosomes. Neurochem. Res. 2004, 29, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Tibbs, G.R.; Barrie, A.P.; Van Mieghem, F.J.E.; McMahon, H.T.; Nicholls, D.G. Repetitive Action Potentials in Isolated Nerve Terminals in the Presence of 4-Aminopyridine: Effects on Cytosolic Free Ca2+ and Glutamate Release. J. Neurochem. 1989, 53, 1693–1699. [Google Scholar] [CrossRef]
- Galvan, M.; Grafe, P.; Bruggencate, G. Ten Convulsant Actions of 4-Aminopyridine on the Guinea-Pig Olfactory Cortex Slice. Brain Res. 1982, 241, 75–86. [Google Scholar] [CrossRef]
- Hamblin, M.R. Shining Light on the Head: Photobiomodulation for Brain Disorders. BBA Clin. 2016, 6, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Nairuz, T.; Sangwoo-Cho; Lee, J.H. Photobiomodulation Therapy on Brain: Pioneering an Innovative Approach to Revolutionize Cognitive Dynamics. Cells 2024, 13, 966. [Google Scholar] [CrossRef]
- Pal, M.M. Glutamate: The Master Neurotransmitter and Its Implications in Chronic Stress and Mood Disorders. Front. Hum. Neurosci. 2021, 15, 722323. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Chen, A.C.-H.; Carroll, J.D.; Hamblin, M.R. Biphasic Dose Response in Low Level Light Therapy. Dose Response 2009, 7, 358–383. [Google Scholar] [CrossRef]
- Amaroli, A.; Ravera, S.; Baldini, F.; Benedicenti, S.; Panfoli, I.; Vergani, L. Photobiomodulation with 808-Nm Diode Laser Light Promotes Wound Healing of Human Endothelial Cells through Increased Reactive Oxygen Species Production Stimulating Mitochondrial Oxidative Phosphorylation. Lasers Med. Sci. 2019, 34, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Sabbieti, M.G.; Marchetti, L.; Zekiy, A.O.; Utyuzh, A.S.; Marchegiani, A.; Laus, F.; Cuteri, V.; Benedicenti, S.; Agas, D. The Effects of 808-Nm near-Infrared Laser Light Irradiation on Actin Cytoskeleton Reorganization in Bone Marrow Mesenchymal Stem Cells. Cell Tissue Res. 2021, 383, 1003–1016. [Google Scholar] [CrossRef]
- Brette, R. Integrative Neuroscience of Paramecium, a “Swimming Neuron”. Eneuro 2021, 8. [Google Scholar] [CrossRef]
- Nakaoka, Y.; Tanaka, H.; Oosawa, F. Ca2+-Dependent Regulation of Beat Frequency of Cilia in Paramecium. J. Cell Sci. 1984, 65, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Ravera, S.; Parker, S.; Panfoli, I.; Benedicenti, A.; Benedicenti, S. 808-Nm Laser Therapy with a Flat-Top Handpiece Photobiomodulates Mitochondria Activities of Paramecium Primaurelia (Protozoa). Lasers Med. Sci. 2016, 31, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Benedicenti, A.; Ferrando, S.; Parker, S.; Selting, W.; Gallus, L.; Benedicenti, S. Photobiomodulation by Infrared Diode Laser: Effects on Intracellular Calcium Concentration and Nitric Oxide Production of Paramecium. Photochem. Photobiol. 2016, 92, 854–862. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential Effects of Mitochondrial Complex I Inhibitors on Production of Reactive Oxygen Species. Biochim. Et Biophys. Acta (BBA)–Bioenerg. 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The Mechanism of Superoxide Production by the Antimycin-Inhibited Mitochondrial Q-Cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef]
- Ramalingam, M.; Huh, Y.J.; Lee, Y. Il The Impairments of α-Synuclein and Mechanistic Target of Rapamycin in Rotenone-Induced SH-SY5Y Cells and Mice Model of Parkinson’s Disease. Front. Neurosci. 2019, 13, 463056. [Google Scholar] [CrossRef]
- Ibarra-Gutiérrez, M.T.; Serrano-García, N.; Orozco-Ibarra, M. Rotenone-Induced Model of Parkinson’s Disease: Beyond Mitochondrial Complex I Inhibition. Mol. Neurobiol. 2023, 60, 1929–1948. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of Toxicity in Rotenone Models of Parkinson’s Disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef] [PubMed]
- Onukwufor, J.O.; Berry, B.J.; Wojtovich, A.P. Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport. Antioxidants 2019, 8, 285. [Google Scholar] [CrossRef]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 273331. [Google Scholar] [CrossRef] [PubMed]
- Kilbride, S.M.; Telford, J.E.; Davey, G.P. Complex I Controls Mitochondrial and Plasma Membrane Potentials in Nerve Terminals. Neurochem. Res. 2021, 46, 100–107. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J. Mitochondrial Complex I Deficiency and Parkinson Disease. Nat. Rev. Neurosci. 2023, 24, 193. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I Deficiency in Parkinson’s Disease Frontal Cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Gómez-Angelats, M.; Cidlowski, J.A. Plasma Membrane Depolarization without Repolarization Is an Early Molecular Event in Anti-Fas-Induced Apoptosis. J. Biol. Chem. 2001, 276, 4304–4314. [Google Scholar] [CrossRef] [PubMed]
- Deri, Z.; Adam--Vizi, V. Detection of Intracellular Free Na+ Concentration of Synaptosomes by a Fluorescent Indicator, Na+-Binding Benzofuran Isophthalate: The Effect of Veratridine, Ouabain, and α-Latrotoxin. J. Neurochem. 1993, 61, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Keller, J.N.; Begley, J.G. Evidence for Synaptic Apoptosis. Exp. Neurol. 1998, 153, 35–48. [Google Scholar] [CrossRef]
- Bicknell, B.; Liebert, A.; Herkes, G. Parkinson’s Disease and Photobiomodulation: Potential for Treatment. J. Pers. Med. 2024, 14, 112. [Google Scholar] [CrossRef]
- Kilbride, S.M.; Telford, J.E.; Tipton, K.F.; Davey, G.P. Partial Inhibition of Complex I Activity Increases Ca-Independent Glutamate Release Rates from Depolarized Synaptosomes. J. Neurochem. 2008, 106, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Trebesova, H.; Grilli, M. Synaptosomes: A Functional Tool for Studying Neuroinflammation. Encyclopedia 2023, 3, 406–418. [Google Scholar] [CrossRef]
- Garcia-Sanz, A.; Badia, A.; Clos, M.V. Superfusion of Synaptosomes to Study Presynaptic Mechanisms Involved in Neurotransmitter Release from Rat Brain. Brain Res. Brain Res. Protoc. 2001, 7, 94–102. [Google Scholar] [CrossRef]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical Progression in Parkinson Disease and the Neurobiology of Axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Tang, R.; Dai, J. Spatiotemporal Imaging of Glutamate-Induced Biophotonic Activities and Transmission in Neural Circuits. PLoS ONE 2014, 9, e85643. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Chai, W.; Wang, Z.; Xiao, F.; Dai, J. Quantum Energy Levels of Glutamate Modulate Neural Biophotonic Signals. Photochem. Photobiol. Sci. 2021, 20, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Dai, J. Biophoton Signal Transmission and Processing in the Brain. J. Photochem. Photobiol. B 2014, 139, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Han, Z.; Wang, Z.; Li, Z.; Xiao, F.; Sun, Y.; Dai, Y.; Tang, R.; Dai, J. Biophotonic Activity and Transmission Mediated by Mutual Actions of Neurotransmitters Are Involved in the Origin and Altered States of Consciousness. Neurosci. Bull. 2018, 34, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Moro, C.; Liebert, A.; Hamilton, C.; Pasqual, N.; Jeffery, G.; Stone, J.; Mitrofanis, J. The Code of Light: Do Neurons Generate Light to Communicate and Repair? Neural Regen. Res. 2022, 17, 1251–1252. [Google Scholar]
- Erboz, A.; Kesekler, E.; Gentili, P.L.; Uversky, V.N.; Coskuner-Weber, O. Electromagnetic radiation and biophoton emission in neuronal communication and neurodegenerative diseases. Prog. Biophys. Mol. Biol. 2025, 195, 87–99. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravera, S.; Farsetti, E.; Maura, G.; Marcoli, M.; Bozzo, M.; Cervetto, C.; Amaroli, A. 810-nm Photobiomodulation Evokes Glutamate Release in Normal and Rotenone-Dysfunctional Cortical Nerve Terminals by Modulating Mitochondrial Energy Metabolism. Cells 2025, 14, 67. https://doi.org/10.3390/cells14020067
Ravera S, Farsetti E, Maura G, Marcoli M, Bozzo M, Cervetto C, Amaroli A. 810-nm Photobiomodulation Evokes Glutamate Release in Normal and Rotenone-Dysfunctional Cortical Nerve Terminals by Modulating Mitochondrial Energy Metabolism. Cells. 2025; 14(2):67. https://doi.org/10.3390/cells14020067
Chicago/Turabian StyleRavera, Silvia, Elisa Farsetti, Guido Maura, Manuela Marcoli, Matteo Bozzo, Chiara Cervetto, and Andrea Amaroli. 2025. "810-nm Photobiomodulation Evokes Glutamate Release in Normal and Rotenone-Dysfunctional Cortical Nerve Terminals by Modulating Mitochondrial Energy Metabolism" Cells 14, no. 2: 67. https://doi.org/10.3390/cells14020067
APA StyleRavera, S., Farsetti, E., Maura, G., Marcoli, M., Bozzo, M., Cervetto, C., & Amaroli, A. (2025). 810-nm Photobiomodulation Evokes Glutamate Release in Normal and Rotenone-Dysfunctional Cortical Nerve Terminals by Modulating Mitochondrial Energy Metabolism. Cells, 14(2), 67. https://doi.org/10.3390/cells14020067