
MAGED2 Enhances Expression and Function of NCC at the Cell Surface via cAMP Signaling Under Hypoxia

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmid Constructs and Site Directed Mutagenesis
2.3. Cell Culture, Hypoxia Induction and Transfections
2.4. Biotinylation
2.5. Immunocytochemistry
2.6. Exocytosis Assay
2.7. Endocytosis Assay
2.8. Lysosomal Inhibition Assays
2.9. Measurement of NCC-Mediated Sodium Uptake Using CoroNa Green
2.10. Ubiquitination Assay
2.11. Western Blotting
2.12. Statistical Analyses
3. Results
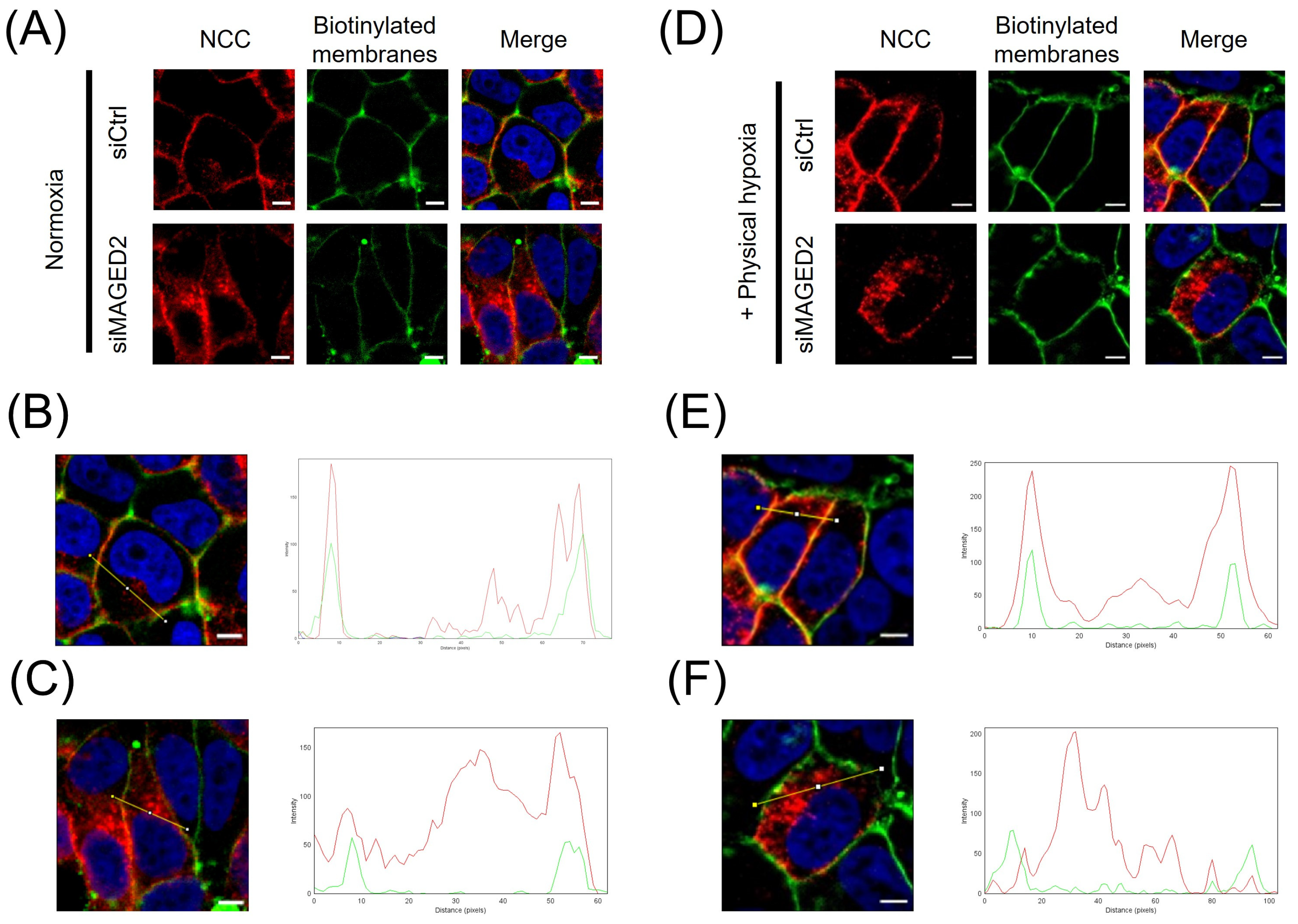
3.1. MAGED2 Regulates Total and Membrane Expression of NCC Under Hypoxic Conditions
3.2. MAGED2 Regulates the Function of NCC Under Hypoxic Conditions
3.3. Forskolin Reverses Reduced Expression of Total and Membrane Bound NCC Caused by MAGED2 Depletion
3.4. MAGED-2 Decreases Exocytotic Insertion of NCC
3.5. MAGED-2 Increases Endocytic Retrieval of NCC
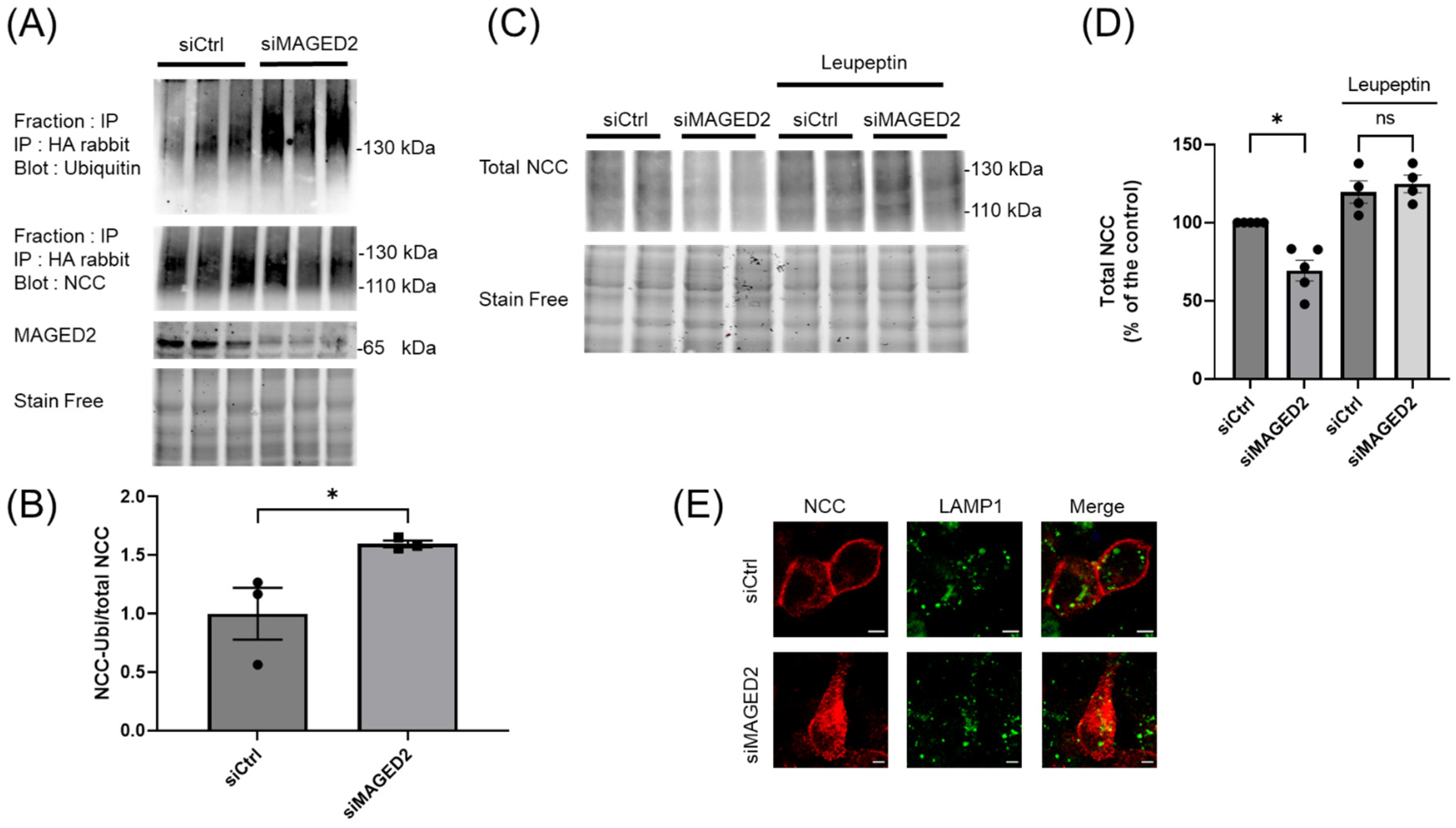
3.6. MAGED2 Depletion Down-Regulates NCC by Enhancing Its Ubiquitination and Lysosomal Degradation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hebert, S.C. Bartter syndrome. Curr. Opin. Nephrol. Hypertens. 2003, 12, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Komhoff, M.; Laghmani, K. Pathophysiology of antenatal Bartter’s syndrome. Curr. Opin. Nephrol. Hypertens. 2017, 26, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Seyberth, H.W.; Weber, S.; Komhoff, M. Bartter’s and Gitelman’s syndrome. Curr. Opin. Pediatr. 2017, 29, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Filser, M.; Bruel, A.; Dard, R.; Quibel, T.; Dubucs, C.; Kwon, T.; Le Tanno, P.; Thevenon, J.; Ziegler, A.; et al. X-linked transient antenatal Bartter syndrome related to MAGED2 gene: Enriching the phenotypic description and pathophysiologic investigation. Genet. Med. 2024, 27, 101217. [Google Scholar] [CrossRef] [PubMed]
- Laghmani, K.; Beck, B.B.; Yang, S.S.; Seaayfan, E.; Wenzel, A.; Reusch, B.; Vitzthum, H.; Priem, D.; Demaretz, S.; Bergmann, K.; et al. Polyhydramnios, Transient Antenatal Bartter’s Syndrome, and Mutations. N. Engl. J. Med. 2016, 374, 1853–1863. [Google Scholar] [CrossRef]
- Legrand, A.; Treard, C.; Roncelin, I.; Dreux, S.; Bertholet-Thomas, A.; Broux, F.; Bruno, D.; Decramer, S.; Deschenes, G.; Djeddi, D.; et al. Prevalence of Novel MAGED2 Mutations in Antenatal Bartter Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 242–250. [Google Scholar] [CrossRef]
- Gee, R.R.F.; Chen, H.; Lee, A.K.; Daly, C.A.; Wilander, B.A.; Tacer, K.F.; Potts, P.R. Emerging roles of the MAGE protein family in stress response pathways. J. Biol. Chem. 2020, 295, 16121–16155. [Google Scholar] [CrossRef]
- Seaayfan, E.; Nasrah, S.; Quell, L.; Kleim, M.; Weber, S.; Meyer, H.; Laghmani, K.; Kömhoff, M. MAGED2 Is Required under Hypoxia for cAMP Signaling by Inhibiting MDM2-Dependent Endocytosis of G-Alpha-S. Cells 2022, 11, 2546. [Google Scholar] [CrossRef]
- Fenton, R.A.; Knepper, M.A. Mouse models and the urinary concentrating mechanism in the new millennium. Physiol. Rev. 2007, 87, 1083–1112. [Google Scholar] [CrossRef]
- Freeburg, P.B.; Abrahamson, D.R. Divergent expression patterns for hypoxia-inducible factor-1beta and aryl hydrocarbon receptor nuclear transporter-2 in developing kidney. J. Am. Soc. Nephrol. 2004, 15, 2569–2578. [Google Scholar] [CrossRef]
- Azlan, N.F.M.; Koeners, M.P.; Zhang, J. Regulatory control of the Na-Cl co-transporter NCC and its therapeutic potential for hypertension. Acta Pharm. Sin. B 2021, 11, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.C.; Wynne, B.M.; Yu, L.; Tomilin, V.; Yue, Q.; Zhou, Y.; Al-Khalili, O.; Mallick, R.; Cai, H.; Alli, A.A.; et al. The sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC) associate. Biochem. J. 2016, 473, 3237–3252. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, K.M. Gordon Syndrome: A continuing story. Pediatr. Nephrol. 2015, 30, 1903–1908. [Google Scholar] [CrossRef]
- Mabillard, H.; Sayer, J.A. The Molecular Genetics of Gordon Syndrome. Genes 2019, 10, 986. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, G.; Zhang, C. Downregulation of Cullin 3 Ligase Signaling Pathways Contributes to Hypertension in Preeclampsia. Front. Cardiovasc. Med. 2021, 8, 654254. [Google Scholar] [CrossRef]
- Seaayfan, E.; Nasrah, S.; Quell, L.; Radi, A.; Kleim, M.; Schermuly, R.T.; Weber, S.; Laghmani, K.; Komhoff, M. Reciprocal Regulation of MAGED2 and HIF-1 alpha Augments Their Expression under Hypoxia: Role of cAMP and PKA Type II. Cells 2022, 11, 3424. [Google Scholar] [CrossRef]
- Xie, A.; Xiao, L.; Zhang, M.; Duan, H.; Ren, Z.; Wang, P.; Jia, Y.; Liu, M.; Lou, J.; Wang, X. Evaluation of Urinary NKCC2 and NCC in Rats with AKI Induced by Candesartan Plus Low-Salt Diet in Rats. J. Am. Soc. Nephrol. 2024, 35. [Google Scholar] [CrossRef]
- Balzer, M.S.; Doke, T.; Yang, Y.W.; Aldridge, D.L.; Hu, H.; Mai, H.; Mukhi, D.; Ma, Z.; Shrestha, R.; Palmer, M.B.; et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat. Commun. 2022, 13, 4018. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Bakhos-Douaihy, D.; Seaayfan, E.; Frachon, N.; Demaretz, S.; Komhoff, M.; Laghmani, K. Diacidic Motifs in the Carboxyl Terminus Are Required for ER Exit and Translocation to the Plasma Membrane of NKCC2. Int. J. Mol. Sci. 2022, 23, 12761. [Google Scholar] [CrossRef] [PubMed]
- Nasrah, S.; Radi, A.; Daberkow, J.K.; Hummler, H.; Weber, S.; Seaayfan, E.; Kömhoff, M. MAGED2 Depletion Promotes Stress-Induced Autophagy by Impairing the cAMP/PKA Pathway. Int. J. Mol. Sci. 2023, 24, 13433. [Google Scholar] [CrossRef] [PubMed]
- Fiszer-Kierzkowska, A.; Vydra, N.; Wysocka-Wycisk, A.; Kronekova, Z.; Jarzab, M.; Lisowska, K.M.; Krawczyk, Z. Liposome-based DNA carriers may induce cellular stress response and change gene expression pattern in transfected cells. BMC Mol. Biol. 2011, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, R.; Stern, P.; Diehn, M.; Shamu, C.; Osario, M.; Zúñiga, M.; Brown, P.O.; Ploegh, H. Degradation of Proteins from the ER of S. cerevisiae Requires an Intact Unfolded Protein Response Pathway. Mol. Cell 2000, 5, 729–735. [Google Scholar] [CrossRef]
- Gamba, G. Regulation of the renal Na+-Cl- cotransporter by phosphorylation and ubiquitylation. Am. J. Physiol. Ren. Physiol. 2012, 303, F1573–F1583. [Google Scholar] [CrossRef]
- Rosenbaek, L.L.; Kortenoeven, M.L.; Aroankins, T.S.; Fenton, R.A. Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J. Biol. Chem. 2014, 289, 13347–13361. [Google Scholar] [CrossRef]
- Piper, R.C.; Dikic, I.; Lukacs, G.L. Ubiquitin-dependent sorting in endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016808. [Google Scholar] [CrossRef]
- Strous, G.J.; Govers, R. The ubiquitin-proteasome system and endocytosis. J. Cell Sci. 1999, 112 Pt 10, 1417–1423. [Google Scholar] [CrossRef]
- Ecelbarger, C.A.; Kim, G.H.; Terris, J.; Masilamani, S.; Mitchell, C.; Reyes, I.; Verbalis, J.G.; Knepper, M.A. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am. J. Physiol. Ren. Physiol. 2000, 279, F46–F53. [Google Scholar] [CrossRef]
- Ecelbarger, C.A.; Yu, S.; Lee, A.J.; Weinstein, L.S.; Knepper, M.A. Decreased renal Na-K-2Cl cotransporter abundance in mice with heterozygous disruption of the G(s)alpha gene. Am. J. Physiol. 1999, 277, F235–F244. [Google Scholar]
- Markadieu, N.; Delpire, E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflug. Arch. 2014, 466, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Mutig, K.; Saritas, T.; Uchida, S.; Kahl, T.; Borowski, T.; Paliege, A.; Bohlick, A.; Bleich, M.; Shan, Q.; Bachmann, S. Short-term stimulation of the thiazide-sensitive Na+-Cl- cotransporter by vasopressin involves phosphorylation and membrane translocation. Am. J. Physiol. Ren. Physiol. 2010, 298, F502–F509. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.B.; Hofmeister, M.V.; Rosenbaek, L.L.; Nielsen, J.; Fenton, R.A. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 2010, 78, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Rana, S.; Mehan, S. Role of adenylyl cyclase activator in controlling experimental diabetic nephropathy in rats. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 144–153. [Google Scholar]
- Almeida, F.C.; Lemonica, I.P. The toxic effects of Coleus barbatus B. on the different periods of pregnancy in rats. J. Ethnopharmacol. 2000, 73, 53–60. [Google Scholar] [CrossRef]
- Donnelly, B.F.; Needham, P.G.; Snyder, A.C.; Roy, A.; Khadem, S.; Brodsky, J.L.; Subramanya, A.R. Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J. Biol. Chem. 2013, 288, 13124–13135. [Google Scholar] [CrossRef]
- Gerits, N.; Mikalsen, T.; Kostenko, S.; Shiryaev, A.; Johannessen, M.; Moens, U. Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. J. Biol. Chem. 2007, 282, 37232–37243. [Google Scholar] [CrossRef]
- Kostenko, S.; Jensen, K.L.; Moens, U. Phosphorylation of heat shock protein 40 (Hsp40/DnaJB1) by mitogen-activated protein kinase-activated protein kinase 5 (MK5/PRAK). Int. J. Biochem. Cell Biol. 2014, 47, 29–37. [Google Scholar] [CrossRef]
- Kortenoeven, M.L.A.; Esteva-Font, C.; Dimke, H.; Poulsen, S.B.; Murali, S.K.; Fenton, R.A. High dietary potassium causes ubiquitin-dependent degradation of the kidney sodium-chloride cotransporter. J. Biol. Chem. 2021, 297, 100915. [Google Scholar] [CrossRef]
- Picard, N.; Trompf, K.; Yang, C.L.; Miller, R.L.; Carrel, M.; Loffing-Cueni, D.; Fenton, R.A.; Ellison, D.H.; Loffing, J. Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J. Am. Soc. Nephrol. 2014, 25, 511–522. [Google Scholar] [CrossRef]
- Penton, D.; Moser, S.; Wengi, A.; Czogalla, J.; Rosenbaek, L.L.; Rigendinger, F.; Faresse, N.; Martins, J.R.; Fenton, R.A.; Loffing-Cueni, D.; et al. Protein Phosphatase 1 Inhibitor-1 Mediates the cAMP-Dependent Stimulation of the Renal NaCl Cotransporter. J. Am. Soc. Nephrol. 2019, 30, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Carbajal-Contreras, H.; Gamba, G.; Castaneda-Bueno, M. The serine-threonine protein phosphatases that regulate the thiazide-sensitive NaCl cotransporter. Front. Physiol. 2023, 14, 1100522. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, Y.; Mori, Y.; Tsuzaki, Y.; Mori, T.; Nomura, N.; Wakabayashi, M.; Takahashi, D.; Zeniya, M.; Kikuchi, E.; Araki, Y.; et al. Impaired degradation of WNK by Akt and PKA phosphorylation of KLHL3. Biochem. Biophys. Res. Commun. 2015, 467, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Mori, T.; Nomura, N.; Khan, M.Z.; Araki, Y.; Zeniya, M.; Sohara, E.; Rai, T.; Sasaki, S.; Uchida, S. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci. Rep. 2014, 34, 195–205. [Google Scholar] [CrossRef]
- Sohara, E.; Uchida, S. Kelch-like 3/Cullin 3 ubiquitin ligase complex and WNK signaling in salt-sensitive hypertension and electrolyte disorder. Nephrol. Dial. Transpl. Transplant. 2016, 31, 1417–1424. [Google Scholar] [CrossRef]
- Maeoka, Y.; Nguyen, L.T.; Sharma, A.; Cornelius, R.J.; Su, X.T.; Gutierrez, M.R.; Carbajal-Contreras, H.; Castaneda-Bueno, M.; Gamba, G.; McCormick, J.A. Dysregulation of the WNK4-SPAK/OSR1 pathway has a minor effect on baseline NKCC2 phosphorylation. Am. J. Physiol. Ren. Physiol. 2024, 326, F39–F56. [Google Scholar] [CrossRef]
- Ismail, N.A.; Baines, D.L.; Wilson, S.M. The phosphorylation of endogenous Nedd4-2 In Na(+)-absorbing human airway epithelial cells. Eur. J. Pharmacol. 2014, 732, 32–42. [Google Scholar] [CrossRef]
- Pohl, P.; Joshi, R.; Petrvalska, O.; Obsil, T.; Obsilova, V. 14-3-3-protein regulates Nedd4-2 by modulating interactions between HECT and WW domains. Commun. Biol. 2021, 4, 899. [Google Scholar] [CrossRef]
- Persaud, A.; Jiang, C.; Liu, Z.; Kefalas, G.; Demian, W.L.; Rotin, D. Elevated intracellular Na(+) and osmolarity stimulate catalytic activity of the ubiquitin ligase Nedd4-2. Proc. Natl. Acad. Sci. USA 2022, 119, e2122495119. [Google Scholar] [CrossRef]
- Rosenbaek, L.L.; Petrillo, F.; van Bemmelen, M.X.; Staub, O.; Murali, S.K.; Fenton, R.A. The E3 ubiquitin-protein ligase Nedd4-2 regulates the sodium chloride cotransporter NCC but is not required for a potassium-induced reduction of NCC expression. Front. Physiol. 2022, 13, 971251. [Google Scholar] [CrossRef]
- Cai, H.; Cebotaru, V.; Wang, Y.H.; Zhang, X.M.; Cebotaru, L.; Guggino, S.E.; Guggino, W.B. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 2006, 69, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Subramanya, A.R.; Liu, J.; Ellison, D.H.; Wade, J.B.; Welling, P.A. WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J. Biol. Chem. 2009, 284, 18471–18480. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhuang, J.; Gu, D.; Wang, H.; Cebotaru, L.; Guggino, W.B.; Cai, H. WNK4 enhances the degradation of NCC through a sortilin-mediated lysosomal pathway. J. Am. Soc. Nephrol. 2010, 21, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, V.; Hallows, K.R. Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 2008, 19, 1845–1854. [Google Scholar] [CrossRef]
- Fouassier, D.; Blanchard, A.; Fayol, A.; Bobrie, G.; Boutouyrie, P.; Azizi, M.; Hulot, J.S. Sequential nephron blockade with combined diuretics improves diastolic function in patients with resistant hypertension. ESC Heart Fail. 2020, 7, 2561–2571. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti-MAGED2 rabbit raised against this peptide (QVQENQDTRPKVKAK) | Eurogentec (Cologne, Germany) | |
| Anti-Gαs | Sigma Aldrich (Schnelldorf, Germany) | 06-237 |
| Anti-Ubiquitin | Invitrogen (Dreieich, Germany) | 13-1600 |
| Anti-HA tag rabbit | Thermo Fisher Scientific (Dreieich, Germany) | 71-5500 |
| Anti-HA tag mouse | Thermo Fisher Scientific (Dreieich, Germany) | 26183 |
| Goat anti-Mouse IgG (H + L), Alexa Fluor Plus 555 | Thermo Fisher Scientific (Dreieich, Germany) | A32727 |
| Streptavidin, Alexa Fluor™ 488 conjugate | Thermo Fisher Scientific (Dreieich, Germany) | S11223 |
| StarBright Blue 520 Goat Anti-Rabbit IgG | Bio-rad (Dreieich, Germany) | 12005869 |
| StarBright Blue 700 Goat Anti-Mouse IgG | Bio-rad (Dreieich, Germany) | 12004158 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EZ-Link™ Sulfo-NHS-LC-Biotin | Thermo Fisher Scientific (Dreieich, Germany) | 21335 |
| Pierce™ Premium Grade Sulfo-NHS-SS-Biotin | Thermo Fisher Scientific (Dreieich, Germany) | PG82077 |
| Pierce™ Sulfo-NHS-Acetate | Thermo Fisher Scientific (Dreieich, Germany) | 26777 |
| L-Glutathion reduziert | Roth (Karlsruhe, Germany) | 6832.3 |
| Leupeptin hemisulfate | AdipoGen Life Sciences (San Diego, CA, USA) | AG-CP3-7000-M005 |
| Mg-132 | Sigma-Aldrich (Schnelldorf, Germany) | 474787 |
| Dynamin-Inhibitor I, Dynasore | Sigma-Aldrich (Schnelldorf, Germany) | 324410 |
| Forskolin | Sigma-Aldrich (Schnelldorf, Germany) | F6886 |
| CoroNa Green, AM | Thermo Fisher Scientific (Dreieich, Germany) | C36676 |
| Streptavidin-Agarose Resin | Thermo Fisher Scientific (Dreieich, Germany) | 20353 |
| Dynabeads Protein G | Thermo Fisher Scientific (Dreieich, Germany) | 10004D |
| Bumetanide | Sigma-Aldrich (Schnelldorf, Germany) | B-3023 |
| Quabain | Sigma-Aldrich (Schnelldorf, Germany) | O-3125 |
| Amiloride | Sigma-Aldrich (Schnelldorf, Germany) | A-7410 |
| Hydrochlorothiazide | Sigma-Aldrich (Schnelldorf, Germany) | H2910 |
| Critical Commercial Assays | ||
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs (Frankfurt am Main, Germany) | E0554S |
| QuikChange Multi Site-Directed Mutagenesis Kit | Agilent Technologies (Santa Clara, CA, USA) | 200515 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | CRL1573 |
| HeLa | Gift from Dr. Vijay Renigunta | |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting Control Pool | Dharmacon (Lafayette, CO, USA) | D-001810-10-05 |
| UGGUUUACAUGUCGACUAA | ||
| UGGUUUACAUGUUGUGUGA | ||
| UGGUUUACAUGUUUUCUGA | ||
| UGGUUUACAUGUUUUCCUA | ||
| ON-TARGETplus Human MAGED2 siRNA—SMARTpool | Dharmacon (Lafayette, CO, USA) | L-017284-01-0005 |
| GGACGAAGCUGAUAUCGGA | ||
| GCUAAAGACCAGACGAAGA | ||
| AGGCGAUGGAAGCGGAUUU | ||
| GAAAAGGACAGUAGCUCGA | ||
| ON-TARGETplus Human GNAS siRNA—SMARTpool | Dharmacon (Lafayette, CO, USA) | L-010825-00-0005 |
| GCAAGUGGAUCCAGUGCUU | ||
| GCAUGCACCUUCGUCAGUA | ||
| AUGAGGAUCCUGCAUGUUA | ||
| CAACCAAAGUGCAGGACAU | ||
| NCC 3×HA primer | Sigma-Aldrich (Schnelldorf, Germany) | |
| CCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTATGGCAGAACTGCCCACA | ||
| ACGTCATAGGGATAGCCAGCGTAATCTGGAACATCGTATGGGTACATGGATCCGAATTCGCCCTATAG | ||
| Recombinant DNA | ||
| pCMV5-HA-1 SLC12A3 (hNCC) | University of Dundee | DU4461 |
| pTargeT | Promega (Walldorf, Germany) | A1410 |
| Software and Algorithms | ||
| ImageJ 1.51j8 | [20] | https://imagej.nih.gov/ij/ (accessed on 25 Fabruary 2022) |
| GraphPad Prism 9 | GraphPad | |
| EndNote 21 | Clarivate Analytics | |
| ZEN Microscopy Software 3.3 | ZEISS Microscopy | |
| BioRender | BioRender | www.biorender.com (accessed on 25 Fabruary 2024) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radi, A.; Nasrah, S.; Auer, M.; Renigunta, A.; Weber, S.; Seaayfan, E.; Kömhoff, M. MAGED2 Enhances Expression and Function of NCC at the Cell Surface via cAMP Signaling Under Hypoxia. Cells 2025, 14, 175. https://doi.org/10.3390/cells14030175
Radi A, Nasrah S, Auer M, Renigunta A, Weber S, Seaayfan E, Kömhoff M. MAGED2 Enhances Expression and Function of NCC at the Cell Surface via cAMP Signaling Under Hypoxia. Cells. 2025; 14(3):175. https://doi.org/10.3390/cells14030175
Chicago/Turabian StyleRadi, Aline, Sadiq Nasrah, Michelle Auer, Aparna Renigunta, Stefanie Weber, Elie Seaayfan, and Martin Kömhoff. 2025. "MAGED2 Enhances Expression and Function of NCC at the Cell Surface via cAMP Signaling Under Hypoxia" Cells 14, no. 3: 175. https://doi.org/10.3390/cells14030175
APA StyleRadi, A., Nasrah, S., Auer, M., Renigunta, A., Weber, S., Seaayfan, E., & Kömhoff, M. (2025). MAGED2 Enhances Expression and Function of NCC at the Cell Surface via cAMP Signaling Under Hypoxia. Cells, 14(3), 175. https://doi.org/10.3390/cells14030175