Abstract
Exon skipping, mediated through antisense oligonucleotides (ASOs), is a promising approach to exclude pathogenic variants from the DYSF gene and treat dysferlinopathies. Understanding the applicability of various exon skipping strategies in the total patient population, an analysis not previously performed, can help guide researchers in prioritizing therapies with the broadest potential impact. Using data from the UMD-DYSF database, we evaluated all reported pathogenic variants in dysferlinopathy patients for the applicability of single- or double-exon skipping approaches to exclude the pathogenic variants while maintaining the open reading frame. A total of 61 theoretically applicable exon skipping strategies were identified, with the potential to address 90.0% of the pathogenic variants reported—44.6% through single-exon skipping and 45.3% through double-exon skipping. The most broadly applicable targets include exons 28 and 29 (9.0%), exons 27 and 28 (6.7%), and exons 50 and 51 (5.4%). While numerous theoretically applicable strategies were identified, it remains unclear if the truncated proteins produced through each exon skipping strategy will have improved functionality to alleviate patient symptoms. Further preclinical studies and clinical trials will be essential to determine the effectiveness of these therapies, potentially expanding access to disease-modifying treatments for dysferlinopathy patients.
1. Introduction
Dysferlinopathies comprise a group of rare muscle disorders caused by pathogenic variants in the DYSF gene that result in a broad spectrum of clinical phenotypes and disease severities [1]. These pathogenic variants result in significant or complete loss of functional dysferlin proteins, which in turn causes skeletal muscle damage and necrosis [2]. One promising therapeutic approach to bypass these variants is exon skipping, where the exon containing the variant, and adjacent exons, if necessary to maintain the open reading frame, are selectively excluded [3]. This strategy can be effectively mediated using antisense oligonucleotides (ASOs), which have already received FDA approval for the treatment of other rare diseases, such as spinal muscular atrophy and Duchenne muscular dystrophy (DMD) [4,5].
While early preclinical trials have shown encouraging results for ASO-mediated exon skipping in dysferlinopathies, further research is necessary before this strategy can be translated to clinical applications [6,7]. Pathogenic variants associated with the disease have been identified in all exons of the DYSF gene, and, due to the variant-specific nature of exon skipping strategies, each exon skipping strategy would only be applicable to a subset of the dysferlinopathy patient population. Thus, evaluating the applicability of all potential exon skipping strategies across the patient population is crucial to allow researchers to prioritize those that could benefit the largest number of individuals.
Currently, a comprehensive evaluation of potential exon skipping targets for dysferlinopathies has not been reported. This study aims to fill that gap by providing an in-depth analysis of the applicability of all possible single- and double-exon skipping approaches that could theoretically target patient pathogenic variants reported in the comprehensive UMD_DYSF database (http://www.umd.be/DYSF/. Accessed on 17 November 2024) [8]. Understanding the applicability of these strategies will help optimize therapeutic development and potentially expand treatment options for patients affected by dysferlinopathies.
2. Background on Dysferlinopathies
Dysferlinopathies are rare autosomal recessive diseases caused by pathogenic variants in the DYSF gene that result in a significant reduction or complete absence of the dysferlin protein [9,10]. Dysferlin is a 237 kDa protein that plays a diverse range of roles and is primarily expressed in striated muscle [11,12,13]. Dysferlin is part of the ferlin protein family, along with myoferlin, otoferlin, Fer1L4, Fer1L5, and Fer1L6. Ferlin proteins are characterized by their large size and multiple tandem C2 domains, which typically form a binding pocket with an affinity for Ca2+ and phospholipids [14,15]. Dysferlin, specifically, is composed of seven C2 domains and is observed to localize predominantly in the sarcolemma or t-tubule membrane in uninjured muscle cells [16,17]. This protein is largely known for its role in muscle membrane repair [18]. Microlesions in the plasma membrane can occur because of mechanical stress associated with activity. Upon damage, dysferlin travels to the cytoplasm where, in a Ca2+-dependent manner, it regulates the trafficking and fusion of vesicles to create a patch for the disrupted membrane [16,19,20]. Dysferlin has also been observed to play an important role in t-tubule maintenance [16,21], regulating Ca2+ levels following mechanical stress [22,23], and has a variety of other functions [24,25,26]. The role of dysferlin in cardiac tissues is less understood at this time, but preliminary studies, and the similarities between cardiac myocytes and skeletal muscle cells, suggest that dysferlin plays a similar role in membrane repair and Ca2+ homeostasis in cardiac tissues [11,27,28,29].
Currently, the exact mechanisms underlying the pathogenesis of dysferlinopathies remain unclear. However, upon injury, dysferlin-deficient tissues have a diminished capacity to repair their membranes, which is believed to result in necrosis and the initiation of an inflammatory response that can damage surrounding tissue [26,30,31]. Muscle biopsies collected from patients with pathogenic DYSF variants typically present with a large variation in fiber size, featuring necrotic and regenerating fibers, alongside increased levels of connective and fatty tissue deposits and inflammatory cells [32,33,34,35]. Patients also present with elevated levels of muscle damage biomarkers, even before the onset of symptoms [36].
There is a diverse spectrum of dysferlinopathies that result from deficient or absent dysferlin proteins due to pathogenic variants in the DYSF gene. The two primary clinical phenotypes are Miyoshi muscular dystrophy (MMD) and limb-girdle muscular dystrophy R2 (LGMDR2), formerly known as LGMD2B. MMD is initially characterized by muscle weakness and atrophy in the distal muscles, particularly in the calves and plantar muscles, which later extends to the thighs and buttocks [37]. In contrast, LGMDR2 patients present with proximal muscle weakness, predominantly affecting the thighs and shoulders [38]. While the age of symptom onset varies significantly among patients, both MMD and LGMDR2 typically manifest around the age of 20 and progress slowly [1,39]. As the diseases advance, patients experience difficulty with walking, stair climbing, and often become reliant on canes or wheelchairs [40]. Less commonly, DYSF variants can also result in other dysferlinopathies such as distal myopathy with anterior tibial onset (DMAT), the proximodistal phenotype, and asymptomatic hyperCKemia. DMAT is associated with the fastest disease progression, beginning with weakness in the anterior tibial muscles and later spreading to the lower and upper limbs [36,37]. Patients with the proximodistal phenotype present with symptoms characteristic of both MMD and LGMDR2, with weakness affecting both distal and proximal muscles simultaneously [41]. A hallmark of all dysferlinopathies is a pronounced elevation in serum creatine kinase (CK) levels [42,43]. In cases of asymptomatic hyperCKemia, patients exhibit elevated CK levels without apparent muscle weakness, although there remains a potential risk for muscle involvement over time [37,44]. Despite attempts to uncover clear genotype–phenotype correlations, predicting disease phenotypes remains challenging due to the variability seen across different patient populations [45,46,47]. Significant phenotypic differences are observed not only among individuals with the same pathogenic DYSF variants but even among family members, suggesting that additional genetic or environmental factors may influence the symptoms and severity of disease experienced by individuals [47,48,49]. This complexity underscores the need for further research to better understand the underlying mechanisms driving these phenotypic differences.
Currently, there are no curative therapies available for dysferlinopathies [2]. Patients primarily rely on symptomatic treatments, such as walking aids, physiotherapy, occupational therapy, and ankle or foot orthoses to improve mobility and overall quality of life [50]. However, numerous studies have focused on evaluating a range of different molecular and genetic therapies aiming to improve the patient’s phenotype and prevent progression [51]. These approaches include strategies like gene therapy, delivering a modified dysferlin gene using AAV vectors [52,53,54], and gene editing, using CRISPR-Cas9 systems to correct specific DYSF variants [55,56]. However, the AAV vectors used to deliver these therapies have significant limitations and safety concerns. Natural exposure to AAVs has resulted in 30-60% of the population developing antibodies against these vectors, which can compromise their effectiveness and raise the risk of adverse immune reactions [57,58,59]. Additional safety risks include the development of innate and adaptive immune responses and the potential genomic consequences of viral genome insertion [60,61]. Moreover, despite the therapeutic potential of AAV vectors, several AAV-related patient fatalities, often associated with high doses, have raised concerns about the clinical safety of these vectors [62,63]. Alternatively, ASO-mediated exon skipping, which uses non-viral approaches to exclude pathogenic variants from the final transcript, shows significant promise and will be discussed in further detail.
3. ASO-Mediated Exon Skipping
ASOs represent a promising therapeutic approach that can be designed to be capable of selectively modulating gene expression at the RNA level [4]. They are short (~20 base pairs in length), synthetic, single-stranded molecules that can bind through Watson–Crick base pairing to a complementary RNA sequence. Upon hybridization, ASOs can either alter the transcript’s splicing or prevent its expression [64,65]. However, delivering unmodified ASOs to target tissues is challenging due to their high susceptibility to degradation by nucleases and lysosomes, as well as their poor cellular uptake [66,67,68]. To address these challenges, chemical modifications have been introduced to enhance both the stability and cellular uptake of ASOs [67,68]. Some ASOs are modified to include a phosphorothioate (PS) backbone and/or the addition of a functional group at the 2′ position of the ribose sugar, which can help protect the ASO from nuclease degradation [69]. Additionally, alternative backbone chemistries, such as the development of phosphorodiamidate morpholino oligomers (PMOs), replacing the natural backbone with morpholino rings linked by phosphorodiamidate bonds, offer advantages like increased targeting affinity and improved safety [5,69]. By selectively incorporating these chemical modifications, ASOs can be tailored to function as versatile therapeutic tools with different mechanisms of action [70]. These mechanisms can be classified into three main categories: (1) RNAse H-dependent mRNA degradation; (2) splicing modulation via steric hindrance of key regulatory sites, such as splice acceptor/donor sites, exonic splicing enhancers (ESEs), or exonic splice silencers (ESSs), thereby blocking spliceosome machinery; and (3) translation modulation by obstructing regulatory elements, like translation inhibitory elements in 5′ UTRs [71,72,73].
ASO-mediated exon skipping has emerged as a promising therapeutic strategy for treating various genetic neurodegenerative and muscular dystrophy disorders by allowing for the exclusion of pathogenic variants from the final mRNA transcript [74,75]. The primary goal of this approach is to generate a truncated, yet partially functional, protein capable of restoring some degree of protein function [76]. This is achieved by using ASOs, with all nucleotides chemically modified, to induce alterations in the transcript’s splicing mechanisms [77]. By targeting specific exons carrying or flanked by small pathogenic variants, ASOs can facilitate the exclusion of these regions while preserving the reading frame [78,79,80]. In addition, exon skipping can also address disruptions in the reading frame caused by large deletions or insertions by promoting the skipping of adjacent exons, which restores a continuous reading frame across the transcript [81].
The location and type of pathogenic variant can significantly affect the success of exon skipping therapies, as the compatibility of the remaining exons after skipping depends on their intron phase, categorized by their position between codons [82,83]. Therefore, ASOs for exon skipping need to be carefully designed to ensure that the exclusion of the target exon or exons preserves the correct reading frame to maintain the functional integrity of the resulting protein [82,83,84]. Symmetrical exons, flanked by introns of the same phase, can generally be skipped without disrupting the protein’s reading frame. In contrast, asymmetrical exons, surrounded by introns of differing phases, present a greater challenge. Skipping only the affected asymmetrical exon is insufficient to restore transcript functionality. In such cases, a multi-exon skipping strategy, where both the mutated exon and adjacent exons are skipped, may be required to maintain a continuous reading frame and ensure proper protein expression (Figure 1) [85].
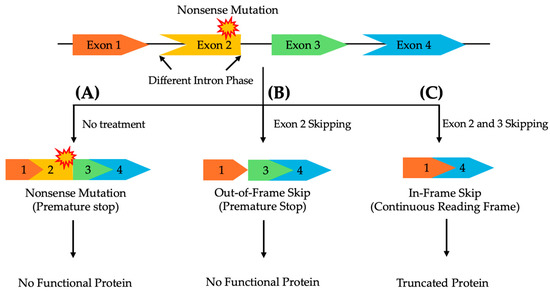
Figure 1.
A schematic representation of multi-exon skipping to exclude pathogenic variants in asymmetrical exons, flanked by differing intron phases, while preserving the reading frame to enable the production of a functional protein. (A) Without treatment, a nonsense mutation in exon 2 prevents the synthesis of a functional protein. (B) Skipping only exon 2 disrupts the reading frame, leading to a non-functional protein. (C) Skipping both exons 2 and 3 restores the reading frame, resulting in a truncated, yet functional, protein.
Given the significant therapeutic potential of ASO-mediated exon skipping, several ASOs have already received FDA approval for rare genetic disorders, with additional clinical trials ongoing to explore the effectiveness of alternative ASO therapies [86]. Notably, several ASOs have been approved for the treatment of DMD, including Eteplirsen, Viltolarsen, Golodirsen, and Casimersen [87]. By promoting the exclusion of specific exons, these therapies have demonstrated effectiveness in restoring partially functional dystrophin protein levels in patients who otherwise lack functional dystrophin [86,88]. Despite their potential, the clinical effectiveness of these therapies for DMD remains controversial at this time. The clinical significance of the minimal functional benefits observed is difficult to determine due to small patient cohorts and reliance on historical controls [79,89,90]. Enhancing ASO delivery and cellular uptake will be essential to improving their therapeutic effect [67].
Furthermore, multi-exon skipping (targeting two or more exons) has recently gained increased attention, as it offers a broader therapeutic application for a wider range of pathogenic variants, including those involving small variants on asymmetrical exons [91,92,93]. Preclinical studies in DMD animal models have shown that combining ASOs targeting different exons (ASO cocktails) is highly effective in restoring dystrophin production [94]. Despite these promising results, further clinical studies are required to confirm the efficacy of this approach in patients.
ASO-Mediated Exon Skipping for the Treatment of Dysferlinopathies
Currently, there are no FDA-approved exon skipping therapies available to patients with dysferlinopathies. However, previous studies have identified patients with variants in the DYSF gene that naturally induce exon skipping without disrupting the reading frame, resulting in partially functional dysferlin and a milder disease phenotype [95,96]. This observation suggests that ASO-mediated exon skipping therapy could be a promising approach for treating dysferlinopathies [95,96]. By targeting the disease-causing exon, and additional adjacent exons if necessary, ASOs can preserve the DYSF reading frame while excluding pathogenic variants, potentially leading to a less severe phenotype [97]. Furthermore, in cases involving large multi-exon deletions in the DYSF gene [98], a strategy similar to current DMD therapies could be applied, where promoting the skipping of adjacent exons could restore the DYSF reading frame [99], potentially enabling the production of a partially functional dysferlin protein.
Preliminary in vitro studies of ASO-mediated exon skipping strategies have shown promising results that warrant further study. Specifically, 2′-O-methyl (2′-OME) PS ASOs targeting the splice donor site of exon 32 were able to promote exon 32 skipping in patient-derived cells harboring variants within this exon [7]. The induced exon skipping led to the production of low levels of a quasi-dysferlin protein, which partially restored membrane repair capacity [7]. In another study, a novel pathogenic variant within intron 50 caused aberrant pseudo-exon inclusion. In this case, 2′-OME PS ASOs targeting the exonic splicing enhancer within intron 50 effectively prevented abnormal splicing, thereby increasing wild-type transcript levels and enhancing dysferlin protein expression in treated cells [6]. Additionally, multi-exon skipping strategies have been evaluated for dysferlinopathies, particularly to bypass pathogenic variants in asymmetrical exons. PMO cocktails targeting exons 28 and 29 successfully induced the skipping of both exons, resulting in a truncated but functional dysferlin protein that restored membrane-resealing capabilities in patient cells [97]. These promising findings indicate that ASOs can affectively promote exon skipping to rescue dysferlin production and restore cellular function. However, further preclinical and clinical studies are essential to translate these advances into viable therapies for this currently underserved patient population.
A critical challenge associated with the effectiveness of these approaches is the possibility that certain exons contain essential regions, and their exclusion may result in a non-functional or improperly folded protein unable to restore its function [87,93]. For instance, while skipping exon 25 maintains the reading frame, it still produces an unstable product that fails to yield a functional protein [94]. Additionally, plasma-membrane-resealing assays have identified exons 19 to 21, 20 to 21, and 46 to 48 as crucial regions for dysferlin-associated membrane repair [95]. Thus, excluding these regions is unlikely to improve symptoms in patients. When feasible, analyzing previously reported in-frame large deletions in patient populations related to the target regions for exclusion may help predict the functionality of the truncated protein generated through exon skipping.
4. The Theoretical Applicability of ASO-Mediated Exon Skipping for the Treatment of Dysferlinopathies
Currently, there are no curative therapies for dysferlinopathies, and existing treatments are primarily focused on managing disease symptoms [2]. This review aims to provide a comprehensive overview of potential exon skipping therapies targeting known pathogenic DYSF variants recorded in the UMD-DYSF database [8]. Given that these therapies are variant-specific, a deeper understanding of each strategy’s applicability to the broader patient population can help guide research efforts toward the most impactful and widely applicable approaches.
4.1. Pathogenic Variants Associated with Dysferlinopathies
Variant analyses reveal a diverse spectrum of disease-causing pathogenic variants throughout the DYSF gene that lead to the complete absence of functional dysferlin in patient muscle tissues [100]. Data from various patient cohorts indicate that most cases involve missense and frameshift variants; however, unlike many genetic disorders, there are no identified mutational hotspots, regions where pathogenic variants frequently occur [8]. Interestingly, seven distinct founder variants have been reported [8]. A study of Portuguese patients from the northern interior region revealed that all seven patients examined carried a missense variant linked to a common founder effect, resulting in exon 49 skipping [101]. Additionally, some studies suggest that pathogenic variants may cluster within certain regions of the DYSF protein in specific populations [102,103]. For example, research on Chinese patients with dysferlinopathies identified a concentration of pathogenic variants within the N-terminal region of the gene, particularly between the C2C and C2B domains [103].
The UMD-DYSF database analyzed in this study is composed of 1174 pathogenic variants, of which 962 are reported in patients affected with dysferlinopathies and 212 are reported in relatives of the affected patients [8]. Only pathogenic variants reported in probands were evaluated during analysis. Of the 962 reported pathogenic variants in patients, 345 (35.9%) were associated with LGMDR2, 342 (35.6%) with MMD, 176 (18.3%) with LGMDR2 or Miyoshi myopathy, 38 (3.9%) with proximodistal phenotype, 20 (2.1%) with isolated hyperCKaemia, 11 (1.1%) with distal myopathy with anterior tibial onset (DMAT), 8 (0.8%) with pseudometabolic disease, 6 (0.6%) with exercise intolerance, 2 (0.2%) with congenital muscular dystrophy, 2 (0.2%) with rigid spine syndrome, 2 (0.2%) with stiffness after exercise, 2 (0.2%) with trunk and lower limb girdle stiffness, and 8 (0.8%) with an unknown diagnosis. Point mutations make up 54%, insertions/deletions make up 29%, and intronic variants make up 17% of the reported patient pathogenic variants in the UMD-DYSF database (Table 1). Among the point mutations, 49% are nonsense variants, while 51% are missense variants.

Table 1.
The frequency of disease-causing mutations in the DYSF gene as reported in the UMD-DYSF database.
4.2. Theoretical Applicability of Exon Skipping for Dysferlinopathies
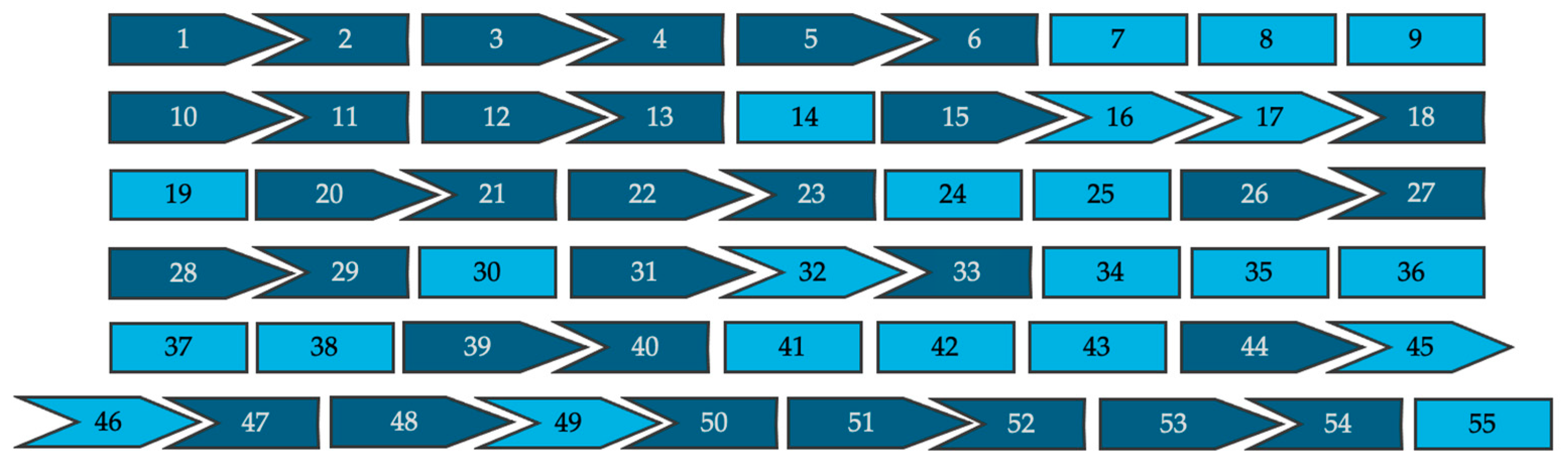
All 962 pathogenic variants reported in affected patients were evaluated for the theoretical applicability of single- and double-exon skipping strategies. These approaches were evaluated for their ability either to exclude intra-exonic disease-causing variants without disrupting the reading frame or to restore reading frame disruptions caused by intronic splice site variants. Splice site variants located nearest the 5′ end of the exon were presumed to result in skipping of the downstream exon and variants nearest the 3′ end were presumed to result in skipping of the upstream exon. Figure 2 is a schematic representation of the 55 exons that make up dysferlin proteins, and their associated intron phase, which can be used as a tool to visualize how the exon’s reading frame will fit together following exon skipping.
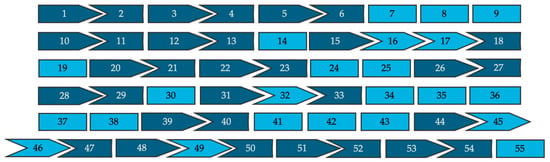
Figure 2.
A visual representation of all 55 exons of the full-length dysferlin transcript and their respective intron phases (not to scale). Symmetrical exons are shown in light blue and asymmetrical exons are shown in dark blue.
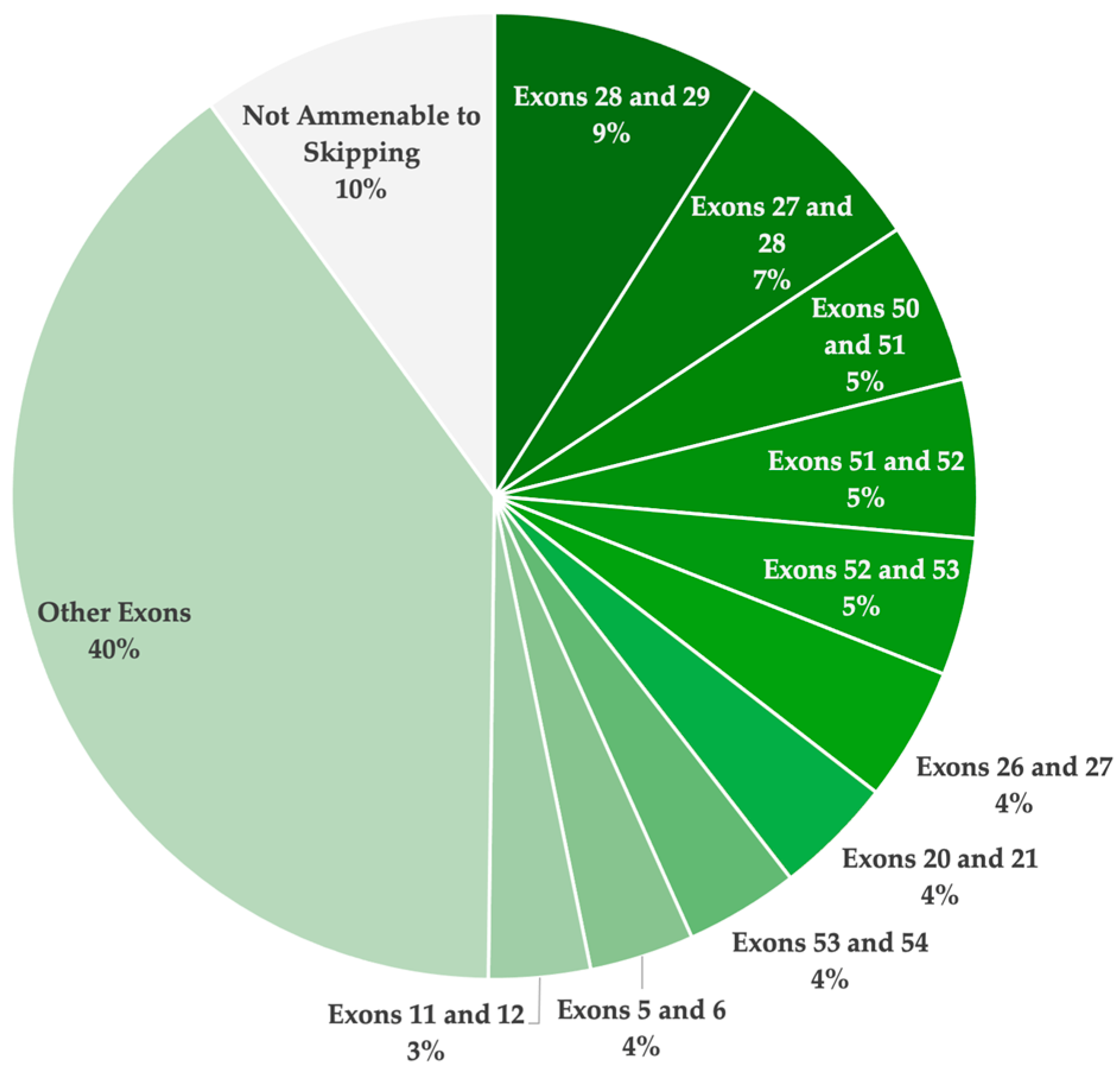
Overall, 61 single- and double-exon skipping strategies were identified to be applicable for the UMD-DYSF patient population (Table 2). Of the 61 strategies, 39 involve skipping a single exon to exclude the pathogenic variants, and 22 of the strategies involve skipping two exons to exclude the variants and maintain the open reading frame. Due to the high distribution of disease-causing variants reported across the DYSF gene, there are no strategies that were found to be applicable for more than 9.0% of the patient population. The highest ranked exon skipping strategies involved exon 28 and 29 (9.0%), exon 27 and 28 (6.7%), and exon 50 and 51 (5.4%). Specifically, for single-exon skipping strategies, the most applicable targets identified include exon 11 (2.9%), exon 24 (2.8%), exon 37 (2.6%), exon 25 (2.6%), and exon 19 (2.6%).
Table 2.
An overview of the applicability of all single- and double-exon skipping approaches for all pathogenic variants reported in patients with dysferlinopathies in the UMD-DYSF database. Each exon skipping strategy was evaluated and ranked based on its potential applicability to the overall patient population analyzed. Exons previously validated in vitro as effective targets for ASO-mediated exon skipping are highlighted in bold. Exons previously identified to contain critical regions essential to dysferlin protein function, which are predicted to yield a non-functional product if skipped, are marked with an asterisk (*).
Many of the pathogenic variants reported were determined to have multiple different applicable strategies that have the potential to exclude the pathogenic variant and maintain or restore the open reading frame. For example, the c.2077delC variant in exon 22, which leads to a premature termination stop codon, can be excluded by targeting exons 21 and 22, or exons 22 and 23. If either of these strategies is translated to the clinic, the other strategy would become less applicable to the total population as a theoretically equally effective therapy would already be accessible to these patients.
Overall, exon skipping was found to be applicable for 90.0% of the reported pathogenic DYSF variants in affected patients, as reported in the UMD-DYSF database (Table 3). Specifically, single-exon skipping has the potential to restore functional dystrophin expression for 44.6% of dysferlinopathy patients. Targeting two exons for exclusion was determined to be applicable for a further 45.3% of patients. The remaining 10% of pathogenic variants were not amenable to single- or double-exon skipping. These included splice site variants leading to the exclusion of an in-frame exon and pathogenic variants located in the first or last exon which cannot be addressed by exon skipping. They also included missense variants in exons 15, 18, 31, 33, and 44, which could potentially be excluded through multi-exon skipping (involving more than two exons) to maintain the reading frame.
Table 3.
A summary of the applicability of single- and double-exon skipping for all 962 pathogenic variants reported in patients in the UMD-DYSF database.
4.3. The Clinical Translation of ASO-Mediated Exon Skipping for the Treatment of Dysferlinopathies
Although numerous strategies were identified to be theoretically applicable for the dysferlinopathy patient population, as reported in the UMD-DYSF database, none of these therapies have yet to receive FDA approval. In total, 61 strategies (39 single- and 22 double-exon skipping) were identified to be applicable. Figure 3 illustrates the total theoretical applicability of single- and double-exon skipping for the treatment of dysferlinopathies, highlighting the top ten exon skipping targets. A total of 44.6% of the reported pathogenic variants were determined to be amenable through single-exon skipping. Furthermore, 90.0% of the patient population was determined to have a pathogenic variant with the potential to be excluded through either single- or double-exon skipping. These findings confirm the incredible potential of ASO-mediated exon skipping to treat most patients with pathogenic DYSF variants resulting in disease.
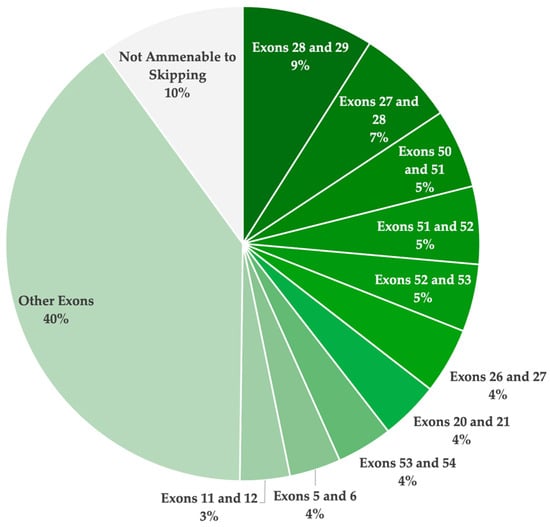
Figure 3.
A pie chart illustrating the overall theoretical applicability of single- and double-exon skipping to exclude pathogenic mutations and/or restore the reading frame, resulting in a truncated yet potentially functional dysferlin protein. This analysis is based on the pathogenic variants reported in the UMD-DYSF database. The ten most highly applicable targets are highlighted.
Despite the identification of numerous promising exon skipping strategies for dysferlinopathies, most approaches remain theoretical, as only a few have been evaluated in preclinical studies. As previously discussed, skipping certain critical regions of dysferlin may fail to improve the phenotype. All proposed strategies require comprehensive evaluation in vitro and/or in animal models before clinical translation [104]. While patient-derived myotubes are the most relevant cell type for assessing therapeutic effectiveness due to the therapy’s direct relevance to muscle pathology [7], fibroblasts from dysferlinopathy patients have also demonstrated utility in evaluating membrane repair capacity following ASO treatment [97]. Given the variant-specific nature of ASOs, ideal animal models would harbor variants amenable to the strategy under study. For example, MMex38 mice, which carry an exon 38 variant, were used to evaluate the efficacy of U7 small nuclear RNA-mediated skipping of exons 37 and 38 [105].
Among the most promising strategies identified for dysferlinopathies are double-exon skipping approaches, which would require a combination, or “cocktail”, of ASOs. However, these multi-exon skipping approaches face additional hurdles for clinical translation, as each ASO, along with their combined use, may necessitate individual toxicology testing, significantly increasing both time and costs associated with their development [106,107]. As a result, reforms in the drug approval process could potentially streamline their path to clinical use [85].
Although several ASOs have received FDA approval for other genetic disorders, as previously discussed, their cellular uptake in patients remains suboptimal, often requiring high-dose frequent injections to achieve therapeutic effect [108]. Depending on the delivery route, ASOs must transverse multiple biological barriers to reach target tissues and influence RNA [109]. To enhance their clinical translation potential, current preclinical studies are investigating systems to optimize ASO delivery and cellular uptake. Lipid-based nanoparticles (LNPs) have shown promise in enhancing ASO stability in the extracellular environment and promoting cellular uptake [110,111]. Other carriers under investigation include extracellular vesicles [112], dendrimers [113,114], metallic nanoparticles [115], and cell-penetrating peptides (CPPs) [109]. Notably, CPPs, which can be conjugated to neutrally charged PMOs, have received particular interest due to their ability to significantly increase ASO uptake in target tissues, where ASOs previously struggled to reach [116]. Continued investigation into delivery systems that can improve ASO efficacy at a reduced dose is expected to improve their potential for clinical translation [117].
5. Conclusions
Given the current lack of disease-modifying therapies for dysferlinopathies, ASO-mediated exon skipping has shown potential in bypassing disease-causing pathogenic variants to generate a truncated dysferlin protein with enhanced functionality [97]. This review identified numerous single- and double-exon skipping targets that could theoretically benefit 90% of patients with dysferlinopathy. However, further research is necessary to confirm whether the truncated proteins produced through these strategies are indeed functional before progressing to clinical trials [96]. Additionally, advancements in ASO chemical modifications, the development of delivery systems to enhance cellular uptake, and reforms to the drug approval process could significantly improve the potential for these therapies to become accessible to patients.
Author Contributions
J.L. and S.H.R.: writing—original draft preparation; T.Y. and M.K.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
T.Y. is supported by the Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, the Canadian Institute of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, the Women and Children’s Health Research Institute (WCHRI), the Heart and Stroke Foundation Canada, and the US Department of Defense. J.L. is supported by scholarships from the University of Alberta Faculty of Medicine and Dentistry and the CIHR Strategic Master’s Award.
Data Availability Statement
The dataset analyzed in this study is available in the UMD-DMD database at http://www.umd.be/DYSF/. The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
We would like to express sincere gratitude to Christophe Béroud and Martin Krahn, curators of the UMD-DYSF database, for their invaluable efforts in maintaining this essential resource, which was instrumental to our study. We also would like to thank the University of Alberta Faculty of Medicine and Dentistry, the Canadian Institute of Health Research, the Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, and the Women and Children’s Health Research Institute for their support.
Conflicts of Interest
T.Y. is a cofounder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. All authors declare that the research was conducted in absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Anwar, S.; He, M.; Lim, K.R.Q.; Maruyama, R.; Yokota, T. A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis. J. Pers. Med. 2021, 11, 46. [Google Scholar] [CrossRef]
- Bouchard, C.; Tremblay, J.P. Portrait of Dysferlinopathy: Diagnosis and Development of Therapy. J. Clin. Med. 2023, 12, 6011. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon Skipping for Nonsense Mutations in Duchenne Muscular Dystrophy: Too Many Mutations, Too Few Patients? Expert. Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef]
- Quemener, A.M.; Centomo, M.L.; Sax, S.L.; Panella, R. Small Drugs, Huge Impact: The Extraordinary Impact of Antisense Oligonucleotides in Research and Drug Development. Molecules 2022, 27, 536. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Dominov, J.A.; Uyan, O.; Sapp, P.C.; McKenna-Yasek, D.; Nallamilli, B.R.R.; Hegde, M.; Brown, R.H. A Novel Dysferlin Mutant Pseudoexon Bypassed with Antisense Oligonucleotides. Ann. Clin. Transl. Neurol. 2014, 1, 703–720. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, F.; Blouin, C.; Wein, N.; Mouly, V.; Courrier, S.; Dionnet, E.; Kergourlay, V.; Mathieu, Y.; Garcia, L.; Butler-Browne, G.; et al. Exon 32 Skipping of Dysferlin Rescues Membrane Repair in Patients’ Cells. J. Neuromuscul. Dis. 2015, 2, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Blandin, G.; Beroud, C.; Labelle, V.; Nguyen, K.; Wein, N.; Hamroun, D.; Williams, B.; Monnier, N.; Rufibach, L.E.; Urtizberea, J.A.; et al. UMD-DYSF, a Novel Locus Specific Database for the Compilation and Interactive Analysis of Mutations in the Dysferlin Gene. Hum. Mutat. 2012, 33, E2317–E2331. [Google Scholar] [CrossRef] [PubMed]
- Cacciottolo, M.; Numitone, G.; Aurino, S.; Caserta, I.R.; Fanin, M.; Politano, L.; Minetti, C.; Ricci, E.; Piluso, G.; Angelini, C.; et al. Muscular Dystrophy with Marked Dysferlin Deficiency Is Consistently Caused by Primary Dysferlin Gene Mutations. Eur. J. Hum. Genet. 2011, 19, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Cheng, Q.; Chen, X.; Yu, Q.; Li, Z. Abnormal Expression of Dysferlin in Blood Monocytes Supports Primary Dysferlinopathy in Patients Confirmed by Genetic Analyses. Front. Neurol. 2020, 11, 540098. [Google Scholar] [CrossRef]
- Quinn, C.J.; Cartwright, E.J.; Trafford, A.W.; Dibb, K.M. On the Role of Dysferlin in Striated Muscle: Membrane Repair, t-Tubules and Ca2+ Handling. J. Physiol. 2024, 602, 1893–1910. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Redpath, G.M.I.; Woolger, N.; Piper, A.K.; Lemckert, F.A.; Lek, A.; Greer, P.A.; North, K.N.; Cooper, S.T. Calpain Cleavage within Dysferlin Exon 40a Releases a Synaptotagmin-like Module for Membrane Repair. Mol. Biol. Cell 2014, 25, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, M.J.; McCord, J.J.; Sutton, R.B. Redefining the Architecture of Ferlin Proteins: Insights into Multi-Domain Protein Structure and Function. PLoS ONE 2022, 17, e0270188. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Davletov, B.A.; Sutton, R.B.; Südhof, T.C.; Rizo, J. Bipartite Ca2+-Binding Motif in C2 Domains of Synaptotagmin and Protein Kinase C. Science 1996, 273, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Therrien, C.; Di Fulvio, S.; Pickles, S.; Sinnreich, M. Characterization of Lipid Binding Specificities of Dysferlin C2 Domains Reveals Novel Interactions with Phosphoinositides. Biochemistry 2009, 48, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Klinge, L.; Laval, S.; Keers, S.; Haldane, F.; Straub, V.; Barresi, R.; Bushby, K. From T-Tubule to Sarcolemma: Damage-Induced Dysferlin Translocation in Early Myogenesis. FASEB J. 2007, 21, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Campbell, K.P. Dysferlin and Muscle Membrane Repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- McDade, J.R.; Michele, D.E. Membrane Damage-Induced Vesicle-Vesicle Fusion of Dysferlin-Containing Vesicles in Muscle Cells Requires Microtubules and Kinesin. Hum. Mol. Genet. 2014, 23, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Klinge, L.; Harris, J.; Sewry, C.; Charlton, R.; Anderson, L.; Laval, S.; Chiu, Y.-H.; Hornsey, M.; Straub, V.; Barresi, R.; et al. Dysferlin Associates with the Developing T-Tubule System in Rodent and Human Skeletal Muscle. Muscle Nerve 2010, 41, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hofhuis, J.; Bersch, K.; Büssenschütt, R.; Drzymalski, M.; Liebetanz, D.; Nikolaev, V.O.; Wagner, S.; Maier, L.S.; Gärtner, J.; Klinge, L.; et al. Dysferlin Mediates Membrane Tubulation and Links T-Tubule Biogenesis to Muscular Dystrophy. J. Cell Sci. 2017, 130, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.P.; Ziman, A.P.; Mueller, A.L.; Muriel, J.M.; Kleinhans-Welte, E.; Gumerson, J.D.; Vogel, S.S.; Ward, C.W.; Roche, J.A.; Bloch, R.J. Dysferlin Stabilizes Stress-Induced Ca2+ Signaling in the Transverse Tubule Membrane. Proc. Natl. Acad. Sci. USA 2013, 110, 20831–20836. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.P.; Ward, C.W.; Bloch, R.J. Dysferlin at Transverse Tubules Regulates Ca2+ Homeostasis in Skeletal Muscle. Front. Physiol. 2014, 5, 89. [Google Scholar] [CrossRef]
- Krajacic, P.; Pistilli, E.E.; Tanis, J.E.; Khurana, T.S.; Lamitina, S.T. FER-1/Dysferlin Promotes Cholinergic Signaling at the Neuromuscular Junction in C. Elegans and Mice. Biol. Open 2013, 2, 1245–1252. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome Up-Regulation and Activation in Dysferlin-Deficient Skeletal Muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Han, R.; Bansal, D.; Miyake, K.; Muniz, V.P.; Weiss, R.M.; McNeil, P.L.; Campbell, K.P. Dysferlin-Mediated Membrane Repair Protects the Heart from Stress-Induced Left Ventricular Injury. J. Clin. Investig. 2007, 117, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Hofhuis, J.; Bersch, K.; Wagner, S.; Molina, C.; Fakuade, F.E.; Iyer, L.M.; Streckfuss-Bömeke, K.; Toischer, K.; Zelarayán, L.C.; Voigt, N.; et al. Dysferlin Links Excitation-Contraction Coupling to Structure and Maintenance of the Cardiac Transverse-Axial Tubule System. EP Eur. 2020, 22, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Frett, E.M.; Levy, J.R.; Rader, E.P.; Lueck, J.D.; Bansal, D.; Moore, S.A.; Ng, R.; Beltrán-Valero de Bernabé, D.; Faulkner, J.A.; et al. Genetic Ablation of Complement C3 Attenuates Muscle Pathology in Dysferlin-Deficient Mice. J. Clin. Investig. 2010, 120, 4366–4374. [Google Scholar] [CrossRef]
- Han, R. Muscle Membrane Repair and Inflammatory Attack in Dysferlinopathy. Skelet. Muscle 2011, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, E.; Rojas-García, R.; de Luna, N.; Pou, A.; Brown, R.H.; Illa, I. Inflammation in Dysferlin Myopathy: Immunohistochemical Characterization of 13 Patients. Neurology 2001, 57, 2136–2138. [Google Scholar] [CrossRef]
- Rekik, S.; Sakka, S.; Romdhane, S.B.; Amer, Y.B.; Lehkim, L.; Farhat, N.; Mahfoudh, K.B.; Authier, F.J.; Dammak, M.; Mhiri, C. Novel Splicing Dysferlin Mutation Causing Myopathy with Intra-Familial Heterogeneity. Mol. Biol. Rep. 2020, 47, 5755–5761. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Eulate, G.; Querin, G.; Moore, U.; Behin, A.; Masingue, M.; Bassez, G.; Leonard-Louis, S.; Laforêt, P.; Maisonobe, T.; Merle, P.-E.; et al. Deep Phenotyping of an International Series of Patients with Late-Onset Dysferlinopathy. Eur. J. Neurol. 2021, 28, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Folland, C.; Johnsen, R.; Botero Gomez, A.; Trajanoski, D.; Davis, M.R.; Moore, U.; Straub, V.; Barresi, R.; Guglieri, M.; Hayhurst, H.; et al. Identification of a Novel Heterozygous DYSF Variant in a Large Family with a Dominantly-Inherited Dysferlinopathy. Neuropathol. Appl. Neurobiol. 2022, 48, e12846. [Google Scholar] [CrossRef] [PubMed]
- Illa, I.; Serrano-Munuera, C.; Gallardo, E.; Lasa, A.; Rojas-García, R.; Palmer, J.; Gallano, P.; Baiget, M.; Matsuda, C.; Brown, R.H. Distal Anterior Compartment Myopathy: A Dysferlin Mutation Causing a New Muscular Dystrophy Phenotype. Ann. Neurol. 2001, 49, 130–134. [Google Scholar] [CrossRef]
- Ivanova, A.; Smirnikhina, S.; Lavrov, A. Dysferlinopathies: Clinical and Genetic Variability. Clin. Genet. 2022, 102, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Tremblay, J.P. Limb-Girdle Muscular Dystrophies Classification and Therapies. J. Clin. Med. 2023, 12, 4769. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nagata, T.; Yokota, T.; Nakamura, A.; Wood, M.J.A.; Partridge, T.; Takeda, S. Highly Efficient in vivo Delivery of PMO into Regenerating Myotubes and Rescue in Laminin-A2 Chain-Null Congenital Muscular Dystrophy Mice. Hum. Mol. Genet. 2013, 22, 4914–4928. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Takahashi, T. Mutational and clinical features of Japanese patients with dysferlinopathy (Miyoshi myopathy and limb girdle muscular dystrophy type 2B). Rinsho Shinkeigaku Clin. Neurol. 2005, 45, 938–942. [Google Scholar]
- Nguyen, K.; Bassez, G.; Bernard, R.; Krahn, M.; Labelle, V.; Figarella-Branger, D.; Pouget, J.; Hammouda, E.H.; Béroud, C.; Urtizberea, A.; et al. Dysferlin Mutations in LGMD2B, Miyoshi Myopathy, and Atypical Dysferlinopathies. Hum. Mutat. 2005, 26, 165. [Google Scholar] [CrossRef]
- Urtizberea, J.A.; Bassez, G.; Leturcq, F.; Nguyen, K.; Krahn, M.; Levy, N. Dysferlinopathies. Neurol. India 2008, 56, 289–297. [Google Scholar] [CrossRef]
- Miyoshi, K.; Kawai, H.; Iwasa, M.; Kusaka, K.; Nishino, H. Autosomal Recessive Distal Muscular Dystrophy as a New Type of Progressive Muscular Dystrophy. Seventeen Cases in Eight Families Including an Autopsied Case. Brain J. Neurol. 1986, 109, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Galassi, G.; Rowland, L.P.; Hays, A.P.; Hopkins, L.C.; Dimauro, S. High Serum Levels of Creatine Kinase: Asymptomatic Prelude to Distal Myopathy. Muscle Nerve 1987, 10, 346–350. [Google Scholar] [CrossRef]
- Umakhanova, Z.R.; Bardakov, S.N.; Mavlikeev, M.O.; Chernova, O.N.; Magomedova, R.M.; Akhmedova, P.G.; Yakovlev, I.A.; Dalgatov, G.D.; Fedotov, V.P.; Isaev, A.A.; et al. Twenty-Year Clinical Progression of Dysferlinopathy in Patients from Dagestan. Front. Neurol. 2017, 8, 77. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Progress and Challenges in Diagnosis of Dysferlinopathy. Muscle Nerve 2016, 54, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Argov, Z.; Sadeh, M.; Mazor, K.; Soffer, D.; Kahana, E.; Eisenberg, I.; Mitrani-Rosenbaum, S.; Richard, I.; Beckmann, J.; Keers, S.; et al. Muscular Dystrophy Due to Dysferlin Deficiency in Libyan Jews. Clinical and Genetic Features. Brain J. Neurol. 2000, 123, 1229–1237. [Google Scholar] [CrossRef]
- Vilchez, J.J.; Gallano, P.; Gallardo, E.; Lasa, A.; Rojas-García, R.; Freixas, A.; De Luna, N.; Calafell, F.; Sevilla, T.; Mayordomo, F.; et al. Identification of a Novel Founder Mutation in the DYSF Gene Causing Clinical Variability in the Spanish Population. Arch. Neurol. 2005, 62, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Aoki, M.; Tateyama, M.; Kondo, E.; Mizuno, T.; Onodera, Y.; Takano, R.; Kawai, H.; Kamakura, K.; Mochizuki, H.; et al. Dysferlin Mutations in Japanese Miyoshi Myopathy: Relationship to Phenotype. Neurology 2003, 60, 1799–1804. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Bushby, K. Therapeutic Possibilities in the Autosomal Recessive Limb-Girdle Muscular Dystrophies. Neurother. J. Am. Soc. Exp. Neurother. 2008, 5, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Poudel, B.H.; Fletcher, S.; Wilton, S.D.; Aung-Htut, M. Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities. Int. J. Mol. Sci. 2024, 25, 5572. [Google Scholar] [CrossRef] [PubMed]
- Lostal, W.; Bartoli, M.; Bourg, N.; Roudaut, C.; Bentaïb, A.; Miyake, K.; Guerchet, N.; Fougerousse, F.; McNeil, P.; Richard, I. Efficient Recovery of Dysferlin Deficiency by Dual Adeno-Associated Vector-Mediated Gene Transfer. Hum. Mol. Genet. 2010, 19, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, P.C.; Griffin, D.A.; Pozsgai, E.R.; Johnson, R.W.; Grose, W.E.; Heller, K.N.; Shontz, K.M.; Montgomery, C.L.; Liu, J.; Clark, K.R.; et al. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015, 2, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Llanga, T.; Nagy, N.; Conatser, L.; Dial, C.; Sutton, R.B.; Hirsch, M.L. Structure-Based Designed Nano-Dysferlin Significantly Improves Dysferlinopathy in BLA/J Mice. Mol. Ther. 2017, 25, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise Correction of Disease Mutations in Induced Pluripotent Stem Cells Derived From Patients With Limb Girdle Muscular Dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef]
- Mou, H.; Smith, J.L.; Peng, L.; Yin, H.; Moore, J.; Zhang, X.-O.; Song, C.-Q.; Sheel, A.; Wu, Q.; Ozata, D.M.; et al. CRISPR/Cas9-Mediated Genome Editing Induces Exon Skipping by Alternative Splicing or Exon Deletion. Genome Biol. 2017, 18, 108. [Google Scholar] [CrossRef]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-Existing Anti-Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Pogoda, J.M.; Provost, R.; Guerrero, J.; Hajjar, R.J.; Zsebo, K.M. Prevalence of AAV1 Neutralizing Antibodies and Consequences for a Clinical Trial of Gene Transfer for Advanced Heart Failure. Gene Ther. 2016, 23, 313–319. [Google Scholar] [CrossRef]
- Weber, T. Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front. Immunol. 2021, 12, 658399. [Google Scholar] [CrossRef]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.L.; Gao, G. Overcoming Innate Immune Barriers That Impede AAV Gene Therapy Vectors. J. Clin. Investig. 2021, 131, e143780. [Google Scholar] [CrossRef] [PubMed]
- Batty, P.; Lillicrap, D. Adeno-Associated Viral Vector Integration: Implications for Long-Term Efficacy and Safety. J. Thromb. Haemost. 2024, 22, 2945–2960. [Google Scholar] [CrossRef] [PubMed]
- Lek, A.; Wong, B.; Keeler, A.; Blackwood, M.; Ma, K.; Huang, S.; Sylvia, K.; Batista, A.R.; Artinian, R.; Kokoski, D.; et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 389, 1203–1210. [Google Scholar] [CrossRef]
- Shen, W.; Liu, S.; Ou, L. rAAV Immunogenicity, Toxicity, and Durability in 255 Clinical Trials: A Meta-Analysis. Front. Immunol. 2022, 13, 1001263. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The Powerful World of Antisense Oligonucleotides: From Bench to Bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.W.M.P.; Aartsma-Rus, A. Opportunities and Challenges for Antisense Oligonucleotide Therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense Oligonucleotides: A Primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef]
- Sang, A.; Zhuo, S.; Bochanis, A.; Manautou, J.E.; Bahal, R.; Zhong, X.-B.; Rasmussen, T.P. Mechanisms of Action of the US Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. BioDrugs 2024, 38, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-H.; Sun, H.; Shen, W.; Wang, S.; Yao, J.; Migawa, M.T.; Bui, H.-H.; Damle, S.S.; Riney, S.; Graham, M.J.; et al. Antisense Oligonucleotides Targeting Translation Inhibitory Elements in 5′ UTRs Can Selectively Increase Protein Levels. Nucleic Acids Res. 2017, 45, 9528–9546. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense Oligonucleotides: The next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, M.C.; van Roon-Mom, W.; Aartsma-Rus, A. Possibilities and Limitations of Antisense Oligonucleotide Therapies for the Treatment of Monogenic Disorders. Commun. Med. 2024, 4, 6. [Google Scholar] [CrossRef]
- Lin, M.; Hu, X.; Chang, S.; Chang, Y.; Bian, W.; Hu, R.; Wang, J.; Zhu, Q.; Qiu, J. Advances of Antisense Oligonucleotide Technology in the Treatment of Hereditary Neurodegenerative Diseases. Evid. Based Complement. Altern. Med. 2021, 2021, 6678422. [Google Scholar] [CrossRef]
- Hwang, J.; Yokota, T. Recent Advancements in Exon-Skipping Therapies Using Antisense Oligonucleotides and Genome Editing for the Treatment of Various Muscular Dystrophies. Expert. Rev. Mol. Med. 2019, 21, e5. [Google Scholar] [CrossRef]
- Matsuo, M. Antisense Oligonucleotide-Mediated Exon-Skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021, 4, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides as Therapeutic Drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Niks, E.H.; Aartsma-Rus, A. Exon Skipping: A First in Class Strategy for Duchenne Muscular Dystrophy. Expert. Opin. Biol. Ther. 2017, 17, 225–236. [Google Scholar] [CrossRef]
- Michaels, W.E.; Pena-Rasgado, C.; Kotaria, R.; Bridges, R.J.; Hastings, M.L. Open Reading Frame Correction Using Splice-Switching Antisense Oligonucleotides for the Treatment of Cystic Fibrosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2114886119. [Google Scholar] [CrossRef]
- Siva, K.; Covello, G.; Denti, M.A. Exon-Skipping Antisense Oligonucleotides to Correct Missplicing in Neurogenetic Diseases. Nucleic Acid. Ther. 2014, 24, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Adkin, C.F.; Meloni, P.L.; Fletcher, S.; Adams, A.M.; Muntoni, F.; Wong, B.; Wilton, S.D. Multiple Exon Skipping Strategies to By-Pass Dystrophin Mutations. Neuromuscul. Disord. 2012, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Carmel, L.; Csuros, M.; Koonin, E.V. Origin and Evolution of Spliceosomal Introns. Biol. Direct 2012, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, S.; de Gusmao, C.M.; Zhao, B.; Chin, D.H.; DiDonato, R.L.; Nguyen, M.A.; Nakayama, T.; Hu, C.A.; Soucy, A.; et al. A Framework for Individualized Splice-Switching Oligonucleotide Therapy. Nature 2023, 619, 828–836. [Google Scholar] [CrossRef]
- Aslesh, T.; Maruyama, R.; Yokota, T. Skipping Multiple Exons to Treat DMD-Promises and Challenges. Biomedicines 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A. Moving towards Successful Exon-Skipping Therapy for Duchenne Muscular Dystrophy. J. Hum. Genet. 2017, 62, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. Recent Trends in Antisense Therapies for Duchenne Muscular Dystrophy. Pharmaceutics 2023, 15, 778. [Google Scholar] [CrossRef] [PubMed]
- Echevarría, L.; Aupy, P.; Goyenvalle, A. Exon-Skipping Advances for Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal Effect of Eteplirsen versus Historical Control on Ambulation in Duchenne Muscular Dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Sheikh, O.; Yokota, T. Pharmacology and Toxicology of Eteplirsen and SRP-5051 for DMD Exon 51 Skipping: An Update. Arch. Toxicol. 2022, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dzierlega, K.; Yokota, T. Optimization of Antisense-Mediated Exon Skipping for Duchenne Muscular Dystrophy. Gene Ther. 2020, 27, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Béroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.-P.; Voelckel, M.-A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon Skipping Leading to an Artificial DMD Protein Lacking Amino Acids from Exons 45 through 55 Could Rescue up to 63% of Patients with Duchenne Muscular Dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takeda, S. Exon-Skipping Therapy for Duchenne Muscular Dystrophy. Neuropathology 2009, 29, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Aoki, Y.; Miskew, B.; Panesar, D.; Touznik, A.; Nagata, T.; Tanihata, J.; Nakamura, A.; Nagaraju, K.; Yokota, T. Long-Term Efficacy of Systemic Multiexon Skipping Targeting Dystrophin Exons 45-55 with a Cocktail of Vivo-Morpholinos in Mdx52 Mice. Mol. Ther. Nucleic Acids 2015, 4, e225. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Singh, K.H.K.; Fokkema, I.F.A.C.; Ginjaar, I.B.; van Ommen, G.-J.; den Dunnen, J.T.; van der Maarel, S.M. Therapeutic Exon Skipping for Dysferlinopathies? Eur. J. Hum. Genet. 2010, 18, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Wein, N.; Avril, A.; Bartoli, M.; Beley, C.; Chaouch, S.; Laforêt, P.; Behin, A.; Butler-Browne, G.; Mouly, V.; Krahn, M.; et al. Efficient Bypass of Mutations in Dysferlin Deficient Patient Cells by Antisense-Induced Exon Skipping. Hum. Mutat. 2010, 31, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.A.; Maruyama, R.; Duddy, W.; Sakurai, H.; Yokota, T. Identification of Novel Antisense-Mediated Exon Skipping Targets in DYSF for Therapeutic Treatment of Dysferlinopathy. Mol. Ther. Nucleic Acids 2018, 13, 596–604. [Google Scholar] [CrossRef]
- Porto, K.J.L.; Mitsui, J.; Ishiura, H.; Kubota, A.; Luspian, K.J.L.; Eduardo, E.; Damian, L.; Toda, T.; Tsuji, S. A Novel Multi-Exon Deletion in the Dysferlin Gene of a Limb-Girdle Muscular Dystrophy Type 2B Filipino Patient. Neurol. Clin. Neurosci. 2020, 8, 419–421. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. The Future of Exon Skipping for Duchenne Muscular Dystrophy. Hum. Gene Ther. 2023, 34, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Klinge, L.; Aboumousa, A.; Eagle, M.; Hudson, J.; Sarkozy, A.; Vita, G.; Charlton, R.; Roberts, M.; Straub, V.; Barresi, R.; et al. New Aspects on Patients Affected by Dysferlin Deficient Muscular Dystrophy. J. Neurol. Neurosurg. Psychiatry 2010, 81, 946–953. [Google Scholar] [CrossRef]
- Santos, R.D.; Duell, P.B.; East, C.; Guyton, J.R.; Moriarty, P.M.; Chin, W.; Mittleman, R.S. Long-Term Efficacy and Safety of Mipomersen in Patients with Familial Hypercholesterolaemia: 2-Year Interim Results of an Open-Label Extension. Eur. Heart J. 2015, 36, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-E.; Kim, H.-S.; Lee, C.-H.; Nam, T.-S.; Choi, Y.-C.; Kim, D.-S. Two Common Mutations (p.Gln832X and c.663+1G>C) Account for about a Third of the DYSF Mutations in Korean Patients with Dysferlinopathy. Neuromuscul. Disord. 2012, 22, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-Q.; Yu, M.; Zhang, W.; Lyu, H.; Yuan, Y.; Wang, Z.-X. Dysferlin Gene Mutation Spectrum in a Large Cohort of Chinese Patients with Dysferlinopathy. Chin. Med. J. 2016, 129, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Egorova, T.V.; Galkin, I.I.; Velyaev, O.A.; Vassilieva, S.G.; Savchenko, I.M.; Loginov, V.A.; Dzhenkova, M.A.; Korshunova, D.S.; Kozlova, O.S.; Ivankov, D.N.; et al. In-Frame Deletion of Dystrophin Exons 8-50 Results in DMD Phenotype. Int. J. Mol. Sci. 2023, 24, 9117. [Google Scholar] [CrossRef] [PubMed]
- Malcher, J.; Heidt, L.; Goyenvalle, A.; Escobar, H.; Marg, A.; Beley, C.; Benchaouir, R.; Bader, M.; Spuler, S.; García, L.; et al. Exon Skipping in a Dysf-Missense Mutant Mouse Model. Mol. Ther. Nucleic Acids 2018, 13, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Yokota, T.; Takeda, S.; Lu, Q.-L.; Partridge, T.A.; Nakamura, A.; Hoffman, E.P. A Renaissance for Antisense Oligonucleotide Drugs in Neurology: Exon Skipping Breaks New Ground. Arch. Neurol. 2009, 66, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Baker, B.F.; Kwoh, T.J.; Cheng, W.; Schulz, D.J.; Xia, S.; Salgado, N.; Bui, H.-H.; Hart, C.E.; Burel, S.A.; et al. Integrated Safety Assessment of 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides in NonHuman Primates and Healthy Human Volunteers. Mol. Ther. 2016, 24, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic Peptide-Mediated Oligonucleotide Therapy Improves Long-Term Survival in Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef]
- Yang, L.; Ma, F.; Liu, F.; Chen, J.; Zhao, X.; Xu, Q. Efficient Delivery of Antisense Oligonucleotides Using Bioreducible Lipid Nanoparticles In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2020, 19, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Pan, Y.; Li, H.; Zhu, Y.; Gao, Y.; Wang, J.; Zhou, Y.; Guan, Z.; Yang, Z. Activity and Tissue Distribution of Antisense Oligonucleotide CT102 Encapsulated with Cytidinyl/Cationic Lipid against Hepatocellular Carcinoma. Mol. Pharm. 2022, 19, 4552–4564. [Google Scholar] [CrossRef] [PubMed]
- Grossen, P.; Portmann, M.; Koller, E.; Duschmalé, M.; Minz, T.; Sewing, S.; Pandya, N.J.; van Geijtenbeek, S.K.; Ducret, A.; Kusznir, E.-A.; et al. Evaluation of Bovine Milk Extracellular Vesicles for the Delivery of Locked Nucleic Acid Antisense Oligonucleotides. Eur. J. Pharm. Biopharm. 2021, 158, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Duong, C.; Oestergaard, M.; Fazio, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. MXD3 Antisense Oligonucleotide with Superparamagnetic Iron Oxide Nanoparticles: A New Targeted Approach for Neuroblastoma. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102127. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Chen, J.; Tai, L.; Liu, C.; Chen, X.; Wei, G.; Lu, W.; Pan, W. Inhibition of Post-Trabeculectomy Fibrosis via Topically Instilled Antisense Oligonucleotide Complexes Co-Loaded with Fluorouracil. Acta Pharm. Sin. B 2020, 10, 1754–1768. [Google Scholar] [CrossRef] [PubMed]
- Beha, M.J.; Ryu, J.S.; Kim, Y.S.; Chung, H.J. Delivery of Antisense Oligonucleotides Using Multi-Layer Coated Gold Nanoparticles to Methicillin-Resistant S. Aureus for Combinatorial Treatment. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 126, 112167. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-Penetrating Peptide-Conjugated Antisense Oligonucleotides Restore Systemic Muscle and Cardiac Dystrophin Expression and Function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).