
A Recombinase-Mediated Cassette Exchange Platform for a Triple Independent Inducible Expression System for Human Pluripotent Stem Cells
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. hPSC Culture and Maintenance
2.2. Genetic Engineering of hPSCs
2.3. hPSC Differentiation into Different Germ Layers
2.4. Endoderm Differentiations Were Carried Out as Previously Described [16]
2.5. Ectoderm Differentiations Were Carried Out as Previously Described [17]
2.6. Mesoderm Differentiations Were Carried Out as Recently Described [18]
2.7. EB Differentiation from hPSCs
2.8. Immunofluorescence Staining
2.9. Flow Cytometry
2.10. RT-qPCR
3. Results
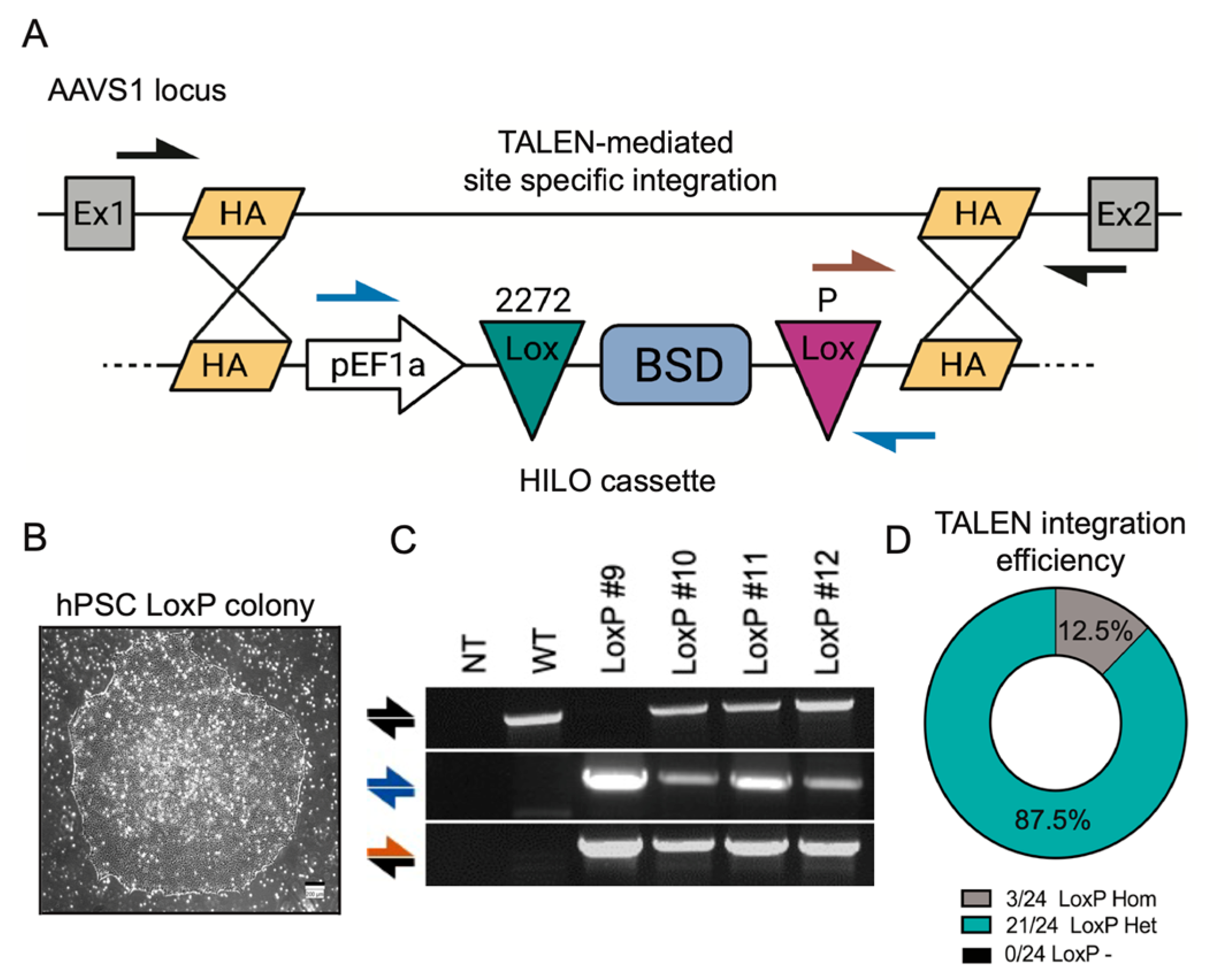
3.1. The Establishment of a RMCE Platform in hPSCs
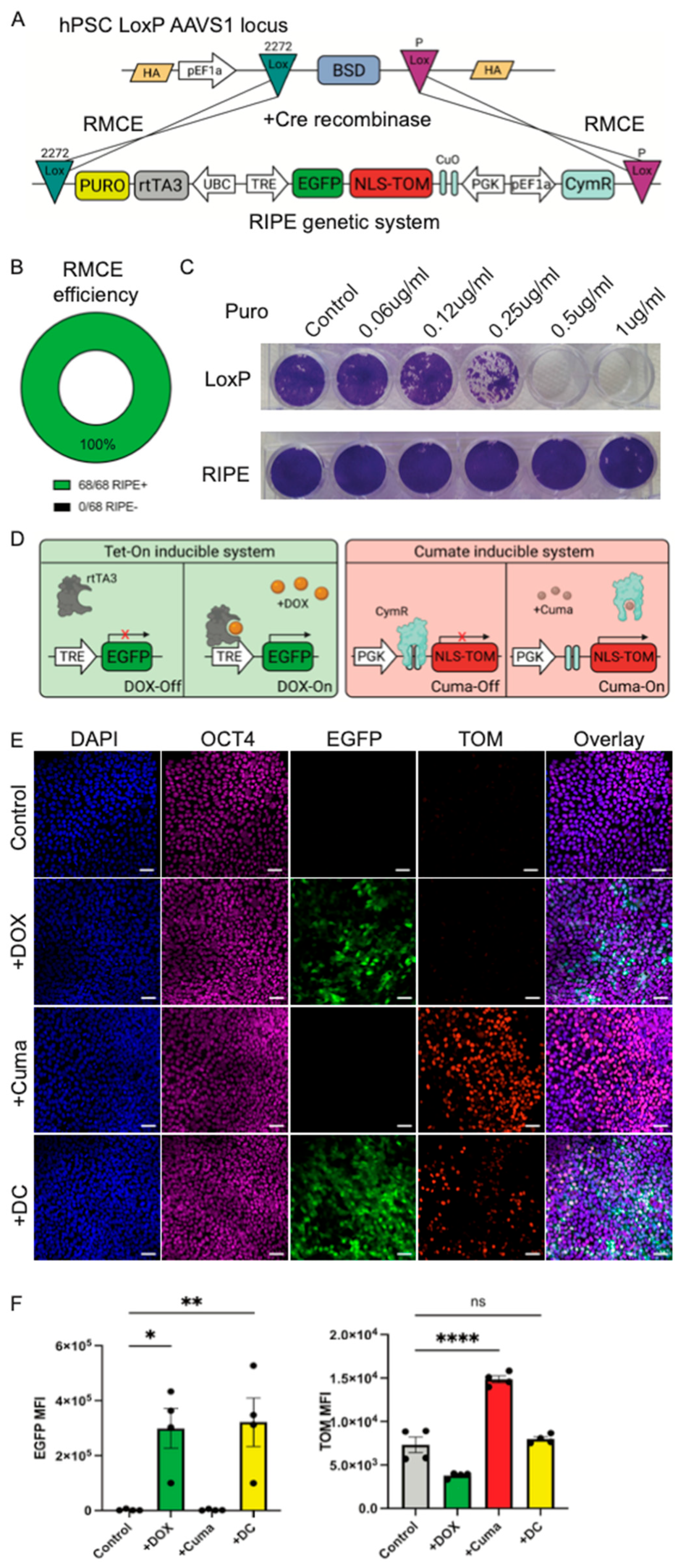
3.2. The Efficient Generation of a Dual Inducible System in hPSCs by RMCE
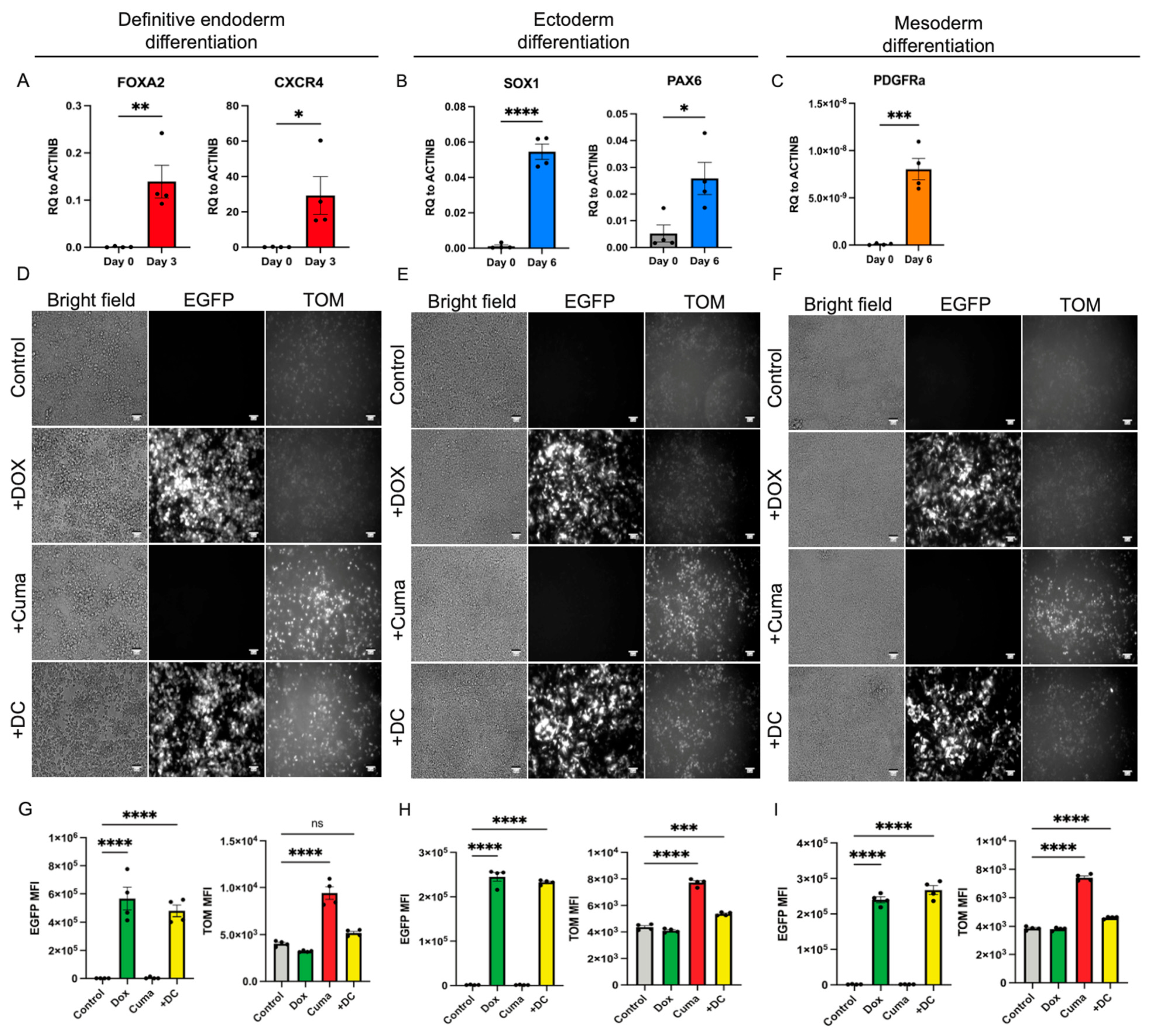
3.3. RIPE System Remains Functional After the Differentiation of the hPSCs into the Cells of the Three Germ Layers
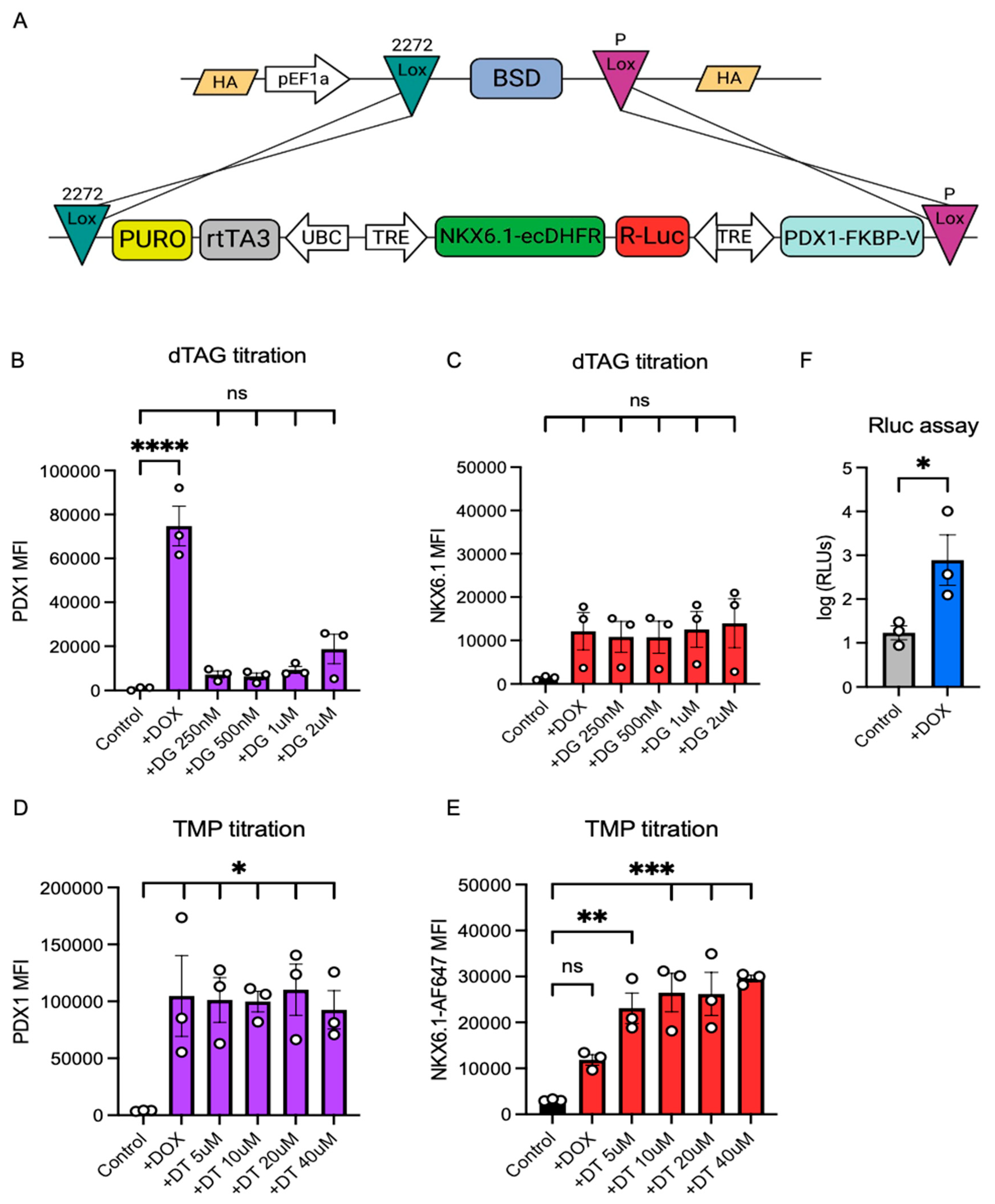
3.4. The Integration of an Improved, Degron-Containing Triple Inducible System (pTriple) into LoxP10 hPSCs Using RMCE
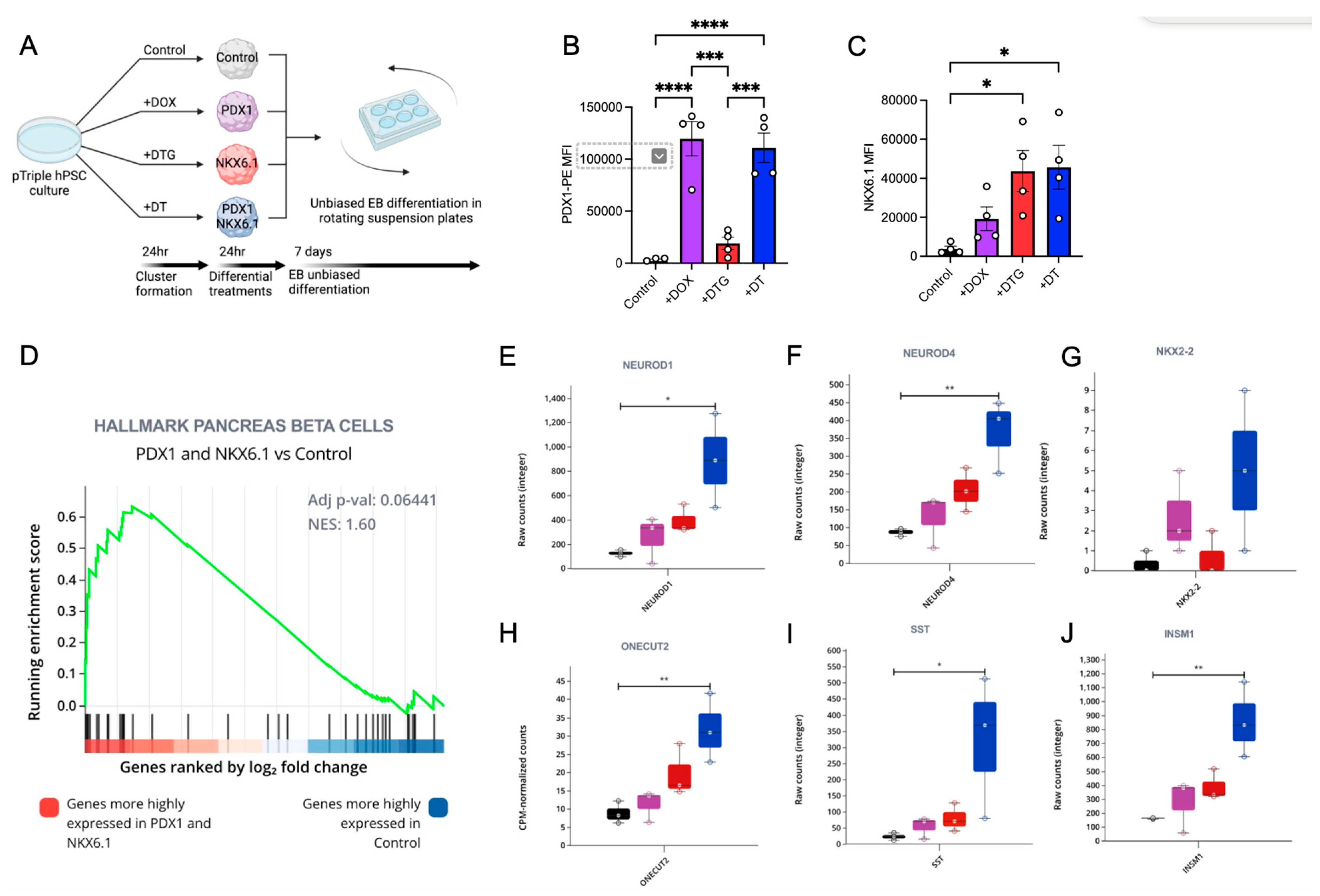
3.5. The Dual, but Not Single, Induction of PDX1 and NKX6.1 Protein Expression Is Sufficient to Induce a Pancreatic Expression Program Including NEUROD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—Opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Sachinidis, A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Hendriks, W.T.; Warren, C.R.; Cowan, C.A. Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions. Cell Stem Cell 2016, 18, 53–65. [Google Scholar] [CrossRef]
- Doss, M.X.; Koehler, C.I.; Gissel, C.; Hescheler, J.; Sachinidis, A. Embryonic stem cells: A promising tool for cell replacement therapy. J. Cell. Mol. Med. 2004, 8, 465–473. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 756–761. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids. 2015, 4, e264. [Google Scholar] [CrossRef]
- Mianné, J.; Bourguignon, C.; Van, C.N.; Fieldès, M.; Nasri, A.; Assou, S.; De Vos, J. Pipeline for the Generation and Characterization of Transgenic Human Pluripotent Stem Cells Using the CRISPR/Cas9 Technology. Cells 2020, 9, 1312. [Google Scholar] [CrossRef]
- Merkle, F.T.; Neuhausser, W.M.; Santos, D.; Valen, E.; Gagnon, J.A.; Maas, K.; Sandoe, J.; Schier, A.F.; Eggan, K. Efficient CRISPR-Cas9-Mediated Generation of Knockin Human Pluripotent Stem Cells Lacking Undesired Mutations at the Targeted Locus. Cell Rep. 2015, 11, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef]
- Schlake, T.; Bode, J. Use of Mutated FLP Recognition Target (FRT) Sites for the Exchange of Expression Cassettes at Defined Chromosomal Loci. Biochemistry 1994, 33, 12746–12751. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Galla, M.; Ernst, E.; Qiao, J.; Voelkel, C.; Schiedlmeier, B.; Zehe, C.; Bode, J. Recombinase-mediated cassette exchange (RMCE): Traditional concepts and current challenges. J. Mol. Biol. 2011, 407, 193–221. [Google Scholar] [CrossRef] [PubMed]
- Sauer, B.; Du, E.I. Functional Expression of the Cre-Lox Site-Specific Recombination System in the Yeast Saccharomyces Cerevisiae. Mol. Cell. Biol. 1987, 7, 2087–2096. [Google Scholar] [PubMed]
- Docherty, F.M.; Riemondy, K.A.; Castro-Gutierrez, R.; Dwulet, J.M.; Shilleh, A.H.; Hansen, M.S.; Williams, S.P.M.; Armitage, L.H.; Santostefano, K.E.; Wallet, M.A.; et al. ENTPD3 Marks Mature Stem Cell–Derived b-Cells Formed by Self-Aggregation In Vitro. Diabetes 2021, 70, 2554–2567. [Google Scholar] [CrossRef]
- Faleo, G.; Russ, H.A.; Wisel, S.; Parent, A.V.; Nguyen, V.; Nair, G.G.; Freise, J.E.; Villanueva, K.E.; Szot, G.L.; Hebrok, M.; et al. Mitigating Ischemic Injury of Stem Cell-Derived Insulin-Producing Cells After Transplant. Stem Cell Rep. 2017, 9, 807–819. [Google Scholar] [CrossRef]
- Castro-Gutierrez, R.; Alkanani, A.; Mathews, C.E.; Michels, A.; Russ, H.A. Protecting Stem Cell Derived Pancreatic Beta-Like Cells from Diabetogenic T Cell Recognition. Front. Endocrinol. 2021, 12, 707881. [Google Scholar] [CrossRef] [PubMed]
- Hudish, L.I.; Bubak, A.; Triolo, T.M.; Niemeyer, C.S.; Lorberbaum, D.S.; Sussel, L.; Nagel, M.; Taliaferro, J.M.; Russ, H.A. Modeling Hypoxia-Induced Neuropathies Using a Fast and Scalable Human Motor Neuron Differentiation System. Stem Cell Rep. 2020, 14, 1033–1043. [Google Scholar] [CrossRef]
- Han, L.; Chaturvedi, P.; Kishimoto, K.; Koike, H.; Nasr, T.; Iwasawa, K.; Giesbrecht, K.; Witcher, P.C.; Eicher, A.; Haines, L.; et al. Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Khandelia, P.; Yap, K.; Makeyev, E.V. Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12799–12804. [Google Scholar] [CrossRef] [PubMed]
- Oceguera-Yanez, F.; Kim, S.-I.; Matsumoto, T.; Tan, G.W.; Xiang, L.; Hatani, T.; Kondo, T.; Ikeya, M.; Yoshida, Y.; Inoue, H.; et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 2016, 101, 43–55. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Pedersen, J.K.; Nelson, S.B.; Jorgensen, M.C.; Henseleit, K.D.; Fujitani, Y.; Wright, C.V.; Sander, M.; Serup, P. Endodermal expression of Nkx6 genes depends differentially on Pdx1. Dev. Biol. 2005, 288, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Breslin, M.B.; Wang, H.W.; Pierce, A.; Aucoin, R.; Lan, M.S. Neurogenin 3 Recruits CBP Co-Activator to Facilitate Histone H3/H4 Acetylation in the Target Gene INSM1. FEBS Lett. 2007, 581, 949–954. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Gutierrez, R.; Arora, A.; Vaeth, K.F.; Taliaferro, J.M.; Russ, H.A. A Recombinase-Mediated Cassette Exchange Platform for a Triple Independent Inducible Expression System for Human Pluripotent Stem Cells. Cells 2025, 14, 184. https://doi.org/10.3390/cells14030184
Castro-Gutierrez R, Arora A, Vaeth KF, Taliaferro JM, Russ HA. A Recombinase-Mediated Cassette Exchange Platform for a Triple Independent Inducible Expression System for Human Pluripotent Stem Cells. Cells. 2025; 14(3):184. https://doi.org/10.3390/cells14030184
Chicago/Turabian StyleCastro-Gutierrez, Roberto, Ankita Arora, Katherine F. Vaeth, J. Matthew Taliaferro, and Holger A. Russ. 2025. "A Recombinase-Mediated Cassette Exchange Platform for a Triple Independent Inducible Expression System for Human Pluripotent Stem Cells" Cells 14, no. 3: 184. https://doi.org/10.3390/cells14030184
APA StyleCastro-Gutierrez, R., Arora, A., Vaeth, K. F., Taliaferro, J. M., & Russ, H. A. (2025). A Recombinase-Mediated Cassette Exchange Platform for a Triple Independent Inducible Expression System for Human Pluripotent Stem Cells. Cells, 14(3), 184. https://doi.org/10.3390/cells14030184