
Comprehensive SUMO Proteomic Analyses Identify HIV Latency-Associated Proteins in Microglia



Abstract
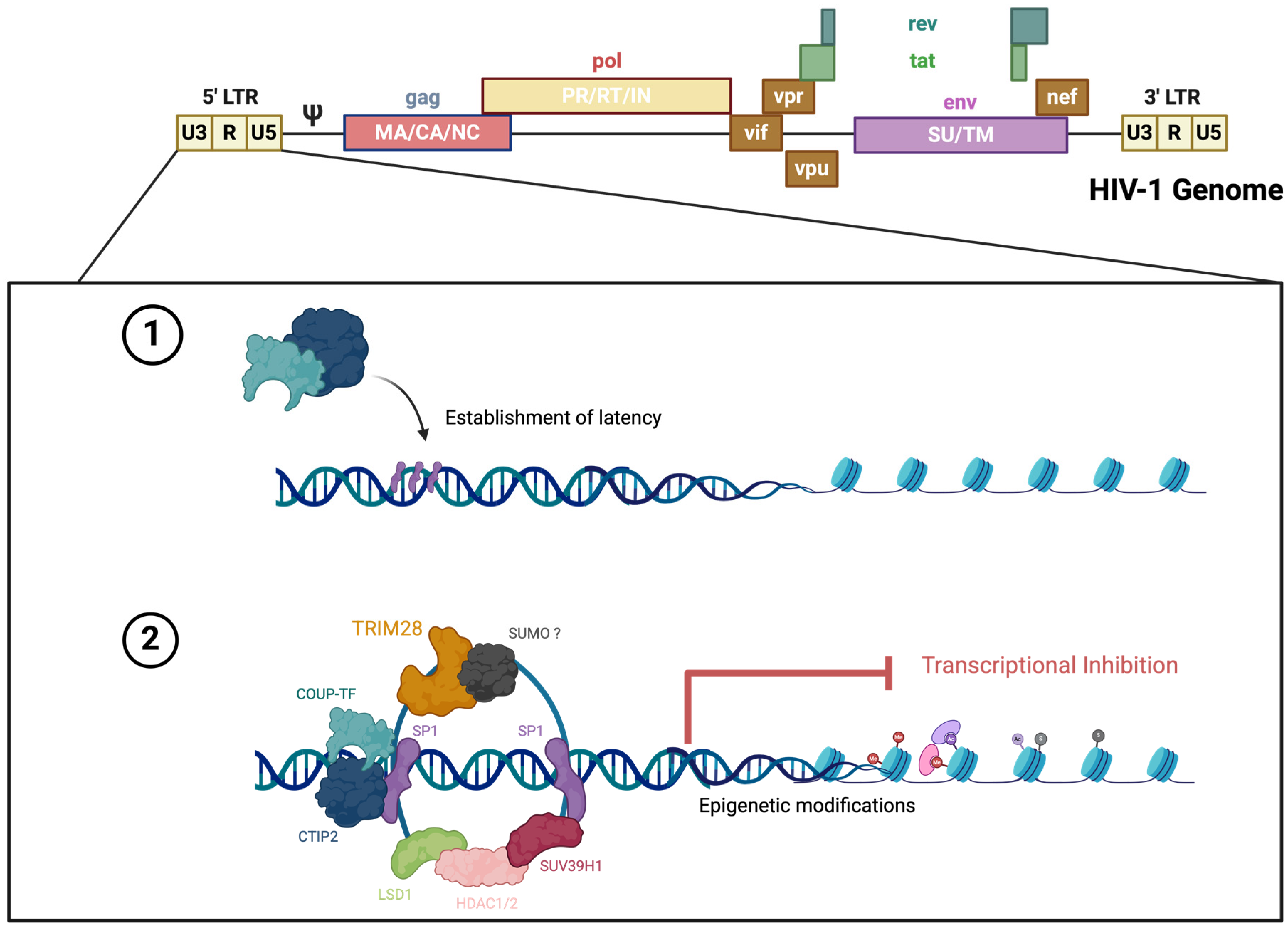
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Confocal Microscopy and Immunofluorescence
2.3. Western Blot Analysis
2.4. Reagents
2.5. Flow Cytometry
2.6. PPI Analysis
2.7. Statistical Analysis
2.8. Illustrations
3. Results
3.1. Viral Reactivation in a Latent HIV Microglia Model Induces a Loss of Global SUMO2/3 Conjugation
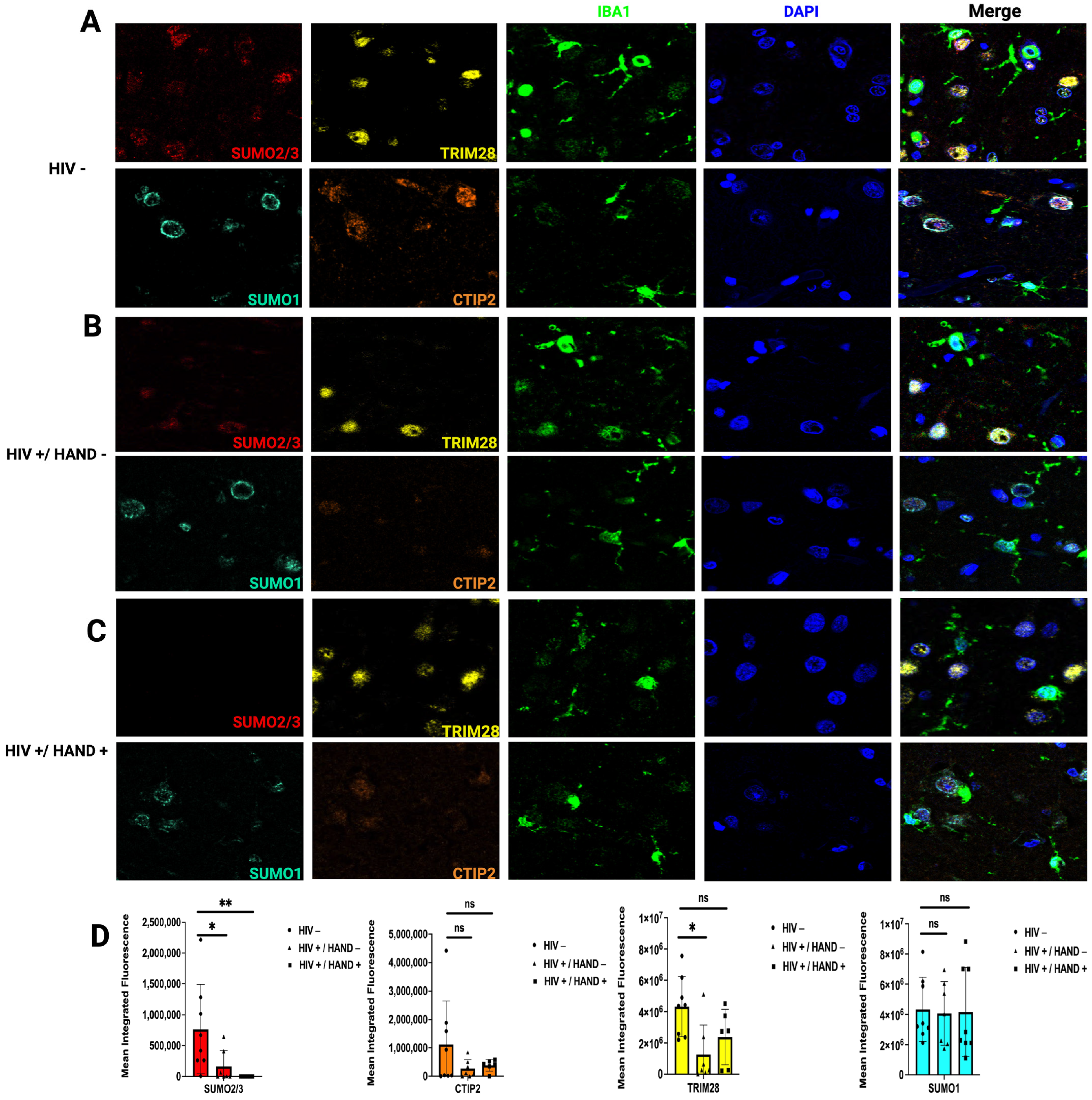
3.2. HIV Infection Decreases SUMO2/3 Protein Expression in Human Brain Tissue
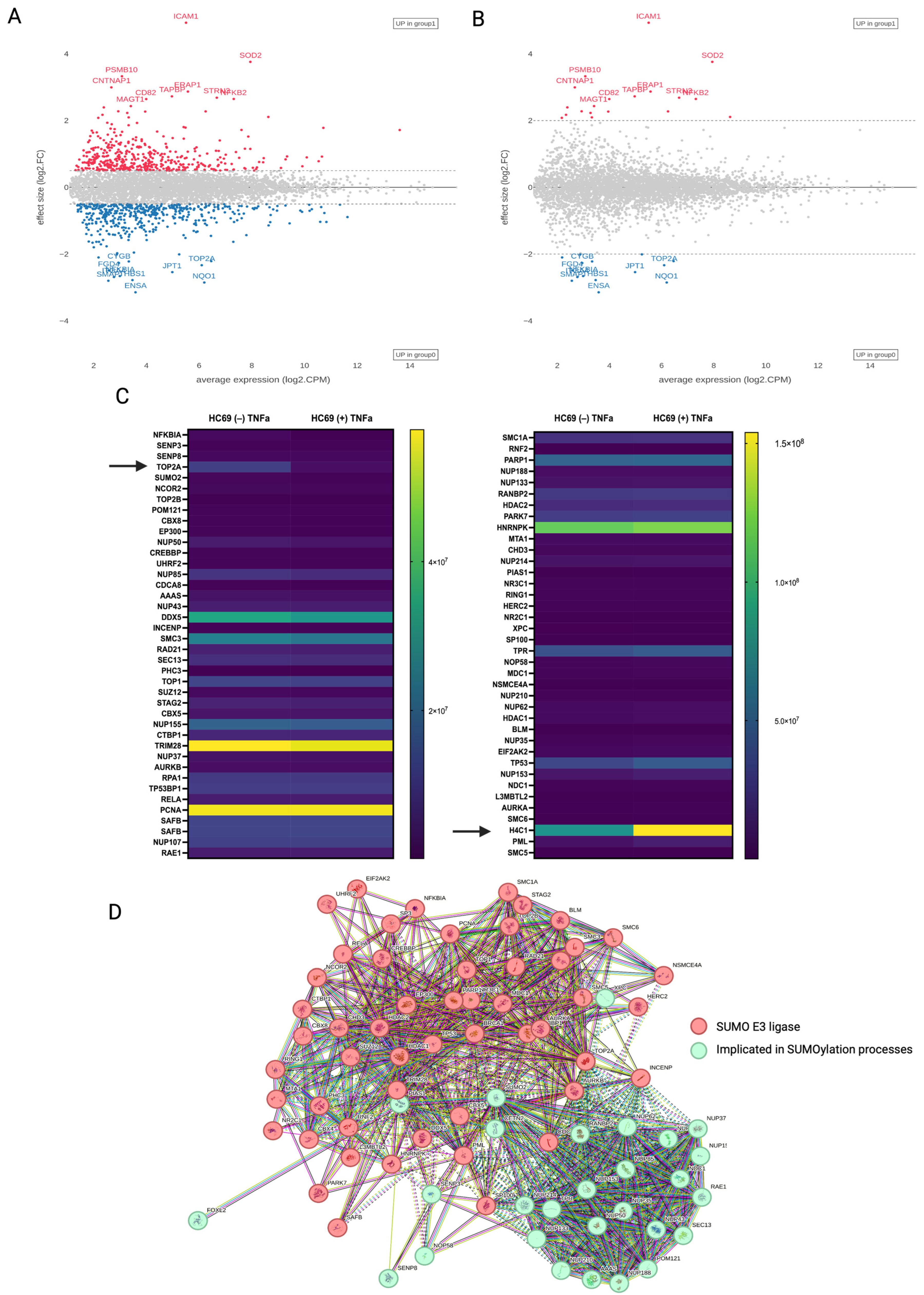
3.3. Quantitative Proteomics of HIV-Induced SUMO2/3 Deconjugation
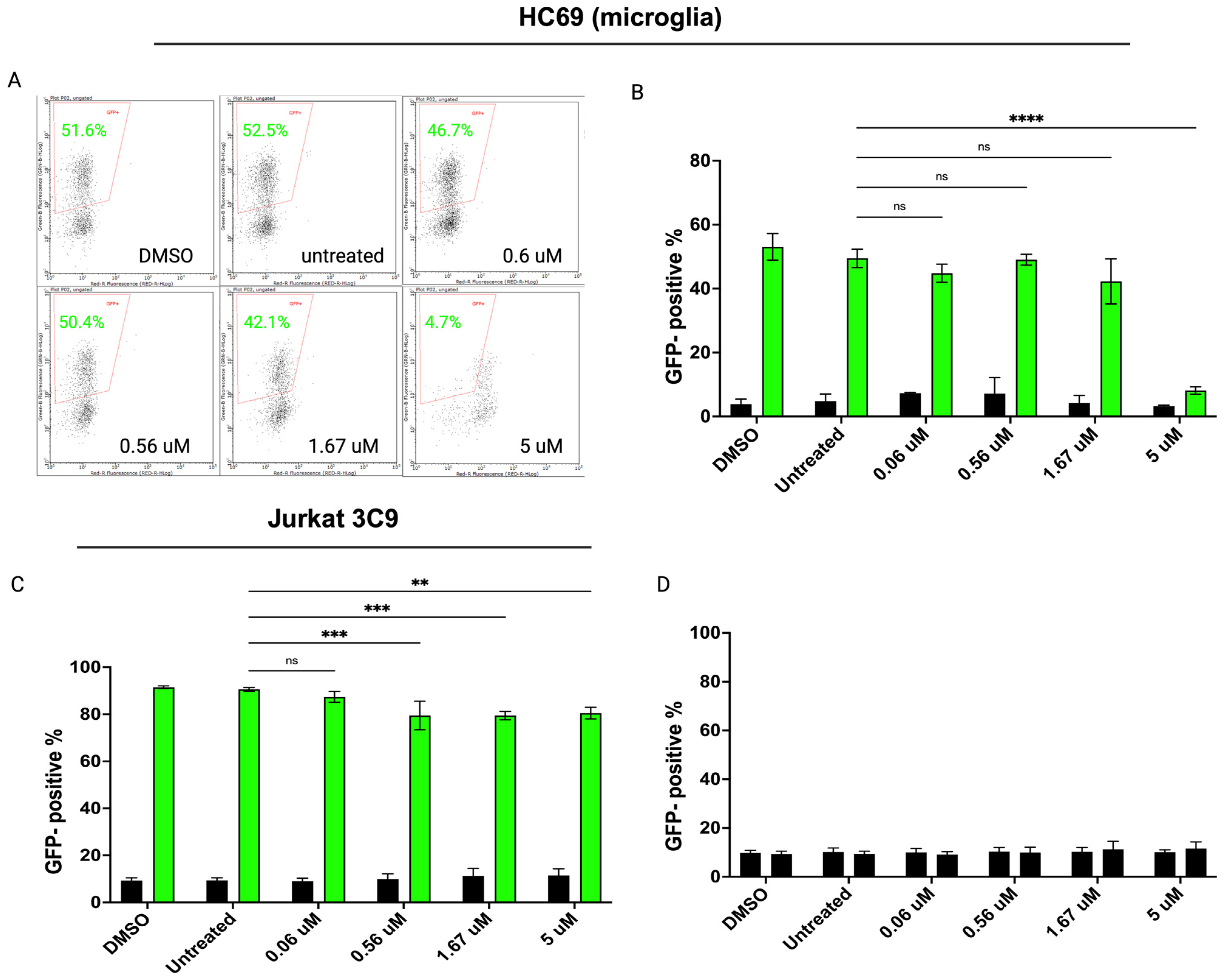
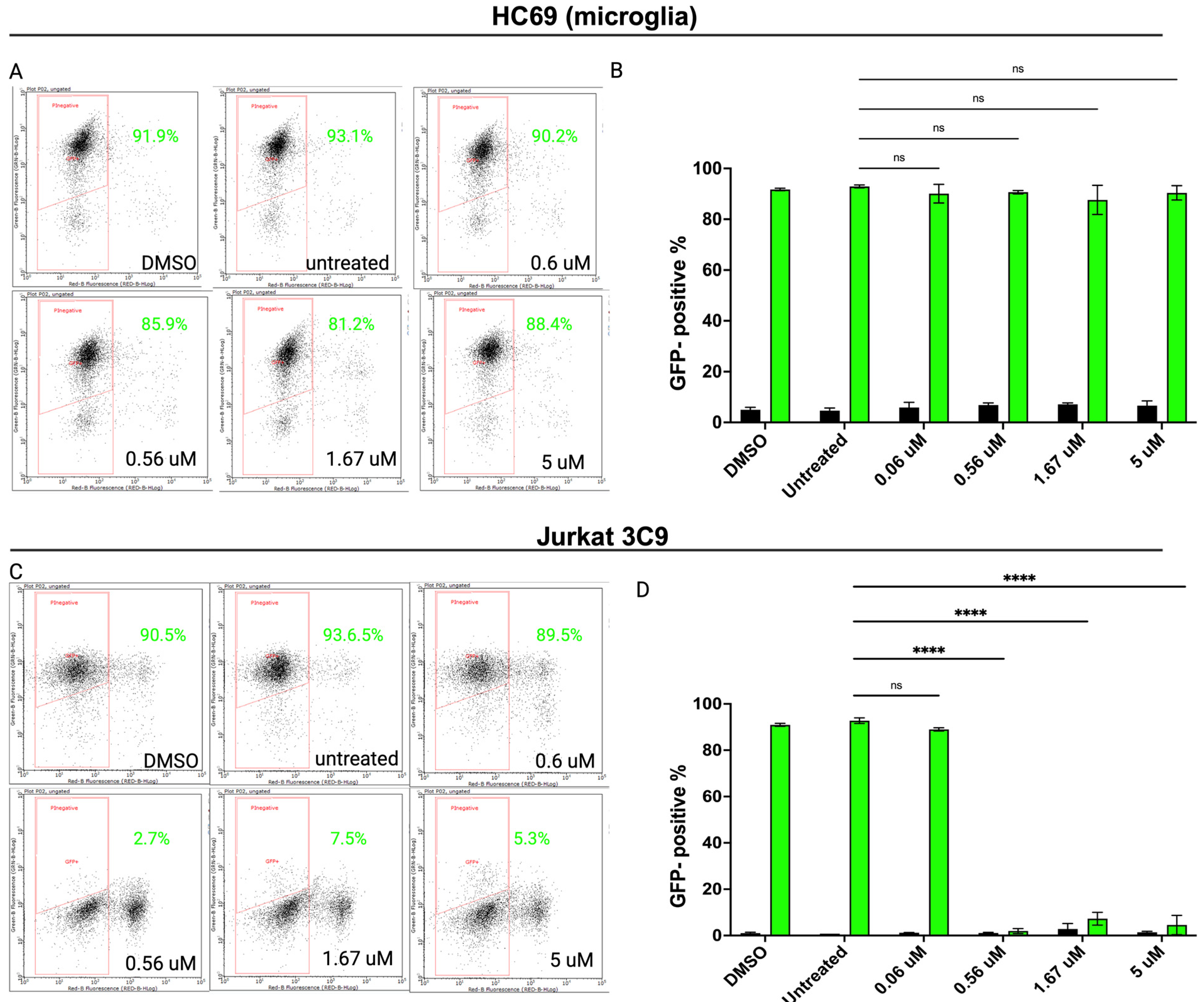
3.4. Inhibiting SUMOylation Blocks TNF-α-Induced HIV Reactivation in Latently Infected Cells
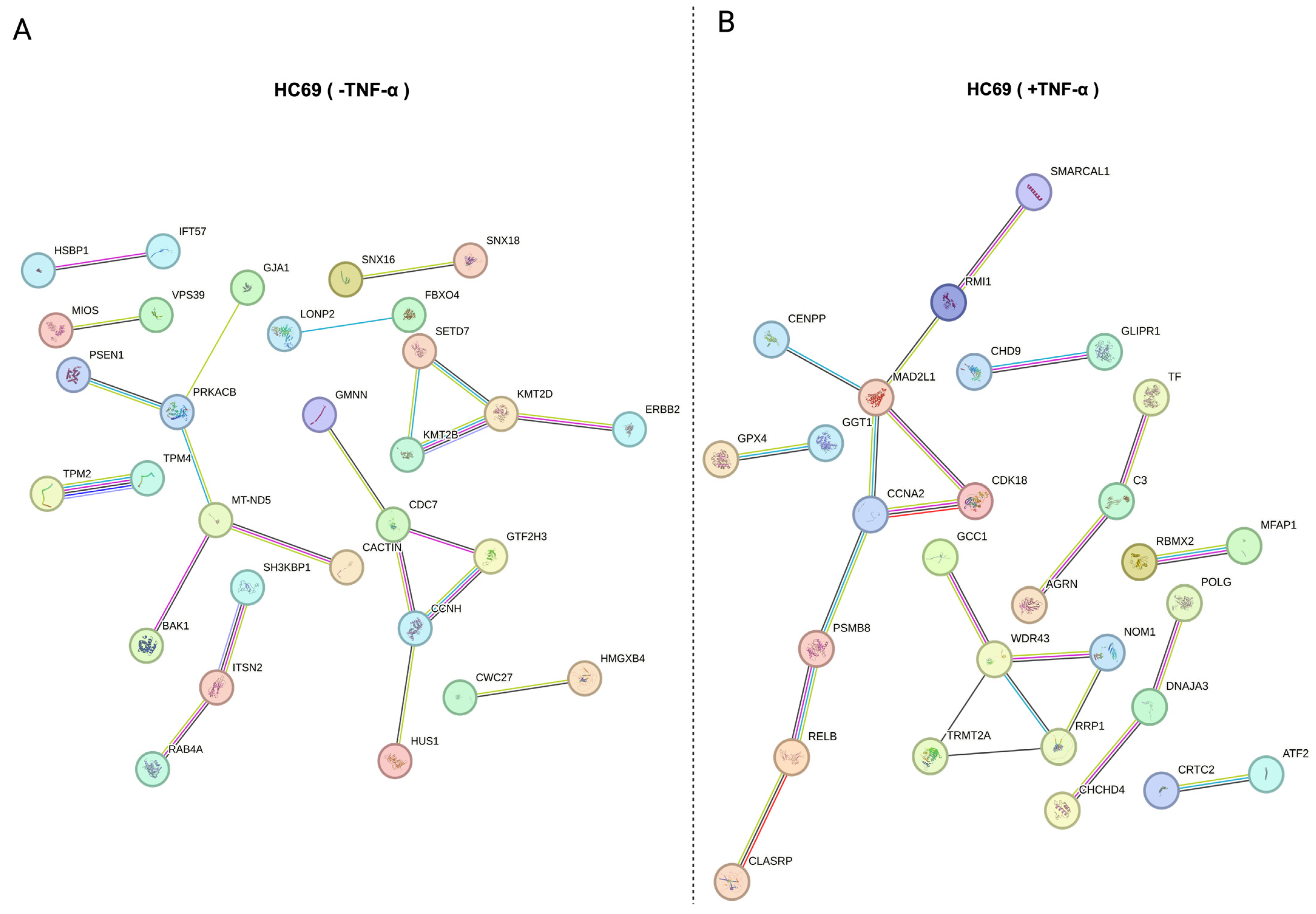
3.5. Protein–Protein Association (PPA) of Distinct Substrate Populations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Menendez, C.M.; Carr, D.J.J. Defining Nervous System Susceptibility During Acute and Latent Herpes Simplex Virus-1 Infection. J. Neuroimmunol. 2017, 308, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood–Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef] [PubMed]
- van den Pol, A.N. Viral infection leading to brain dysfunction: More prevalent than appreciated? Neuron 2009, 64, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Keppler, O.T.; Schölz, C. Post-translational Modification-Based Regulation of HIV Replication. Front. Microbiol. 2018, 9, 2131. [Google Scholar] [CrossRef]
- Le Douce, V.; Herbein, O.; Rohr, G.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010, 7, 32. [Google Scholar] [CrossRef]
- Sarracino, A.; Gharu, L.; Kula, A.; Pasternak, A.O.; Avettand-Fenoel, V.; Rouzioux, C.; Bardina, M.; De Wit, S.; Benkirane, M.; Berkhout, B. Posttranscriptional Regulation of HIV-1 Gene Expression during Replication and Reactivation from Latency by Nuclear Matrix Protein MATR3. mBio 2018, 9, e02158-18. [Google Scholar] [CrossRef]
- Khoury, G.; Mota, T.M.; Li, S.; Tumpach, C.; Lee, M.Y.; Jacobson, J.; Harty, L.; Anderson, J.L.; Lewin, S.R.; Purcell, D.F.J. HIV latency reversing agents act through Tat post translational modifications. Retrovirology 2018, 15, 36. [Google Scholar] [CrossRef]
- Ott, M.; Geyer, M.; Zhou, Q. The Control of HIV Transcription: Keeping RNA Polymerase II on Track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef]
- Ehrentraut, S.F.; Colgan, S.P. Implications of Protein Post-translational Modifications in IBD. Inflamm. Bowel Dis. 2012, 18, 1378–1388. [Google Scholar] [CrossRef]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-translational Modifications in Alzheimer’s Disease. ChemBioChem 2020, 21, 1052–1079. [Google Scholar] [CrossRef]
- Si, Y.; Zhang, Y.; Chen, Z.; Zhou, R.; Zhang, Y.; Hao, D.; Yan, D. Posttranslational Modification Control of Inflammatory Signaling. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; pp. 37–61. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Imbert, F.; Leavitt, G.; Langford, D. SUMOylation and Viral Infections of the Brain. Pathogens 2022, 11, 818. [Google Scholar] [CrossRef] [PubMed]
- Imbert, F.; Langford, D. Viruses, SUMO, and immunity: The interplay between viruses and the host SUMOylation system. J. Neurovirol. 2021, 27, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lee, C.-C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5, a Novel Poly-SUMO Isoform, Regulates PML Nuclear Bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein Modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef]
- Su, H.L.; Li, S.S.L. Molecular features of human ubiquitin-like SUMO genes and their encoded proteins. Gene 2002, 296, 65–73. [Google Scholar] [CrossRef]
- Mabb, A.M.; Miyamoto, S. SUMO and NF-κB ties. Cell. Mol. Life Sci. 2007, 64, 1979–1996. [Google Scholar] [CrossRef]
- Sahin, U.; Ferhi, O.; Carnec, X.; Zamborlini, A.; Peres, L.; Jollivet, F.; Vitaliano-Prunier, A.; de Thé, H.; Lallemand-Breitenbach, V. Interferon controls SUMO availability via the Lin28 and let-7 axis to impede virus replication. Nat. Commun. 2014, 5, 4187. [Google Scholar] [CrossRef]
- Chen, N.C.; Partridge, A.T.; Sell, C.; Torres, C.; Martín-García, J. Fate of microglia during HIV-1 infection: From activation to senescence?: Microglial Dysfunction During HIV-1 Infection. Glia 2017, 65, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Lu, Q.; Farrell, M.; Lappin, J.M.; Shi, J.; Lu, L.; Bao, Y. Global prevalence and burden of HIV-associated neurocognitive disorder: A meta-analysis. Neurology 2020, 95, e2610–e2621. [Google Scholar] [CrossRef] [PubMed]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Dobrowolski, C.; Karn, J. The Glucocorticoid Receptor Is a Critical Regulator of HIV Latency in Human Microglial Cells. J. Neuroimmune Pharmacol. 2018, 14, 94. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, C.; Liu, X.; Ma, X.; Bian, X.; Xiao, X.; Gao, R.; Sun, Y.; Wu, W.; Zhao, P. Dexamethasone inhibits stemness maintenance and enhances chemosensitivity of hepatocellular carcinoma stem cells by inducing deSUMOylation of HIF-1α and Oct4. Int. J. Oncol. 2020, 57, 780. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, B.R.; Dao, T.T.P.; Kim, J.M.; Kim, Y.J.; Son, H.; Jo, S.; Kim, D.; Kim, J.; Suh, Y.J.; et al. TAK-981, a SUMOylation inhibitor, suppresses AML growth immune-independently. Blood Adv. 2023, 7, 3155–3168. [Google Scholar] [CrossRef]
- Lam, V.; Roleder, C.; Liu, T.; Bruss, N.; Best, S.; Wang, X.; Phillips, T.; Shouse, G.; Berger, A.J.; Alinari, L.; et al. T Cell-intrinsic Immunomodulatory Effects of TAK-981 (Subasumstat), a SUMO-activating Enzyme Inhibitor, in Chronic Lymphocytic Leukemia. Mol. Cancer Ther. 2023, 22, 1040–1051. [Google Scholar] [CrossRef]
- Zhou, P.; Chen, X.; Li, M.; Tan, J.; Zhang, Y.; Yuan, W.; Zhou, J.; Wang, G. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019, 513, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Joung, H.; Liu, H. 2-D08 mediates notable anticancer effects through multiple cellular pathways in uterine leiomyosarcoma cells. Oncol. Rep. 2024, 52, 97. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, S.; Fath, E.K.; Biel, N.; Ray, A.; Moss, C.R.; Patel, A.; Patel, S.; Hilding, L.; Varn, M.; Ross, T.; et al. Epstein-Barr Virus Latent Membrane Protein-1 Induces the Expression of SUMO-1 and SUMO-2/3 in LMP1-positive Lymphomas and Cells. Sci. Rep. 2019, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.Y.; Ahn, S.H.; Hochstrasser, M. SUMO and cellular adaptive mechanisms. Exp. Mol. Med. 2020, 52, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, H.; Hinchey, J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 2000, 275, 6252–6258. [Google Scholar] [CrossRef]
- Tirard, M.; Hsiao, H.H.; Nikolov, M.; Urlaub, H.; Melchior, F.; Brose, N. In vivo localization and identification of SUMOylated proteins in the brain of His6-HA-SUMO1 knock-in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21122–21127. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Bellefroid, M.; Daouad, F.; Martinelli, V.; Van Assche, J.; Wallet, C.; Rodari, A.; De Rovere, M.; Fahrenkrog, B.; Schwartz, C.; et al. Inhibition of HIV-1 gene transcription by KAP1 in myeloid lineage. Sci. Rep. 2021, 11, 2692. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Fujinaga, K.; Gross, J.D.; Frankel, A.D. Enhanced NF-κB activation via HIV-1 Tat-TRAF6 cross-talk. Sci. Adv. 2024, 10, eadi4162. [Google Scholar] [CrossRef]
- Sullivan, M.N.; Brill, S.A.; Mangus, L.M.; Jeong, Y.J.; Solis, C.V.; Knight, A.C.; Colantuoni, C.; Keceli, G.; Paolocci, N.; Queen, S.E.; et al. Upregulation of Superoxide Dismutase 2 by Astrocytes in the SIV/Macaque Model of HIV-Associated Neurologic Disease. J. Neuropathol. Exp. Neurol. 2020, 79, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shang, H.; Jiang, Y. ICAM-1 in HIV infection and underlying mechanisms. Cytokine 2020, 125, 154830. [Google Scholar] [CrossRef] [PubMed]
- Sugden, S.M.; Pham, T.N.Q.; Cohen, E.A. HIV-1 Vpu Downmodulates ICAM-1 Expression, Resulting in Decreased Killing of Infected CD4+ T Cells by NK Cells. J. Virol. 2017, 91, e02442-16. [Google Scholar] [CrossRef] [PubMed]
- Suárez, H.; Rocha-Perugini, V.; Álvarez, S.; Yáñez-Mó, M. Tetraspanins, Another Piece in the HIV-1 Replication Puzzle. Front. Immunol. 2018, 9, 1811. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ding, X.; Li, Z.; Rao, F.; Xu, H.; Lu, J.; Ma, X.; Zhang, M.; Xie, Z. Comprehensive analyses identify potential biomarkers for encephalitis in HIV infection. Sci. Rep. 2023, 13, 18418. [Google Scholar] [CrossRef]
- Bradley, T.; Ferrari, G.; Haynes, B.F.; Margolis, D.M.; Browne, E.P. Single-Cell Analysis of Quiescent HIV Infection Reveals Host Transcriptional Profiles that Regulate Proviral Latency. Cell Rep. 2018, 25, 107–117.e3. [Google Scholar] [CrossRef]
- Mauro, E.; Lesbats, P.; Lapaillerie, D.; Chaignepain, S.; Maillot, B.; Oladosu, O.; Robert, X.; Fiorini, F.; Kieffer, B.; Bouaziz, S.; et al. Human H4 tail stimulates HIV-1 integration through binding to the carboxy-terminal domain of integrase. Nucleic Acids Res. 2019, 47, 3607–3618. [Google Scholar] [CrossRef]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell. Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef]
- Sun, Y.; Nitiss, J.L.; Pommier, Y. SUMO: A Swiss Army Knife for Eukaryotic Topoisomerases. Front. Mol. Biosci. 2022, 9, 871161. [Google Scholar] [CrossRef]
- Tian, T.; Bu, M.; Chen, X.; Ding, L.; Yang, Y.; Han, J.; Feng, X.H.; Xu, P.; Liu, T.; Ying, S.; et al. The ZATT-TOP2A-PICH Axis Drives Extensive Replication Fork Reversal to Promote Genome Stability. Molecular Cell. 2021, 81, 198–211.e6. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Melchior, F. Ubiquitin-Related Modifier SUMO1 and Nucleocytoplasmic Transport. Traffic 2002, 3, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.; Judge, M.A.; Pastor, L.; Fuente-Soro, L.; Jairoce, C.; Carter, K.W.; Anderson, D.; Mandomando, I.; Clifford, H.D.; Naniche, D.; et al. Gene dysregulation in acute HIV-1 infection—Early transcriptomic analysis reveals the crucial biological functions affected. Front. Cell Infect. Microbiol. 2023, 13, 1074847. [Google Scholar] [CrossRef] [PubMed]
- Lista, M.J.; Jousset, A.C.; Cheng, M.; Saint-André, V.; Perrot, E.; Rodrigues, M.; Di Primo, C.; Gadelle, D.; Toccafondi, E.; Segeral, E.; et al. DNA topoisomerase 1 represses HIV-1 promoter activity through its interaction with a guanine quadruplex present in the LTR sequence. Retrovirology 2023, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Kuzin, V.; Cameron, D.P.; Sanford, S.; Jha, R.K.; Nie, Z.; Rosello, M.T.; Holewinski, R.; Andresson, T.; Wisniewski, J.; et al. MYC assembles and stimulates topoisomerases 1 and 2 in a ‘topoisome’. Mol. Cell 2022, 82, 140–158.e12. [Google Scholar] [CrossRef]
- Taylor, E.M.; Copsey, A.C.; Hudson, J.J.R.; Vidot, S.; Lehmann, A.R. Identification of the proteins, including MAGEG1, that make up the human SMC5-6 protein complex. Mol. Cell. Biol. 2008, 28, 1197–1206. [Google Scholar] [CrossRef]
- Reynoso, R.; Wieser, M.; Ojeda, D.; Bönisch, M.; Kühnel, H.; Bolcic, F.; Quendler, H.; Grillari, J.; Grillari-Voglauer, R.; Quarleri, J. HIV-1 Induces Telomerase Activity in Monocyte-Derived Macrophages, Possibly Safeguarding One of Its Reservoirs. J. Virol. 2012, 86, 10327–10337. [Google Scholar] [CrossRef]
- Irwan, I.D.; Bogerd, H.P.; Cullen, B.R. Epigenetic silencing by the SMC5/6 complex mediates HIV-1 latency. Nat. Microbiol. 2022, 7, 2101–2113. [Google Scholar] [CrossRef]
- Zamborlini, A.; Coiffic, A.; Beauclair, G.; Delelis, O.; Paris, J.; Koh, Y.; Magne, F.; Giron, M.L.; Tobaly-Tapiero, J.; Deprez, E.; et al. Impairment of Human Immunodeficiency Virus Type-1 Integrase SUMOylation Correlates with an Early Replication Defect*. J. Biol. Chem. 2011, 286, 21013–21022. [Google Scholar] [CrossRef]
- Zheng, Y.; Jayappa, K.D.; Ao, Z.; Qiu, X.; Su, R.C.; Yao, X. Noncovalent SUMO-interaction motifs in HIV integrase play important roles in SUMOylation, cofactor binding, and virus replication. Virol. J. 2019, 16, 42. [Google Scholar] [CrossRef]
- Gurer, C.; Berthoux, L.; Luban, J. Covalent modification of human immunodeficiency virus type 1 p6 by SUMO-1. J. Virol. 2005, 79, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Mete, B.; Pekbilir, E.; Bilge, B.N.; Georgiadou, P.; Çelik, E.; Sutlu, T.; Tabak, F.; Sahin, U. Human immunodeficiency virus type 1 impairs sumoylation. Life Sci. Alliance 2022, 5, 202101103. [Google Scholar] [CrossRef] [PubMed]
- Langston, S.P.; Grossman, S.; England, D.; Afroze, R.; Bence, N.; Bowman, D.; Bump, N.; Chau, R.; Chuang, B.C.; Claiborne, C.; et al. Discovery of TAK-981, a First-in-Class Inhibitor of SUMO-Activating Enzyme for the Treatment of Cancer. J. Med. Chem. 2021, 64, 2501–2520. [Google Scholar] [CrossRef] [PubMed]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative Elongation Factor Is Required for the Maintenance of Proviral Latency but Does Not Induce Promoter-Proximal Pausing of RNA Polymerase II on the HIV Long Terminal Repeat. Mol. Cell. Biol. 2014, 34, 1911. [Google Scholar] [CrossRef]
- Kim, Y.S.; Nagy, K.; Keyser, S.; Schneekloth, J.S. An Electrophoretic Mobility Shift Assay Identifies a Mechanistically Unique Inhibitor of Protein Sumoylation. Chem. Biol. 2013, 20, 604–613. [Google Scholar] [CrossRef]
- Georges, A.; Benayoun, B.A.; Marongiu, M.; Dipietromaria, A.; L’Hôte, D.; Todeschini, A.L.; Auer, J.; Crisponi, L.; Veitia, R.A. SUMOylation of the Forkhead Transcription Factor FOXL2 Promotes Its Stabilization/Activation through Transient Recruitment to PML Bodies. PLoS ONE 2011, 6, e25463. [Google Scholar] [CrossRef]
- Marongiu, M.; Deiana, M.; Meloni, A.; Marcia, L.; Puddu, A.; Cao, A.; Schlessinger, D.; Crisponi, L. The Forkhead Transcription Factor Foxl2 Is Sumoylated in Both Human and Mouse: Sumoylation Affects Its Stability, Localization, and Activity. PLoS ONE 2010, 5, e9477. [Google Scholar] [CrossRef]
- Kuo, F.T.; Bentsi-Barnes, I.K.; Barlow, G.M.; Bae, J.; Pisarska, M.D. Sumoylation of Forkhead L2 by Ubc9 is required for its activity as a transcriptional repressor of the Steroidogenic Acute Regulatory gene. Cell. Signal. 2009, 21, 1935–1944. [Google Scholar] [CrossRef]
- Jefferys, S.R.; Burgos, S.D.; Peterson, J.J.; Selitsky, S.R.; Turner, A.W.; James, L.I.; Tsai, Y.H.; Coffey, A.R.; Margolis, D.M.; Parker, J.; et al. Epigenomic characterization of latent HIV infection identifies latency regulating transcription factors. PLoS Pathog. 2021, 17, e1009346. [Google Scholar] [CrossRef]
- Barroso-Gomila, O.; Trulsson, F.; Muratore, V.; Canosa, I.; Merino-Cacho, L.; Cortazar, A.R.; Pérez, C.; Azkargorta, M.; Iloro, I.; Carracedo, A.; et al. Identification of proximal SUMO-dependent interactors using SUMO-ID. Nat. Commun. 2021, 12, 6671. [Google Scholar] [CrossRef]
- Appikonda, S.; Thakkar, K.N.; Shah, P.K.; Dent, S.Y.R.; Andersen, J.N.; Barton, M.C. Cross-talk between chromatin acetylation and SUMOylation of tripartite motif–containing protein 24 (TRIM24) impacts cell adhesion. J. Biol. Chem. 2018, 293, 7476–7485. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.M.; Dahabieh, M.; Malcolm, T.; Sadowski, I. TRIM24 controls induction of latent HIV-1 by stimulating transcriptional elongation. Commun. Biol. 2023, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.M.; Brumme, Z.L.; Sadowski, I. Inhibition of the TRIM24 bromodomain reactivates latent HIV-1. Sci. Rep. 2023, 13, 556. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926. [Google Scholar] [CrossRef]
- Ross, S.; Best, J.L.; Zon, L.I.; Gill, G. SUMO-1 Modification Represses Sp3 Transcriptional Activation and Modulates Its Subnuclear Localization. Mol. Cell 2002, 10, 831–842. [Google Scholar] [CrossRef]
- Rohr, O.; Aunis, D.; Schaeffer, E. COUP-TF and Sp1 Interact and Cooperate in the Transcriptional Activation of the Human Immunodeficiency Virus Type 1 Long Terminal Repeat in Human Microglial Cells. J. Biol. Chem. 1997, 272, 31149–31155. [Google Scholar] [CrossRef]
- Gottwein, E.; Kräusslich, H.G. Analysis of Human Immunodeficiency Virus Type 1 Gag Ubiquitination. J. Virol. 2005, 79, 9134–9144. [Google Scholar] [CrossRef]
- Zheng, Y.; Yao, X. Posttranslational Modifications of HIV-1 Integrase by Various Cellular Proteins during Viral Replication. Viruses 2013, 5, 1787. [Google Scholar] [CrossRef]
- Chen, S.; Yang, X.; Cheng, W.; Ma, Y.; Shang, Y.; Cao, L.; Chen, S.; Chen, Y.; Wang, M.; Guo, D. Immune regulator ABIN1 suppresses HIV-1 transcription by negatively regulating the ubiquitination of Tat. Retrovirology 2017, 14, 12. [Google Scholar] [CrossRef]
- Jin, Y.J.; Cai, C.Y.; Zhang, X.; Burakoff, S.J. Lysine 144, a Ubiquitin Attachment Site in HIV-1 Nef, Is Required for Nef-Mediated CD4 Down-Regulation1. J. Immunol. 2008, 180, 7878–7886. [Google Scholar] [CrossRef]
- Arora, S.; Verma, S.; Banerjea, A.C. HIV-1 Vpr Redirects Host Ubiquitination Pathway. J. Virol. 2014, 88, 9141–9152. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Major Function |
|---|---|
| Nef | Modulation of cellular receptors including CD4, CD28, and MHC I |
| Rev | Nuclear export of incompletely spliced viral mRNA |
| Tat | Transcriptional activation of HIV-1LTR |
| Vif | Stimulation of reverse transcription |
| Vpr | Nuclear import of pre-integration complex |
| Vpu | Promotes virion release through CD4 degradation in the ER |
| Name | p-Value | Adjusted p-Value |
|---|---|---|
| Protein Phosphatase 2A Binding (GO: 0051721) | 0.0007355 | 0.04096 |
| NF-kappaB Binding (GO: 0051059) | 0.0009417 | 0.04096 |
| Small GTPase Binding (GO: 0031267) | 0.005024 | 0.1110 |
| Protein Homodimerization Activity (GO: 0042803) | 0.005581 | 0.1110 |
| GTPase Binding (GO: 0051020) | 0.007053 | 0.1110 |
| Cytochrome-B5 Redcutase Activity, Acting On NAD(P)H (GO: 0004128) | 0.008720 | 0.1110 |
| Interleukin-6 Receptor Binding (GO: 0005138) | 0.01046 | 0.1110 |
| NADH Dehydrogenase Activity (GO: 0003954) | 0.01219 | 0.1110 |
| Armadillo Repeat Domain Binding (GO: 0070016) | 0.01219 | 0.1110 |
| Protein Binding Involved In Heterotypic Cell-Cell Adhesion (GO: 0086080) | 0.01564 | 0.1110 |
| Name | p-Value | Adjusted p-Value |
|---|---|---|
| Histone H3 Methyltransferase Activity (GO: 0140938) | 0.0006025 | 0.06901 |
| Histone H3K4 Methyltransferase Activity (GO: 0042800) | 0.001580 | 0.06901 |
| Protein Heterodimerization Activity (GO: 0046982) | 0.001851 | 0.06901 |
| Protein-Lysine N-methyltransferase Activity (GO: 0016279) | 0.001972 | 0.06901 |
| Aspartic Endopeptidase Activity, intramembrane Cleaving (GO: 0042500) | 0.02279 | 0.3049 |
| cAMP-dependent Protein Kinase Activity (GO: 0004691) | 0.02279 | 0.3049 |
| Keratin Filament Binding (GO: 1990254) | 0.02279 | 0.3049 |
| Pyruvate Transmembrane Transporter Activity (GO: 0050833) | 0.02279 | 0.3049 |
| K48-linked Polyubiqutin Modification-Dependent Protein Binding (GO: 0036435) | 0.02279 | 0.3049 |
| Unmethylated CpG Binding (GO: 0045322) | 0.03175 | 0.3049 |
| Name | p-Value | Adjusted p-Value |
|---|---|---|
| SH2 Domain Binding (GO: 0042169) | 0.006489 | 0.1818 |
| 3′-5- Exonucease Activity (GO: 0008408) | 0.01169 | 0.1818 |
| Serine-Type Endopeptidase Inhibitor Activty (GO: 0004867) | 0.01820 | 0.1818 |
| Oligosaccharyl trasferase Activity (GO: 0004576) | 0.01935 | 0.1818 |
| Ceraminde 1-Phosphate Binding (GO: 1902387) | 0.01935 | 0.1818 |
| Ceramide 1-Phosphate Transfer Activity (GO: 1902388) | 0.01935 | 0.1818 |
| Protein Phosphatase 2B Binding (GO: 0030346) | 0.01935 | 0.1818 |
| Acid Phosphatase Activity (GO: 0003993) | 0.02318 | 0.1818 |
| Galactosidase Activity (GO: 0015925) | 0.02318 | 0.1818 |
| Beta-Galactosidase (CMP) Alpha-2,3-Sialytransferase Activity (GO: 0003836) | 0.02318 | 0.1818 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imbert, F.; Langford, D. Comprehensive SUMO Proteomic Analyses Identify HIV Latency-Associated Proteins in Microglia. Cells 2025, 14, 235. https://doi.org/10.3390/cells14030235
Imbert F, Langford D. Comprehensive SUMO Proteomic Analyses Identify HIV Latency-Associated Proteins in Microglia. Cells. 2025; 14(3):235. https://doi.org/10.3390/cells14030235
Chicago/Turabian StyleImbert, Fergan, and Dianne Langford. 2025. "Comprehensive SUMO Proteomic Analyses Identify HIV Latency-Associated Proteins in Microglia" Cells 14, no. 3: 235. https://doi.org/10.3390/cells14030235
APA StyleImbert, F., & Langford, D. (2025). Comprehensive SUMO Proteomic Analyses Identify HIV Latency-Associated Proteins in Microglia. Cells, 14(3), 235. https://doi.org/10.3390/cells14030235