
ATF3-SLC7A7 Axis Regulates mTORC1 Signaling to Suppress Lipogenesis and Tumorigenesis in Hepatocellular Carcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Maintenance
2.2. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.3. Vector Construction
2.4. Lentivirus Production and Infection
2.5. Transwell Assay
2.6. Wound Healing Assay
2.7. Colony Formation Assay
2.8. MTT Assay
2.9. Immunoblotting
2.10. Confocal Assay
2.11. Nile Red Staining Assay
2.12. Oil Red O Staining Assay
2.13. Triglyceride Quantification Assay
2.14. Free Fatty Acid Quantification Assay
2.15. Chromatin Immunoprecipitation (ChIP) Assay
2.16. Luciferase Reporter Assay
2.17. Mouse Xenograft Model
2.18. Immunohistochemistry
2.19. RNA-Seq Analysis
2.20. Gene Expression Analysis
2.21. Kaplan–Meier (KM) Survival Analysis
2.22. Correlation Analysis
2.23. Statistical Analysis
3. Results
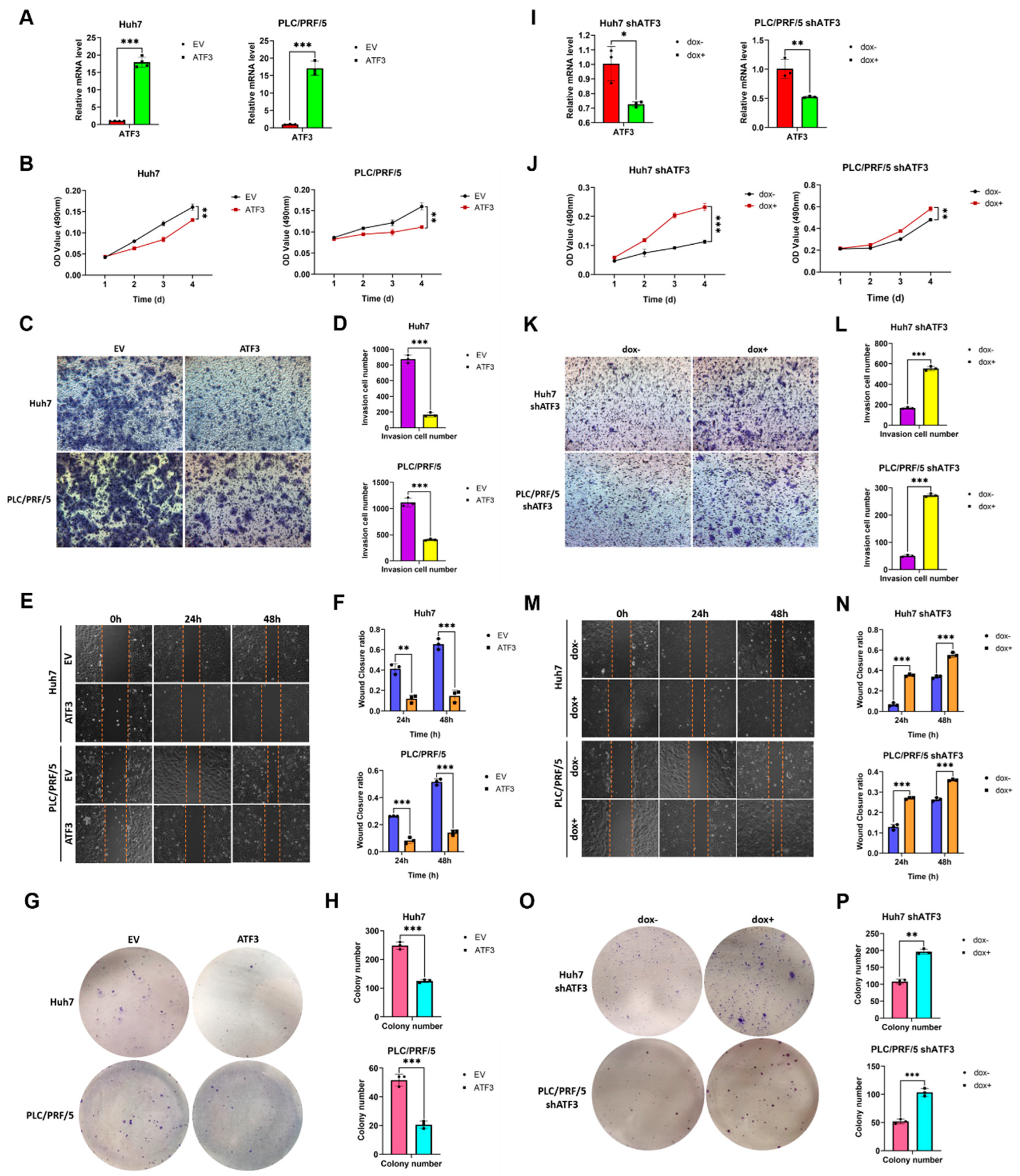
3.1. ATF3 Suppresses the Amplification and Invasion of HCC Cells
3.2. ATF3 Is Correlated with Better Prognosis and Downregulated Lipid Synthesis in HCC
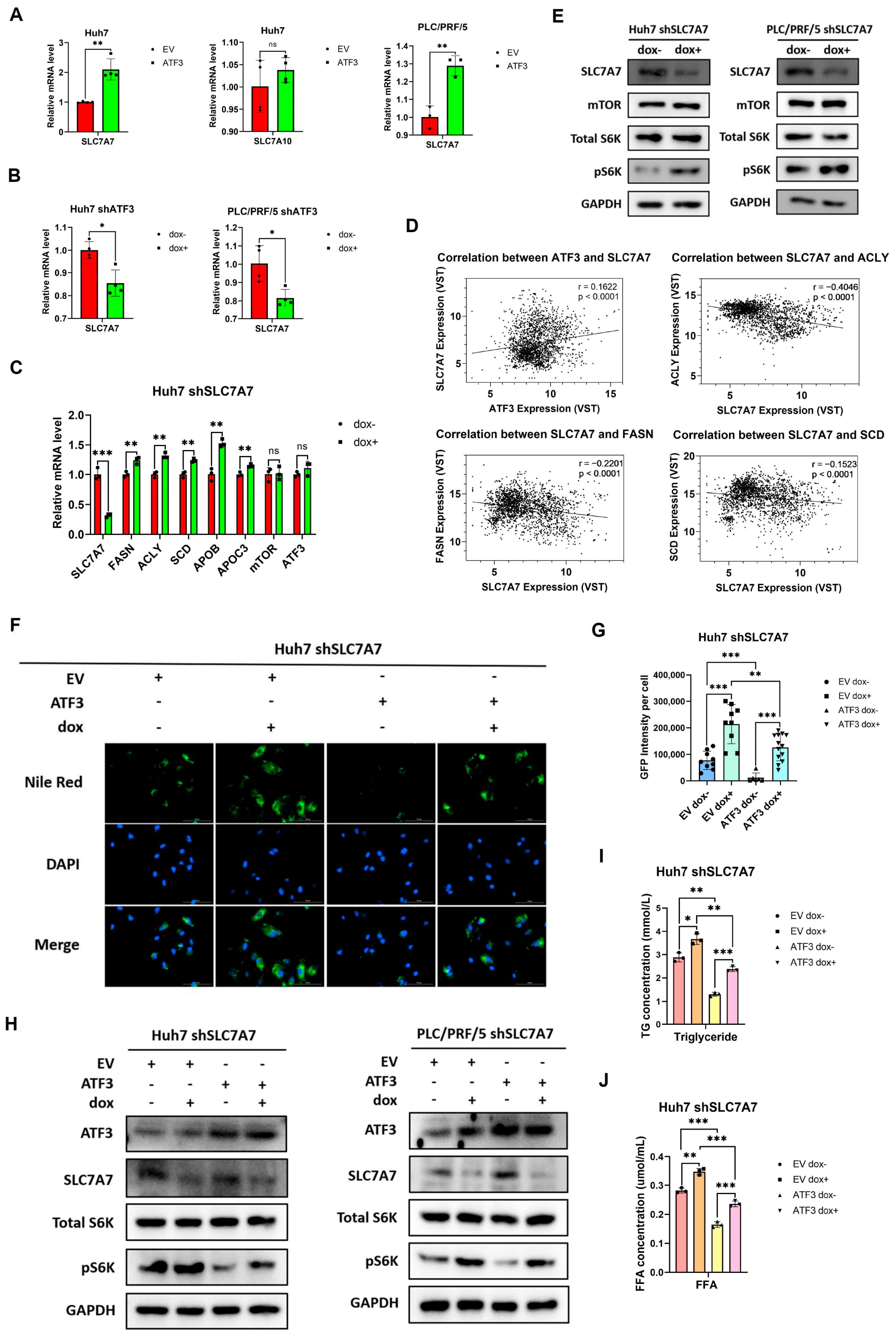
3.3. ATF3 Inhibits Lipid Biosynthesis in HCC Cells
3.4. ATF3 Represses Lipid Biosynthesis by Inhibiting mTORC1 Signaling
3.5. SLC7A7 Is Involved in the Inhibition of ATF3 on mTORC1 Signaling
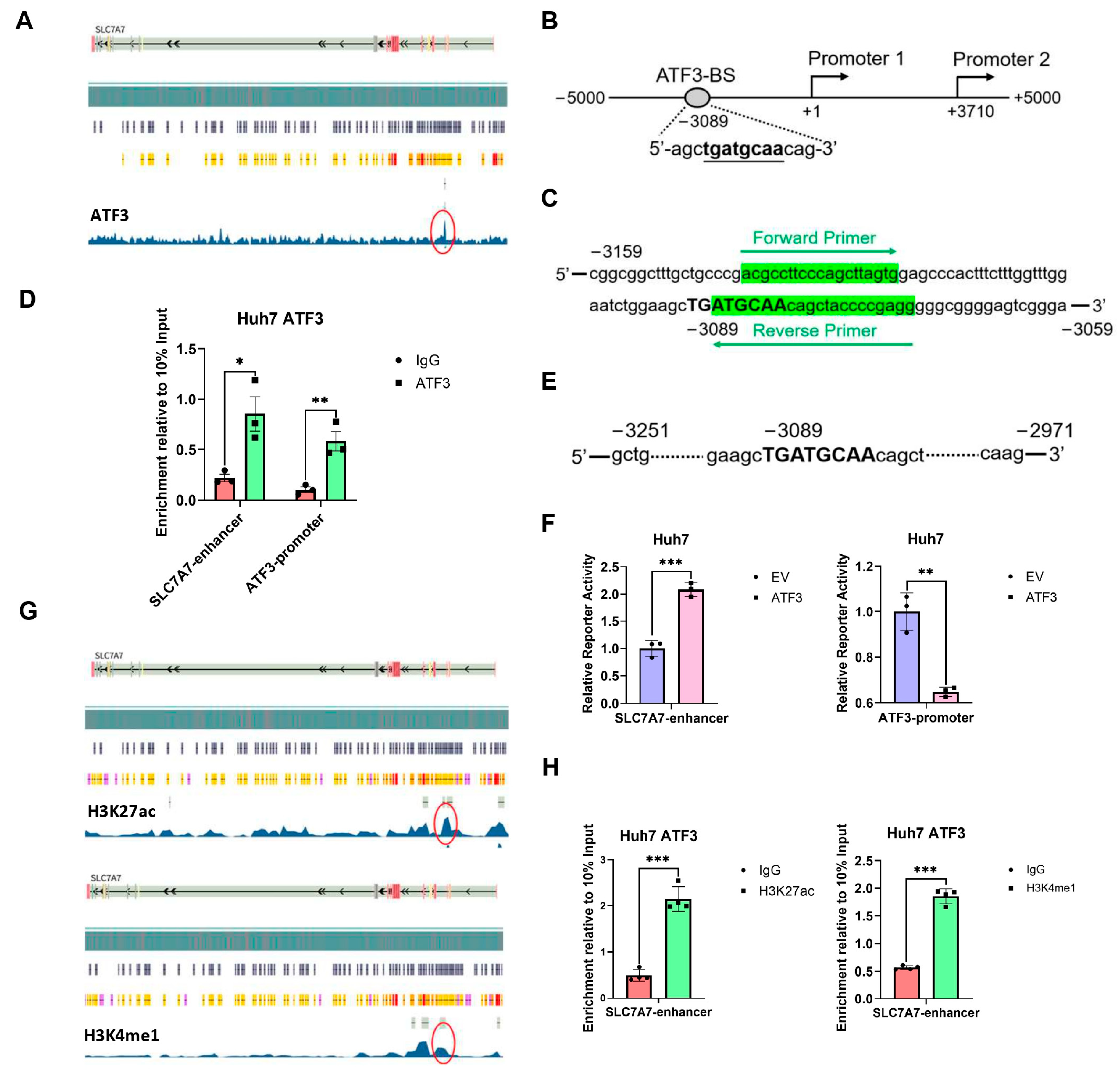
3.6. ATF3 Binds to SLC7A7 Enhancer and Activates Its Transcription
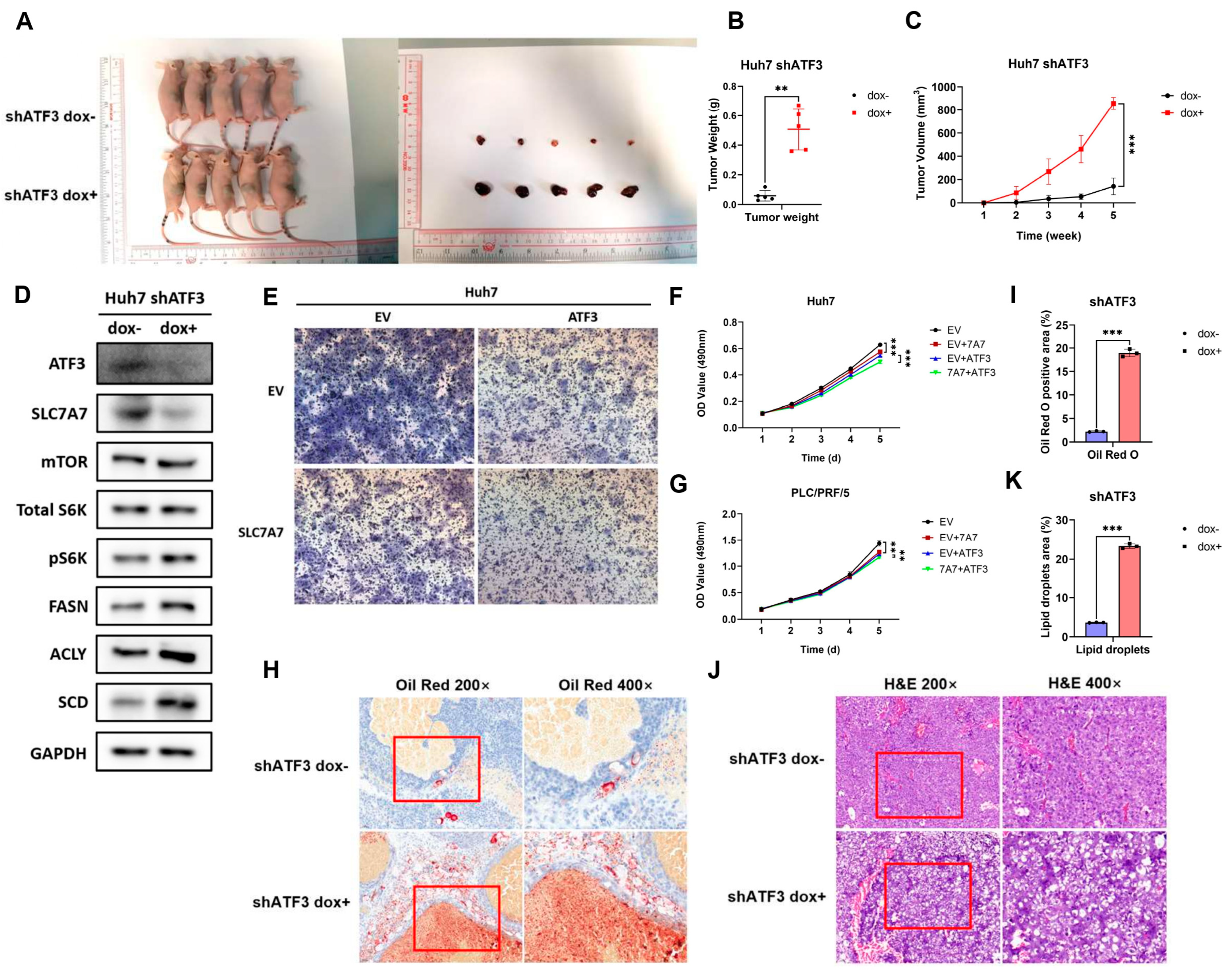
3.7. ATF3 Represses Tumor Growth and Lipogenesis In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Tsilimigras, D.I.; Ruff, S.M.; Mohseni, A.; Kamel, I.R.; Cloyd, J.M.; Pawlik, T.M. Management of Hepatocellular Carcinoma: A Review. JAMA Surg. 2023, 158, 410–420. [Google Scholar] [CrossRef]
- Ganesan, P.; Kulik, L.M. Hepatocellular Carcinoma: New Developments. Clin. Liver Dis. 2023, 27, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e812. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Guo, X.; Liu, X.; Lu, C.; Wang, A.; Wang, X.; Wang, W.; Chen, H.; Qin, W.; Liu, X.; et al. USP22 regulates lipidome accumulation by stabilizing PPARgamma in hepatocellular carcinoma. Nat. Commun. 2022, 13, 2187. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Yu, C.; Peng, C.; Feng, X.; Cheng, Q.; Wu, W.; Lu, Y.; et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021, 502, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Roessler, S.; Zhao, X.; Yu, Z.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.X.; Ye, Q.H.; Jia, H.L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075.e1061. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, J.; Calvisi, D.F.; Chen, X. Role of Lipogenesis Rewiring in Hepatocellular Carcinoma. Semin. Liver Dis. 2022, 42, 77–86. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Owen, J.L.; Zhang, Y.; Bae, S.H.; Farooqi, M.S.; Liang, G.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 16184–16189. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, H.; Jiang, B.; Ni, X.; Jiang, L.; Li, C.; Wang, X.; Zhang, F.; Ke, B.; Lu, L. Loss of ATF3 exacerbates liver damage through the activation of mTOR/p70S6K/HIF-1alpha signaling pathway in liver inflammatory injury. Cell Death Dis. 2018, 9, 910. [Google Scholar] [CrossRef]
- Ku, H.C.; Cheng, C.F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Li, L.; Song, S.; Fang, X.; Cao, D. Role of ATF3 as a prognostic biomarker and correlation of ATF3 expression with macrophage infiltration in hepatocellular carcinoma. BMC Med. Genom. 2021, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.F.; Ku, H.C.; Cheng, J.J.; Chao, S.W.; Li, H.F.; Lai, P.F.; Chang, C.C.; Don, M.J.; Chen, H.H.; Lin, H. Adipocyte browning and resistance to obesity in mice is induced by expression of ATF3. Commun. Biol. 2019, 2, 389. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, F.; Tong, Y.; Shi, D.; Zhang, J. CHD4 R975H mutant activates tumorigenic pathways and promotes stemness and M2-like macrophage polarization in endometrial cancer. Sci. Rep. 2024, 14, 18617. [Google Scholar] [CrossRef]
- Fu, C.; Donovan, W.P.; Shikapwashya-Hasser, O.; Ye, X.; Cole, R.H. Hot Fusion: An efficient method to clone multiple DNA fragments as well as inverted repeats without ligase. PLoS ONE 2014, 9, e115318. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ge, C.; Liu, Z.; Li, L.; Zhao, F.; Tian, H.; Chen, T.; Li, H.; Yao, M.; Li, J. ATF3 inhibits the tumorigenesis and progression of hepatocellular carcinoma cells via upregulation of CYR61 expression. J. Exp. Clin. Cancer Res. 2018, 37, 263. [Google Scholar] [CrossRef]
- Gyorffy, B. Transcriptome-level discovery of survival-associated biomarkers and therapy targets in non-small-cell lung cancer. Br. J. Pharmacol. 2024, 181, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.E.; Bishop, A.J.R. Correlation AnalyzeR: Functional predictions from gene co-expression correlations. BMC Bioinform. 2021, 22, 206. [Google Scholar] [CrossRef]
- Lin, L.; Yao, Z.; Bhuvaneshwar, K.; Gusev, Y.; Kallakury, B.; Yang, S.; Shetty, K.; He, A.R. Transcriptional regulation of STAT3 by SPTBN1 and SMAD3 in HCC through cAMP-response element-binding proteins ATF3 and CREB2. Carcinogenesis 2014, 35, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Kong, L.; Wang, S.; Xia, M.; Zhang, Y.; Wu, J.; Yang, F.; Zuo, S.; Wei, J. Oncolytic adenovirus encoding apolipoprotein A1 suppresses metastasis of triple-negative breast cancer in mice. J. Exp. Clin. Cancer Res. 2024, 43, 102. [Google Scholar] [CrossRef] [PubMed]
- Ieda, A.; Wada, M.; Moriyasu, Y.; Okuno, Y.; Zaima, N.; Moriyama, T. Ellagic Acid Suppresses ApoB Secretion and Enhances ApoA-1 Secretion from Human Hepatoma Cells, HepG2. Molecules 2021, 26, 3885. [Google Scholar] [CrossRef]
- Ren, L.; Yi, J.; Yang, Y.; Li, W.; Zheng, X.; Liu, J.; Li, S.; Yang, H.; Zhang, Y.; Ge, B.; et al. Systematic pan-cancer analysis identifies APOC1 as an immunological biomarker which regulates macrophage polarization and promotes tumor metastasis. Pharmacol. Res. 2022, 183, 106376. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, Z.; Xia, Y.; Shao, F.; Xia, W.; Wei, Y.; Li, X.; Qian, X.; Lee, J.H.; Du, L.; et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature 2020, 580, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; Cavaliere, P.M.; et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017, 171, 1545–1558.e1518. [Google Scholar] [CrossRef]
- Zhou, X.; Clister, T.L.; Lowry, P.R.; Seldin, M.M.; Wong, G.W.; Zhang, J. Dynamic Visualization of mTORC1 Activity in Living Cells. Cell Rep. 2015, 10, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Qiu, D.; Liang, X.; Huang, Y.; Wang, Y.; Jia, X.; Li, K.; Zhao, J.; Du, C.; Qiu, X.; et al. Lipotoxicity-induced STING1 activation stimulates MTORC1 and restricts hepatic lipophagy. Autophagy 2022, 18, 860–876. [Google Scholar] [CrossRef] [PubMed]
- Fotiadis, D.; Kanai, Y.; Palacin, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Aspects Med. 2013, 34, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Asaoka, Y.; Nagai, Y.; Namae, M.; Furutani-Seiki, M.; Nishina, H. SLC7 family transporters control the establishment of left-right asymmetry during organogenesis in medaka by activating mTOR signaling. Biochem. Biophys. Res. Commun. 2016, 474, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Jersin, R.A.; Tallapragada, D.S.P.; Madsen, A.; Skartveit, L.; Fjaere, E.; McCann, A.; Lawrence-Archer, L.; Willems, A.; Bjune, J.I.; Bjune, M.S.; et al. Role of the Neutral Amino Acid Transporter SLC7A10 in Adipocyte Lipid Storage, Obesity, and Insulin Resistance. Diabetes 2021, 70, 680–695. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, F.; Huang, Y.; He, L.; Li, Y.; Wan, Y.C.E.; Ding, Y.; Chan, K.M.; Xie, T.; Sun, H.; et al. ATF3 induction prevents precocious activation of skeletal muscle stem cell by regulating H2B expression. Nat. Commun. 2023, 14, 4978. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, C.D.; Liang, G.; Okamoto, Y.; Allen, A.E.; Hai, T. Transcriptional autorepression of the stress-inducible gene ATF3. J. Biol. Chem. 2000, 275, 16865–16870. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kim, Y.W.; Kang, J.; Kim, A. Histone H3K4me1 and H3K27ac play roles in nucleosome eviction and eRNA transcription, respectively, at enhancers. FASEB J. 2021, 35, e21781. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Ishizawa, T.; Hasegawa, K.; Aoki, T.; Takahashi, M.; Inoue, Y.; Sano, K.; Imamura, H.; Sugawara, Y.; Kokudo, N.; Makuuchi, M. Neither multiple tumors nor portal hypertension are surgical contraindications for hepatocellular carcinoma. Gastroenterology 2008, 134, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Pope, E.D., 3rd; Kimbrough, E.O.; Vemireddy, L.P.; Surapaneni, P.K.; Copland, J.A., 3rd; Mody, K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert. Opin. Ther. Targets 2019, 23, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Lu, D.; Hai, T.; Boyd, D.D. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005, 24, 2425–2435. [Google Scholar] [CrossRef]
- Liu, S.; Li, Z.; Lan, S.; Hao, H.; Baz, A.A.; Yan, X.; Gao, P.; Chen, S.; Chu, Y. The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses. Int. J. Mol. Sci. 2024, 25, 824. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Casano, A.M.; Henke, K.; Richter, K.; Peri, F. The SLC7A7 Transporter Identifies Microglial Precursors prior to Entry into the Brain. Cell Rep. 2015, 11, 1008–1017. [Google Scholar] [CrossRef]
- Ji, X.; Yang, X.; Wang, N.; Kang, M.; Wang, Y.; Rong, L.; Fang, Y.; Xue, Y. Function of SLC7A7 in T-Cell Acute Lymphoblastic Leukemia. Cell Physiol. Biochem. 2018, 48, 731–740. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhu, F.; Tong, Y.; Huang, Y.; Zhang, J. ATF3-SLC7A7 Axis Regulates mTORC1 Signaling to Suppress Lipogenesis and Tumorigenesis in Hepatocellular Carcinoma. Cells 2025, 14, 253. https://doi.org/10.3390/cells14040253
Zhang Q, Zhu F, Tong Y, Huang Y, Zhang J. ATF3-SLC7A7 Axis Regulates mTORC1 Signaling to Suppress Lipogenesis and Tumorigenesis in Hepatocellular Carcinoma. Cells. 2025; 14(4):253. https://doi.org/10.3390/cells14040253
Chicago/Turabian StyleZhang, Qinglin, Fengzhi Zhu, Yin Tong, Yunxing Huang, and Jiangwen Zhang. 2025. "ATF3-SLC7A7 Axis Regulates mTORC1 Signaling to Suppress Lipogenesis and Tumorigenesis in Hepatocellular Carcinoma" Cells 14, no. 4: 253. https://doi.org/10.3390/cells14040253
APA StyleZhang, Q., Zhu, F., Tong, Y., Huang, Y., & Zhang, J. (2025). ATF3-SLC7A7 Axis Regulates mTORC1 Signaling to Suppress Lipogenesis and Tumorigenesis in Hepatocellular Carcinoma. Cells, 14(4), 253. https://doi.org/10.3390/cells14040253