
Immune Modulation in Alzheimer’s Disease: From Pathogenesis to Immunotherapy





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune System Modifications in AD
2.1. Microglia and AD Pathology
2.1.1. Microglia and Aβ
2.1.2. Microglia and Tau
2.2. Peripheral Blood Immune Cells
2.2.1. Monocytes
2.2.2. Neutrophils
2.2.3. NK Cells
2.3. Adaptive Immunity
2.3.1. T Cells
2.3.2. B Cells
2.4. Immune Microenvironment in the Brain Parenchyma
3. Immunotherapy for AD
3.1. Immunotherapy Based on Targeting Aβ
3.1.1. Active Immunotherapy
3.1.2. Passive Immunotherapy
3.2. Limitations in Amyloid-Targeting Therapies
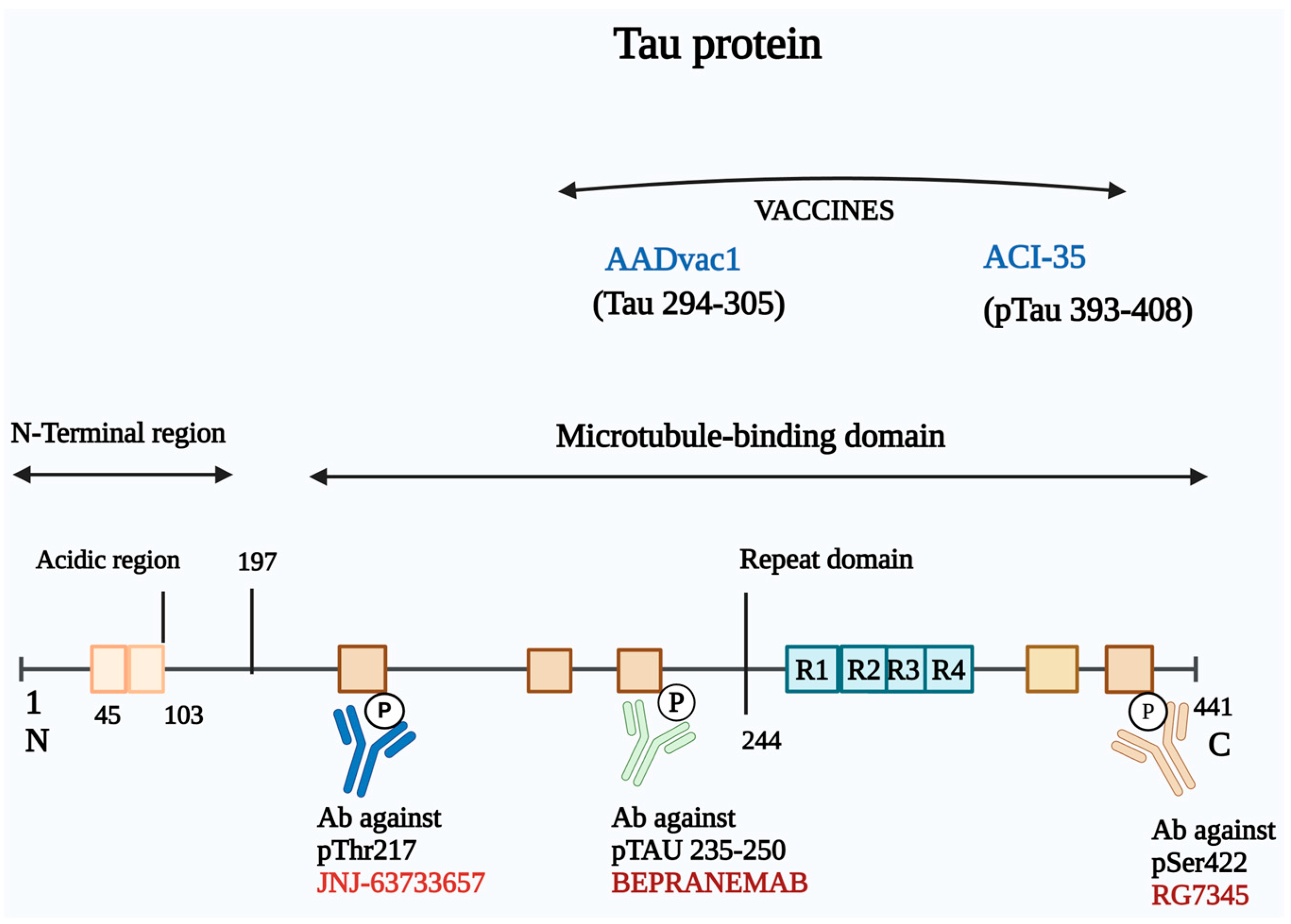
3.3. Immunotherapy Based on Targeting Tau
3.3.1. Active Immunotherapy
3.3.2. Passive Immunotherapy
3.4. Immunotherapy Targeting Microglia
4. Challenges in Immunotherapy of AD
4.1. Limitations of Animal Models
4.2. Suboptimal Immune Responses
4.3. Complexities in Preventive Clinical Trials
4.4. Insufficient Characterization of Immune Responses
4.5. Identification of Therapeutic Targets
4.6. Challenges in Delivery Drugs Across the Blood–Brain Barrier
4.7. Mitigating Adverse Effects
4.8. Need for Comprehensive Treatment Approaches
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, Y.-X.; Jiang, X.-J.; Lu, B.; Gao, Q.; Chen, Y.-F.; Wu, D.-B.; Zeng, W.-Y.; Yang, L.; Li, H.-H.; Yu, B. Roles of Gut Microbiota inPathogenesis of Alzheimer’s Disease and Therapeutic Effects of Chinese Medicine. Chin. J. Integr. Med. 2022, 28, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef] [PubMed]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef] [PubMed]
- Barber, R.C. The genetics of Alzheimer’s disease. Scientifica 2012, 2012, 246210. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-D.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef]
- Chen, C.; Liao, J.; Xia, Y.; Liu, X.; Jones, R.; Haran, J.; McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 2022, 71, 2233–2252. [Google Scholar] [CrossRef]
- Askarova, S.; Umbayev, B.; Masoud, A.-R.; Kaiyrlykyzy, A.; Safarova, Y.; Tsoy, A.; Olzhayev, F.; Kushugulova, A. The Links Between the Gut Microbiome, Aging, Modern Lifestyle and Alzheimer’s Disease. Front. Cell. Infect. Microbiol. 2020, 10, 104. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Alawieyah Syed Mortadza, S.; Sim, J.A.; Neubrand, V.E.; Jiang, L.H. A critical role of TRPM2 channel in Aβ42-induced microglial activation and generation of tumor necrosis factor-α. Glia 2018, 66, 562–575. [Google Scholar] [CrossRef]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [PubMed]
- Hartmann, T.; Bieger, S.C.; Bruhl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease Ab40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Sweeney, D.; Wang, R.; Thinakaran, G.; Lo, A.C.; Sisodia, S.S.; Greengard, P.; Gandy, S. Generation of Alzheimer b-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. USA 1997, 94, 3748–3752. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Eckman, E.A. Endothelin-converting enzymes degrade intracellular b-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J. Biol. Chem. 2013, 288, 5606–5615. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Alonso, A.D.C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef]
- Almeida, C.G.; Takahashi, R.H.; Gouras, G.K. b-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 2006, 26, 4277–4288. [Google Scholar] [CrossRef]
- Gruenberg, J.; Stenmark, H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 317–323. [Google Scholar] [CrossRef]
- Cherry, J.; Olschowka, J.; O’Banion, M. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Lu, H.; Yang, Q.; Wu, H.; Wang, J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol. Neurobiol. 2017, 54, 1874–1886. [Google Scholar] [CrossRef]
- Felsky, D.; Roostaei, T.; Nho, K.; Risacher, S.; Bradshaw, E.; Petyuk, V.; Schneider, J.A.; Saykin, A.; Bennett, D.A.; De Jager, P.L. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat. Commun. 2019, 10, 409. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Götzl, J.K.; Brendel, M.; Werner, G.; Parhizkar, S.; Monasor, L.S.; Kleinberger, G.; Colombo, A.; Deussing, M.; Wagner, M.; Winkelmann, J.; et al. Opposite microglial activation stages upon loss of PGRN or TREM2 result in reduced cerebral glucose metabolism. EMBO Mol. Med. 2019, 11, e9711. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tang, G.; Zhou, C.; Guo, H.; Hu, Z.; Hu, Q.; Li, G. Revisiting the intersection of microglial activation and neuroinflammation in Alzheimer’s disease from the perspective of ferroptosis. Chem. Biol. Interact. 2023, 375, 110387. [Google Scholar] [CrossRef]
- Yao, K.; Zu, H.B. Microglial polarization: Novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, H.; Liu, X.; Zhang, W.; Zhang, S.; Jiao, B. APP, PSEN1, and PSEN2 variants in Alzheimer’s disease: Systematic re-evaluation according to ACMG guidelines. Front. Aging Neurosci. 2021, 13, 695808. [Google Scholar] [CrossRef]
- Huda, T.I.; Diaz, M.J.; Gozlan, E.C.; Chobrutskiy, A.; Chobrutskiy, B.I.; Blanck, G. Immunogenomics Parameters for Patient Stratification in Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 88, 619–629. [Google Scholar] [CrossRef]
- Babić Leko, M.; Nikolac Perković, M.; Klepac, N.; Švob Štrac, D.; Borovečki, F.; Pivac, N.; Hof, P.R.; Šimić, G. IL-1β, IL-6, IL-10, and TNFα single nucleotide polymorphisms in humans influence the susceptibility to Alzheimer’s disease pathology. J. Alzheimer’s Dis. 2020, 75, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Gauthier, S.; Cummings, J. The utility of the new research diagnostic criteria for Alzheimer’s disease. Int. Psychogeriatr. 2013, 25, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Dhami, M.; Raj, K.; Singh, S. Relevance of gut microbiota to Alzheimer’s Disease (AD): Potential effects of probiotic in management of AD. Aging Health Res. 2023, 3, 100128. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802, Erratum in Sci. Rep. 2017, 7, 46856. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 108. [Google Scholar] [CrossRef]
- Li, Q.; Wu, Y.; Chen, J.; Xuan, A.; Wang, X. Microglia and immunotherapy in Alzheimer’s disease. Acta Neurol. Scand. 2022, 145, 273–278. [Google Scholar] [CrossRef]
- Davis, I.D. An overview of cancer immunotherapy. Immunol. Cell Biol. 2000, 78, 179–195. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, lifestyle stress, and neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef]
- Ji, K.; Akgul, G.; Wollmuth, L.; Tsirka, S. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.; Friedman, B.; Dejanovic, B.; Sheng, M. Microglia in brain development, homeostasis, and neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.; Walker, D.; Brachova, L.; Beach, T.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 13, 856376. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, Z.; Wang, D.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; Can, D.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Zhang, P.F.; Hu, H.; Tan, L.; Yu, J.T. Microglia Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3388–3404. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.; Cardona, A.; Ransohoff, R.; Lamb, B. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Llorens-Martín, M.; Perea, J.; Jurado-Arjona, J.; Rabano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Sheng, J.; Jones, R.; Zhou, X.; McGinness, J.; Van Eldik, L.; Mrak, R.E.; Griffin, W.S. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: Potential significance for tau protein phosphorylation. Neurochem. Int. 2001, 39, 341–348. [Google Scholar] [CrossRef]
- Heneka, M.; Kummer, M.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Stancu, I.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- García-Culebras, A.; Cuartero, M.I.; Peña-Martínez, C.; Moraga, A.; Vázquez-Reyes, S.; de Castro-Millán, F.J.; Cortes-Canteli, M.; Lizasoain, I.; Moro, M.A. Myeloid cells in vascular dementia and Alzheimer’s disease: Possible therapeutic targets? Br. J. Pharmacol. 2024, 181, 777–798. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Dani, N.; Herbst, R.H.; McCabe, C.; Green, G.S.; Kaiser, K.; Head, J.P.; Cui, J.; Shipley, F.B.; Jang, A.; Dionne, D.; et al. A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell 2021, 184, 3056–3074.e21. [Google Scholar] [CrossRef] [PubMed]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef]
- Mehdi Jorfi, M.; Maaser-Hecker, A.; Tanzi, R.E. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023, 15, 6. [Google Scholar] [CrossRef]
- Pietronigro, E.C.; Della Bianca, V.; Zenaro, E.; Constantin, G. NETosis in Alzheimer’s disease. Front. Immunol. 2017, 8, 211. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Chen, M.; Inestrosa, N.C.; Ross, G.S.; Fernandez, H.L. Platelets are the primary source of amyloid beta peptide in human blood. Biochem. Biophys. Res. Commun. 1995, 213, 96–103. [Google Scholar] [CrossRef]
- Bassendine, M.F.; Taylor-Robinson, S.D.; Fertleman, M.; Khan, M.; Neely, D. Is Alzheimer’s disease a liver disease of the brain? J. Alzheimer’s Dis. 2020, 75, 1–14. [Google Scholar] [CrossRef]
- Kitazume, S.; Yoshihisa, A.; Yamaki, T.; Oikawa, M.; Tachida, Y.; Ogawa, K.; Imamaki, R.; Hagiwara, Y.; Kinoshita, N.; Takeishi, Y.; et al. Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets: A novel biomarker for acute coronary syndrome. J. Biol. Chem. 2012, 287, 40817–40825. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Kokjohn, T.A.; Watson, M.D.; Woods, A.S.; Cotter, R.J.; Sue, L.I.; Kalback, W.M.; Emmerling, M.R.; Beach, T.G.; Roher, A.E. Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am. J. Pathol. 2000, 156, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, W.E.; Melchor, J.P. Disruption of pathologic amyloid betaprotein fibril assembly on the surface of cultured human cerebrovascular smooth muscle cells. Amyloid 2001, 8 (Suppl. S1), 20–27. [Google Scholar] [PubMed]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Auffray, C.; Sieweke, M.; Geissmann, F. Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef]
- Gren, S.; Rasmussen, T.; Janciauskiene, S.; Håkansson, K.; Gerwien, J.; Grip, O. A single-cell gene-expression profile reveals inter-cellular heterogeneity within human monocyte subsets. PLoS ONE 2015, 10, e0144351. [Google Scholar] [CrossRef]
- Thome, A.; Faridar, A.; Beers, D.; Thonhoff, J.; Zhao, W.; Wen, S.; Pascual, B.; Masdeu, J.C.; Appel, S.H. Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 61. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Croxford, A.L.; Becher, B.; Landreth, G.E. Plaque-associated myeloid cells derive from resident microglia in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20191374. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Naert, G.; Rivest, S. A deficiency in CCR2+ monocytes: The hidden side of Alzheimer’s disease. J. Mol. Cell Biol. 2013, 5, 284–293. [Google Scholar] [CrossRef]
- Rego, S.; Sanchez, G.; Da Mesquita, S. Current views on meningeal lymphatics and immunity in aging and Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 55. [Google Scholar] [CrossRef]
- Martin, E.; Boucher, C.; Fontaine, B.; Delarasse, C. Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer’s disease models: Effects of aging and amyloid pathology. Aging Cell 2017, 16, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Bai, Y.D.; Yu, Z.Y.; Li, H.Y.; Liu, J.; Tan, C.R.; Zeng, G.H.; Tu, Y.F.; Sun, P.Y.; Jia, Y.J.; et al. Improving blood monocyte energy metabolism enhances its ability to phagocytose amyloid-β and prevents Alzheimer’s disease type pathology and cognitive deficits. Neurosci. Bull. 2023, 39, 1775–1788. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008, 14, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Mocsai, A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J. Exp. Med. 2013, 210, 1283–1299. [Google Scholar] [CrossRef]
- Stock, A.; Kasus-Jacobi, A.; Pereira, H. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 240. [Google Scholar] [CrossRef]
- Wu, C.; Bawa, K.; Ouk, M.; Leung, N.; Yu, D.; Lanctôt, K.L.; Herrmann, N.; Pakosh, M.; Swardfager, W. Neutrophil activation in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta analysis of protein markers in blood and cerebrospinal fluid. Ageing Res. Rev. 2020, 62, 101130. [Google Scholar] [CrossRef]
- Park, J.; Baik, S.; Mook-Jung, I.; Irimia, D.; Cho, H. Mimicry of central-peripheral immunity in Alzheimer’s disease and discovery of neurodegenerative roles in neutrophil. Front. Immunol. 2019, 10, 2231. [Google Scholar] [CrossRef]
- Le Page, A.; Bourgade, K.; Lamoureux, J.; Frost, E.; Pawelec, G.; Larbi, A.; Witkowski, J.M.; Dupuis, G.; Fülöp, T. NK cells are activated in amnestic mild cognitive impairment but not in mild Alzheimer’s disease patients. J. Alzheimer’s Dis. 2015, 46, 93–107. [Google Scholar] [CrossRef]
- Solerte, S.B.; Cravello, L.; Ferrari, E.; Fioravanti, M. Overproduction of IFNgamma and TNF-alpha from natural killer (NK) cells is associated with abnormal NK reactivity and cognitive derangement in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 917, 331–340. [Google Scholar] [CrossRef]
- Zhang, Y.; Fung, I.; Sankar, P.; Chen, X.; Robison, L.; Ye, L.; D’Souza, S.S.; Salinero, A.E.; Kuentzel, M.L.; Chittur, S.V.; et al. Depletion of NK cells improves cognitive function in the alzheimer disease mouse model. J. Immunol. 2020, 205, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Villacampa, N.; Heneka, M.T. Microglia: You’ll Never Walk Alone! Immunity 2018, 48, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; Pan, S. Discovery and validation of key biomarkers based on immune infiltrates in Alzheimer’s disease. Front. Genet. 2021, 12, 658323. [Google Scholar] [CrossRef]
- Spani, C.; Suter, T.; Derungs, R.; Ferretti, M.T.; Welt, T.; Wirth, F.; Gericke, C.; Nitsch, R.M.; Kulic, L. Reduced β-amyloid pathology in an APP transgenic mouse model of Alzheimer’s disease lacking functional B and T cells. Acta Neuropathol. Commun. 2015, 3, 71. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Fu, Z.; Kipnis, J. The meningeal lymphatic system: A new player in neurophysiology. Neuron 2018, 100, 375–388. [Google Scholar] [CrossRef]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 18, 7967. [Google Scholar] [CrossRef]
- Baik, S.H.; Cha, M.Y.; Hyun, Y.M.; Cho, H.; Hamza, B.; Kim, D.K.; Han, S.H.; Choi, H.; Kim, K.H.; Moon, M.; et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Maes, M.; Puri, B.K. Could Alzheimer’s disease originate in the periphery and if so how so? Mol. Neurobiol. 2019, 56, 406–434. [Google Scholar] [CrossRef]
- Lueg, G.; Gross, C.C.; Lohmann, H.; Johnen, A.; Kemmling, A.; Deppe, M.; Groger, J.; Minnerup, J.; Wiendl, H.; Meuth, S.G.; et al. Clinical relevance of specific t-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol. Aging 2015, 36, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 2019, 4, eaau8380. [Google Scholar] [CrossRef] [PubMed]
- Stojić-Vukanić, Z.; Hadžibegović, S.; Nicole, O.; Nacka-Aleksić, M.; Leštarević, S.; Leposavić, G. CD8+ T cell-mediated mechanisms contribute to the progression of neurocognitive impairment in both multiple sclerosis and Alzheimer’s disease? Front. Immunol. 2020, 11, 566225. [Google Scholar] [CrossRef]
- Seifert, M.; Küppers, R. Human memory B cells. Leukemia 2016, 30, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Oliver, A.; Kearney, J. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 2001, 14, 617–629. [Google Scholar] [CrossRef]
- Bulati, M.; Buffa, S.; Martorana, A.; Gervasi, F.; Camarda, C.; Azzarello, D.M.; Monastero, R.; Caruso, C.; Colonna-Romano, G. Double negative (IgG+IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J. Alzheimer’s Dis. 2015, 44, 1241–1251. [Google Scholar] [CrossRef]
- Sabatino, J.; Pröbstel, A.; Zamvil, S. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat. Rev. Neurosci. 2019, 20, 728–745. [Google Scholar] [CrossRef]
- Nataf, S.; Guillen, M.; Pays, L. Common neurodegeneration-associated proteins are physiologically expressed by human B lymphocytes and are interconnected via the Inflammation/Autophagy-Related proteins TRAF6 and SQSTM1. Front. Immunol. 2019, 10, 2704. [Google Scholar] [CrossRef]
- Dodel, R.; Hampel, H.; Depboylu, C.; Lin, S.; Gao, F.; Schock, S.; Jäckel, S.; Wei, X.; Buerger, K.; Höft, C.; et al. Human antibodies against amyloid beta peptide: A potential treatment for Alzheimer’s disease. Ann. Neurol. 2002, 52, 253–256. [Google Scholar] [CrossRef]
- Kim, K.; Wang, X.; Ragonnaud, E.; Bodogai, M.; Illouz, T.; DeLuca, M.; McDevitt, R.A.; Gusev, F.; Okun, E.; Rogaev, E.; et al. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat. Commun. 2021, 12, 2185. [Google Scholar] [CrossRef]
- Schwartz, M.; Kipnis, J.; Rivest, S.; Prat, A. How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 2013, 33, 17587–17596. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef]
- Rexach, J.E.; Polioudakis, D.; Yin, A.; Swarup, V.; Chang, T.S.; Nguyen, T.; Sarkar, A.; Chen, L.; Huang, J.; Lin, L.C.; et al. Tau pathology drives dementia risk-associated gene networks toward chronic inflammatory states and immunosuppression. Cell Rep. 2020, 33, 108398. [Google Scholar] [CrossRef]
- Rosenzweig, N.; Dvir-Szternfeld, R.; Tsitsou-Kampeli, A.; Keren-Shaul, H.; Ben-Yehuda, H.; Weill-Raynal, P.; Cahalon, L.; Kertser, A.; Baruch, K.; Amit, I.; et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat. Commun. 2019, 10, 465. [Google Scholar] [CrossRef]
- Aljassabi, A.; Zieneldien, T.; Kim, J.; Regmi, D.; Cao, C. Alzheimer’s Disease Immunotherapy: Current Strategies and Future Prospects. J. Alzheimer’s Dis. 2024, 98, 755–772. [Google Scholar] [CrossRef]
- Wraith, D.C. The Future of Immunotherapy: A 20-Year Perspective. Front. Immunol. 2017, 8, 1668. [Google Scholar] [CrossRef]
- Vashisth, K.; Sharma, S.; Ghosh, S.; Babu, M.A.; Ghosh, S.; Iqbal, D.; Kamal, M.; Almutary, A.G.; Jha, S.K.; Ojha, S.; et al. Immunotherapy in Alzheimer’s Disease: Current Status and Future Directions. J. Alzheimer’s Dis. 2024, 101, S23–S39. [Google Scholar] [CrossRef]
- Hur, J.Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef]
- Mullard, A. BACE failures lower AD expectations, again. Nat. Rev. Drug Discov. 2018, 17, 385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Rovira, M.B.; Guerra, M.L.S.; Rey, M.J.; Costa-Juss, F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004, 14, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Black, R.; Thal, L.J.; Fox, N.C.; Daniels, M.; McLennan, G.; Tompkins, C.; Leibman, C.; Pomfret, M.; Grundman, M. Long-term follow-up of patients immunized with AN1792: Reduced functional decline in antibody responders. Curr. Alzheimer Res. 2009, 6, 144–151. [Google Scholar] [CrossRef]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Vogt, A.-C.S.; Jennings, G.T.; Mohsen, M.O.; Vogel, M.; Bachmann, M.F. Alzheimer’s disease: A brief history of immunotherapies targeting amyloid β. Int. J. Mol. Sci. 2023, 24, 3895. [Google Scholar] [CrossRef]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- Haeberlein, S.B.; von Hehn, C.; Tian, Y.; Chalkias, S.; Muralidharan, K.K.; Chen, T.; Wu, S.; Skordos, L.; Nisenbaum, L.; Rajagovindan, R.; et al. Emerge and engage topline results: Phase 3 studies of aducanumab in early Alzheimer’s disease. Alzheimer Dement. 2020, 16, e047259. [Google Scholar] [CrossRef]
- Schneider, L.S. Aducanumab Trials EMERGE But Don’t ENGAGE. J. Prev. Alzheimer’s Dis. 2022, 9, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef]
- Sperling, R.A.; Donohue, M.C.; Raman, R.; Rafii, M.S.; Johnson, K.; Masters, C.L.; van Dyck, C.H.; Iwatsubo, T.; Marshall, G.A.; Yaari, R.; et al. Trial of solanezumab in preclinical Alzheimer’s disease. N. Engl. J. Med. 2023, 389, 1096–1107. [Google Scholar] [CrossRef]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimer Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef]
- Dash, U.C.; Bhol, N.K.; Swain, S.K.; Samal, R.R.; Nayak, P.K.; Raina, V.; Panda, S.K.; Kerry, R.G.; Duttaroy, A.K.; Jena, A.B. Oxidative stress and inflammation in the pathogenesis of neurological disorders: Mechanisms and implications. Acta Pharm. Sin. B 2024. [Google Scholar] [CrossRef]
- Rummel, N.G.; Butterfield, D.A. Altered metabolism in Alzheimer disease brain: Role of oxidative stress. Antioxid. Redox Signal 2022, 36, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Aborode, A.T.; Pustake, M.; Awuah, W.A.; Alwerdani, M.; Shah, P.; Yarlagadda, R.; Ahmad, S.; Silva Correia, I.F.; Chandra, A.; Nansubuga, E.P.; et al. Targeting oxidative stress mechanisms to treat Alzheimer’s and Parkinson’s disease: A critical review. Oxid. Med. Cell. Longev. 2022, 2022, 7934442. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 639–649. [Google Scholar] [CrossRef]
- Augustyniak, A.; Bartosz, G.; Čipak, A.; Duburs, G.; Horáková, L.; Łuczaj, W.; Majekova, M.; Odysseos, A.D.; Rackova, L.; Skrzydlewska, E.; et al. Natural and synthetic antioxidants: An updated overview. Free Radic. Res. 2010, 44, 1216–1262. [Google Scholar] [CrossRef]
- Martinez, B.; Peplow, P. Neuroprotection by immunomodulatory agents in animal models of Parkinson’s disease. Neural Regen. Res. 2018, 13, 1493–1506. [Google Scholar]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and therapeutic perspectives of oxidative stress and neurodegenerative diseases: A narrative review. Adv. Ther. 2019, 37, 113–139. [Google Scholar] [CrossRef]
- Kenche, V.B.; Barnham, K.J. Alzheimer’s disease &metals: Therapeutic opportunities. Br. J. Pharmacol. 2011, 163, 211–219. [Google Scholar] [PubMed]
- Feng, Z.; Qin, C.; Chang, Y.; Zhang, J.T. Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer’s disease. Free Radic. Biol. Med. 2006, 40, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Budzynska, B.; Boguszewska-Czubara, A.; Kruk-Slomka, M.; Skalicka-Wozniak, K.; Michalak, A.; Musik, I.; Biala, G. Effects of imperatorin on scopolamine-induced cognitive impairment and oxidative stress in mice. Psychopharmacology 2015, 232, 931–942. [Google Scholar] [CrossRef]
- Javed, H.; Khan, M.M.; Khan, A.; Vaibhav, K.; Ahmad, A.; Khuwaja, G.; Ahmed, E.; Raza, S.S.; Ashafaq, M.; Tabassum, R.; et al. S-allyl cysteine attenuates oxidative stress associated cognitive impairment and neurodegeneration in mouse model of streptozotocin-induced experimental dementia of Alzheimer’s type. Brain Res. 2011, 1389, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in Alzheimer’s disease: Interplay mechanisms and clinical translation. J. Neuroinflamm. 2023, 20, 165. [Google Scholar] [CrossRef]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P.; et al. FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against Tau protein pathology in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 108. [Google Scholar] [CrossRef]
- Bittar, A.; Bhatt, N.; Kayed, R. Advances and considerations in AD Tautargeted immunotherapy. Neurobiol. Dis. 2020, 134, 104707. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Tortelli, R.; Galizia, I.; Prete, C.; Daniele, A.; et al. Tau-based therapeutics for Alzheimer’s disease: Active and passive immunotherapy. Immunotherapy 2016, 8, 1119–1134. [Google Scholar] [CrossRef]
- Novak, P.; Kovacech, B.; Katina, S.; Schmidt, R.; Scheltens, P.; Kontsekova, E.; Ropele, S.; Fialova, L.; Kramberger, M.; Paulenka-Ivanovova, N.; et al. ADAMANT: A placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nat. Aging 2021, 1, 521–534. [Google Scholar] [CrossRef]
- Ayalon, G.; Lee, S.H.; Adolfsson, O.; Foo-Atkins, C.; Atwal, J.K.; Blendstrup, M.; Booler, H.; Bravo, J.; Brendza, R.; Brunstein, F.; et al. Antibody semorinemab reduces Tau pathology in a transgenic mouse model and engages Tau in patients with Alzheimer’s disease. Sci. Transl. Med. 2021, 13, eabb2639. [Google Scholar] [CrossRef]
- Monteiro, C.; Toth, B.; Brunstein, F.; Bobbala, A.; Datta, S.; Ceniceros, R.; Sanabria Bohorquez, S.M.; Anania, V.G.; Wildsmith, K.R.; Schauer, S.P.; et al. Randomized phase II study of the safety and efficacy of semorinemab in participants with mild-to-moderate Alzheimer disease: Lauriet. Neurology 2023, 101, e1391–e1401. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat. Rev. Drug Discov. 2021, 20, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sopko, R.; Golonzhka, O.; Arndt, J.; Quan, C.; Czerkowicz, J.; Cameron, A.; Smith, B.; Murugesan, Y.; Gibbons, G.; Kim, S.J.; et al. Characterization of tau binding by gosuranemab. Neurobiol. Dis. 2020, 146, 105120. [Google Scholar] [CrossRef]
- Dam, T.; Boxer, A.L.; Golbe, L.I.; H¨oglinger, G.U. Safety and efficacy of anti-tau monoclonal antibody gosuranemab in progressive supranuclear palsy: A phase 2, randomized, placebo-controlled trial. Nat. Med. 2021, 27, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Mikytuck, B.; Suh, E.; Gibbons, G.S.; Van Deerlin, V.M.; Vaishnavi, S.N.; Spindler, M.A.; Massimo, L.; Grossman, M.; Trojanowski, J.Q.; et al. Tau immunotherapy is associated with glial responses in FTLD-tau. Acta Neuropathol. 2021, 142, 243–257. [Google Scholar] [CrossRef] [PubMed]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and clinical development of ABBV-8E12, a humanized anti-Tau antibody, for treatment of Alzheimer’s disease and other Tauopathies. J. Prev. Alzheimer Dis. 2017, 4, 236–241. [Google Scholar]
- Albert, M.; Mairet-Coello, G.; Danis, C.; Lieger, S.; Caillierez, R.; Carrier, S.; Skrobala, E.; Landrieu, I.; Michel, A.; Schmitt, M.; et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019, 142, 1736–1750. [Google Scholar] [CrossRef]
- Roberts, M.; Sevastou, I.; Imaizumi, Y.; Mistry, K.; Talma, S.; Dey, M.; Gartlon, J.; Ochiai, H.; Zhou, Z.; Akasofu, S.; et al. Preclinical characterisation of E2814, a high-affinity antibody targeting the microtubule-binding repeat domain of tau for passive immunotherapy in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 13. [Google Scholar] [CrossRef]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging microglia biology defnes novel therapeutic approaches for Alzheimer’s disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef]
- Pascoal, A.T.; Benedet, L.A.; Ashton, J.N.; Kang, S.M.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Ransohof, R.M. How neuroinfammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Claudio, C.A. Early and late CNS infammation in Alzheimer’s disease: Two extremes of a continuum? Trends Pharmacol. Sci. 2017, 38, 956–966. [Google Scholar]
- Song, C.; Shi, J.; Zhang, P.; Zhang, Y.; Xu, J.; Zhao, L.; Zhang, R.; Wang, H.; Chen, H. Immunotherapy for Alzheimer’s disease: Targeting β-amyloid and beyond. Transl. Neurodegener. 2022, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fuid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Schindowski, K.; Eckert, A.; Peters, J.; Gorriz, C.; Schramm, U.; Weinandi, T.; Maurer, K.; Frölich, L.; Müller, W.E. Increased T-cell reactivity and elevated levels of CD8+ memory T-cells in Alzheimer’s disease-patients and T-cell hyporeactivity in an Alzheimer’s disease-mouse model: Implications for immunotherapy. Neuromol. Med. 2007, 9, 340–354. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium oligomannate: Frst approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinfammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Guo, X.; Yan, L.; Zhang, D.; Zhao, Y. Passive immunotherapy for Alzheimer’s disease. Ageing Res. Rev. 2024, 94, 102192. [Google Scholar] [CrossRef]
- Xu, Q.-Q.; Yang, W.; Zhong, M.; Lin, Z.-X.; E Gray, N.; Xian, Y.-F. Animal models of Alzheimer’s disease: Preclinical insights and challenges. Acta Mater. Medica 2023, 2, 192–215. [Google Scholar] [CrossRef]
- Spencer, B.; Masliah, E. Immunotherapy for Alzheimer’s disease: Past, present and future. Front. Aging Neurosci. 2014, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Langbaum, J.B.; Zissimopoulos, J.; Au, R.; Bose, N.; Edgar, C.J.; Ehrenberg, E.; Fillit, H.; Hill, C.V.; Hughes, L.; Irizarry, M.; et al. Recommendations to address key recruitment challenges of Alzheimer’s disease clinical trials. Alzheimer’s Dement. 2023, 19, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Clay, T.M.; Hobeika, A.C.; Mosca, P.J.; Lyerly, H.K.; Morse, M.A. Assays for monitoring cellular immune responses to active immunotherapy of cancer. Clin. Cancer Res. 2001, 7, 1127–1135. [Google Scholar] [PubMed]
- Upton, D.H.; Ung, C.; George, S.M.; Tsoli, M.; Kavallaris, M.; Ziegler, D.S. Challenges and opportunities to penetrate the blood-brain barrier for brain cancer therapy. Theranostics 2022, 12, 4734–4752. [Google Scholar] [CrossRef]
- Goulet, D.R.; Atkins, W.M. Considerations for the design of antibody-based therapeutics. J. Pharm. Sci. 2019, 109, 74–103. [Google Scholar] [CrossRef]
- Salloway, S.P.; Sevingy, J.; Budur, K.; Pederson, J.T.; DeMattos, R.B.; Von Rosenstiel, P.; Paez, A.; Evans, R.; Weber, C.J.; Hendrix, J.A.; et al. Advancing combination therapy for Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12073. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balkhi, S.; Di Spirito, A.; Poggi, A.; Mortara, L. Immune Modulation in Alzheimer’s Disease: From Pathogenesis to Immunotherapy. Cells 2025, 14, 264. https://doi.org/10.3390/cells14040264
Balkhi S, Di Spirito A, Poggi A, Mortara L. Immune Modulation in Alzheimer’s Disease: From Pathogenesis to Immunotherapy. Cells. 2025; 14(4):264. https://doi.org/10.3390/cells14040264
Chicago/Turabian StyleBalkhi, Sahar, Anna Di Spirito, Alessandro Poggi, and Lorenzo Mortara. 2025. "Immune Modulation in Alzheimer’s Disease: From Pathogenesis to Immunotherapy" Cells 14, no. 4: 264. https://doi.org/10.3390/cells14040264
APA StyleBalkhi, S., Di Spirito, A., Poggi, A., & Mortara, L. (2025). Immune Modulation in Alzheimer’s Disease: From Pathogenesis to Immunotherapy. Cells, 14(4), 264. https://doi.org/10.3390/cells14040264