
Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cardiac Structural Remodeling in Heart Failure
3. Molecular Mechanisms Underlying Heart Failure
3.1. Mitochondrial Dysfunction
3.1.1. Mitochondrial Biogenesis in the Heart
3.1.2. Mitochondrial Biogenesis Impairments in Heart Failure
3.1.3. Mitochondrial Fusion and Fission in the Heart
3.1.4. Mitochondrial Dynamic Abnormalities in Heart Failure
3.1.5. Therapeutically Targeting Mitochondrial Biogenesis and Dynamics in Heart Failure
3.1.6. Cardiac Energy Production in the Heart
3.1.7. Metabolic Shifts in Heart Failure
3.1.8. Oxidative Stress in Heart Failure
3.1.9. Targeting Metabolic and Oxidative Stress in Heart Failure
4. Cardiac Lipotoxicity
4.1. Lipid Metabolism in the Heart
4.2. Cardiac Lipotoxicity in Heart Failure
4.3. Targeting Abnormal Lipid Metabolism in Heart Failure
5. Protein Quality Control
5.1. Endoplasmic Reticulum and the Unfolded Protein Response in the Heart
5.2. Adaptive and Maladaptive ER Stress During the Development of Heart Failure
5.3. Targeting ER Stress in Heart Failure
5.4. Golgi Apparatus in Heart Failure
6. Autophagy
6.1. Autophagy in Cardiac Physiology
6.2. The Dual Role of Autophagy in Heart Failure
6.3. Targeting Autophagy in Heart Failure
6.4. Impaired Mitophagy in Heart Failure
7. Inflammation
7.1. Pro-Inflammatory Cytokines in the Heart
7.1.1. HMGB1
7.1.2. GDF15
7.1.3. CRP
7.1.4. IL-11
7.2. Macrophage Infiltration in the Heart
7.3. Anti-Inflammatory Cytokines in the Heart
7.4. Targeting Myocardial Inflammation in Heart Failure
8. Programmed Cell Death
8.1. Apoptosis in Cardiac Physiology
8.2. Apoptosis in Heart Failure
8.3. Targeting Apoptosis in Heart Failure
8.4. Ferroptosis in Cardiac Physiology
8.5. Ferroptosis in Heart Failure
8.6. Targeting Ferroptosis in Heart Failure
8.7. Pyroptosis in Cardiac Physiology
8.8. Pyroptosis in Heart Failure
8.9. Targeting Pyroptosis in Heart Failure
9. Endothelial Dysfunction
9.1. Endothelial Function in Cardiac Physiology
9.2. Endothelial Dysfunction in Heart Failure
9.3. Mechanisms Underlying Endothelial Dysfunction in Heart Failure
9.4. Hypertension in Heart Failure
9.5. Diagnosing Endothelial Dysfunction
9.6. Targeting Endothelial Dysfunction in Heart Failure
10. Dysfunctional Cardiac Conductivity
10.1. Adrenergic Signaling and Calcium Handling in the Heart
10.2. Physiology and Pathological Role of Sarcomeres in the Heart
10.3. Physiological and Pathological Roles of Junction Proteins in the Heart
10.3.1. Gap Junctions
10.3.2. Desmosomes
10.3.3. Adherens Junctions
10.4. Targeting Defective Cardiac Contractility in Heart Failure
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AngII | Angiotensin II |
| ATP | Adenosine Triphosphate |
| Ca2+ | Calcium Ion |
| DCM | Diabetic Cardiomyopathy |
| EF | Ejection Fraction |
| eNOS | Endothelial Nitric Oxide Synthase |
| ER | Endoplasmic Reticulum |
| FA | Fatty Acid |
| FAO | Fatty Acid Oxidation |
| GA | Golgi Apparatus |
| GJ | Gap Junction |
| HF | Heart Failure |
| HFpEF | Heart Failure with Preserved Ejection Fraction |
| HFrEF | Heart Failure with Reduced Ejection Fraction |
| I/R | Ischemia/Reperfusion |
| ID | Intercalated Discs |
| IL | Interleukin |
| LV | Left Ventricle |
| MI | Myocardial Infarction |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric Oxide |
| OXPHOS | Oxidative Phosphorylation |
| PPAR | Peroxisome Proliferator-activated Receptor |
| ROS | Reactive Oxygen Species |
| SERCA | Sarcoendoplasmic Reticulum Calcium ATPase |
| SR | Sarcoplasmic Reticulum |
| TAC | Transverse Aortic Constriction |
| TCA | Tricarboxylic Acid Cycle |
| TG | Triglyceride |
| TNFα | Tumor Necrosis Factor α |
| UPR | Unfolded Protein Response |
| β-AR | β-Adrenergic Receptor |
References
- Aune, E.; McMurray, J.; Lundgren, P.; Sattar, N.; Israelsson, J.; Nordberg, P.; Herlitz, J.; Rawshani, A. Clinical characteristics and survival in patients with heart failure experiencing in hospital cardiac arrest. Sci. Rep. 2022, 12, 5685. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Cleland, J.G. Causes and treatment of oedema in patients with heart failure. Nat. Rev. Cardiol. 2013, 10, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Poelzl, G.; Ess, M.; Von der Heidt, A.; Rudnicki, M.; Frick, M.; Ulmer, H. Concomitant renal and hepatic dysfunctions in chronic heart failure: Clinical implications and prognostic significance. Eur. J. Intern. Med. 2013, 24, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, K.; Kornej, J.; Shantsila, E.; Lip, G.Y.H. Heart Failure and Stroke. Curr. Heart Fail. Rep. 2018, 15, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Emmons-Bell, S.; Johnson, C.; Roth, G. Prevalence, incidence and survival of heart failure: A systematic review. Heart 2022, 108, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, e263–e421. [Google Scholar] [CrossRef] [PubMed]
- Screever, E.M.; van der Wal, M.H.L.; van Veldhuisen, D.J.; Jaarsma, T.; Koops, A.; van Dijk, K.S.; Warink-Riemersma, J.; Coster, J.E.; Westenbrink, B.D.; van der Meer, P.; et al. Comorbidities complicating heart failure: Changes over the last 15 years. Clin. Res. Cardiol. 2023, 112, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, M.; Pirozzi, F.; Hirsch, E.; Ghigo, A. Mechanisms underlying the cross-talk between heart and cancer. J. Physiol. 2020, 598, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V. Sex and Gender Differences in Heart Failure. Int. J. Heart Fail. 2020, 2, 157–181. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Kavazis, A.N. Pathological vs. physiological cardiac hypertrophy. J. Physiol. 2015, 593, 3767. [Google Scholar] [CrossRef]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Schotten, U. Electrophysiological Consequences of Cardiac Fibrosis. Cells 2021, 10, 3220. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef]
- Patten, I.S.; Arany, Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol. Metab. 2012, 23, 90–97. [Google Scholar] [CrossRef]
- De Vitto, H.; Bode, A.M.; Dong, Z. The PGC-1/ERR network and its role in precision oncology. NPJ Precis. Oncol. 2019, 3, 9. [Google Scholar] [CrossRef]
- Yin, Z.; Zhao, Y.; He, M.; Li, H.; Fan, J.; Nie, X.; Yan, M.; Chen, C.; Wang, D.W. MiR-30c/PGC-1beta protects against diabetic cardiomyopathy via PPARalpha. Cardiovasc. Diabetol. 2019, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes. Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Chaube, B.; Bhat, M.K. AMPK, a key regulator of metabolic/energy homeostasis and mitochondrial biogenesis in cancer cells. Cell Death Dis. 2016, 7, e2044. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Tokudome, S.; Shimizu, N.; Yoshikawa, N.; Ogawa, C.; Shirakawa, K.; Endo, J.; Katayama, T.; Yuasa, S.; Ieda, M.; et al. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor gamma coactivator 1alpha. J. Biol. Chem. 2007, 282, 25970–25980. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jaeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: Modulation by p38 MAPK. Genes. Dev. 2004, 18, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Kelly, T.J.; Lerin, C.; Haas, W.; Gygi, S.P.; Puigserver, P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 2009, 284, 19945–19952. [Google Scholar] [CrossRef] [PubMed]
- Lerin, C.; Rodgers, J.T.; Kalume, D.E.; Kim, S.H.; Pandey, A.; Puigserver, P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006, 3, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, T.; Chen, C.K.; Wang, S.W.; Pan, P.; Ju, Z.; Wang, J.; Takada, S.; Klein, W.H.; Mao, C.A. Essential roles of mitochondrial biogenesis regulator Nrf1 in retinal development and homeostasis. Mol. Neurodegener. 2018, 13, 56. [Google Scholar] [CrossRef]
- Huo, L.; Scarpulla, R.C. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol. Cell. Biol. 2001, 21, 644–654. [Google Scholar] [CrossRef]
- Qi, X.M.; Zhang, W.Z.; Zuo, Y.Q.; Qiao, Y.B.; Zhang, Y.L.; Ren, J.H.; Li, Q.S. Nrf2/NRF1 signaling activation and crosstalk amplify mitochondrial biogenesis in the treatment of triptolide-induced cardiotoxicity using calycosin. Cell Biol. Toxicol. 2024, 41, 2. [Google Scholar] [CrossRef]
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010, 647, 37–74. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Elston, M.; Nicholson, C.K.; Gundewar, S.; Jha, S.; Elrod, J.W.; Ramachandran, A.; Lefer, D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 2010, 122, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Zhang, L.F.; Cui, Y.J.; Chang, Y.M.; Jiang, C.W.; Meng, Z.Z.; Xu, P.; Liu, H.Y.; Wang, D.Y.; Cao, X.B. The Protective Effects of Salidroside from Exhaustive Exercise-Induced Heart Injury by Enhancing the PGC-1 alpha -NRF1/NRF2 Pathway and Mitochondrial Respiratory Function in Rats. Oxid. Med. Cell Longev. 2015, 2015, 876825. [Google Scholar] [CrossRef]
- De la Cruz Lopez, K.G.; Toledo Guzman, M.E.; Sanchez, E.O.; Garcia Carranca, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. USA 2009, 106, 22229–22232. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Schieke, S.M.; Phillips, D.; McCoy, J.P., Jr.; Aponte, A.M.; Shen, R.F.; Balaban, R.S.; Finkel, T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Mialet-Perez, J.; Belaidi, E. Interplay between hypoxia inducible Factor-1 and mitochondria in cardiac diseases. Free Radic. Biol. Med. 2024, 221, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 2009, 24, 97–106. [Google Scholar] [CrossRef]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes. Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- O’Hagan, K.A.; Cocchiglia, S.; Zhdanov, A.V.; Tambuwala, M.M.; Cummins, E.P.; Monfared, M.; Agbor, T.A.; Garvey, J.F.; Papkovsky, D.B.; Taylor, C.T.; et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2188–2193. [Google Scholar] [CrossRef] [PubMed]
- Regueira, T.; Lepper, P.M.; Brandt, S.; Ochs, M.; Vuda, M.; Takala, J.; Jakob, S.M.; Djafarzadeh, S. Hypoxia inducible factor-1 alpha induction by tumour necrosis factor-alpha, but not by toll-like receptor agonists, modulates cellular respiration in cultured human hepatocytes. Liver Int. 2009, 29, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Garbacz, W.G.; Xie, W. Constitutive activities of estrogen-related receptors: Transcriptional regulation of metabolism by the ERR pathways in health and disease. Biochim. Biophys. Acta 2015, 1852, 1912–1927. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Kopp, R.P.; Kelly, D.P. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J. Biol. Chem. 2002, 277, 40265–40274. [Google Scholar] [CrossRef]
- Huss, J.M.; Imahashi, K.; Dufour, C.R.; Weinheimer, C.J.; Courtois, M.; Kovacs, A.; Giguere, V.; Murphy, E.; Kelly, D.P. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007, 6, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890. [Google Scholar] [CrossRef]
- Canto, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef]
- Suwa, M.; Egashira, T.; Nakano, H.; Sasaki, H.; Kumagai, S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J. Appl. Physiol. (1985) 2006, 101, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Rinaldi, B.; Corbi, G.; Conti, V.; Stiuso, P.; Boccuti, S.; Rengo, G.; Rossi, F.; Filippelli, A. Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res. 2008, 11, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, W.; Yao, K.; Wang, H.; Liu, B.; Li, T.; Song, L.; Diao, D.; Mao, G.; Huang, P.; et al. Fine-Tuning of PGC1alpha Expression Regulates Cardiac Function and Longevity. Circ. Res. 2019, 125, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, e101. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ. Res. 2011, 109, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Bowen, T.S.; Rolim, N.; Dieterlen, M.T.; Werner, S.; Hoshi, T.; Fischer, T.; Mangner, N.; Linke, A.; Schuler, G.; et al. Skeletal Muscle Alterations Are Exacerbated in Heart Failure With Reduced Compared With Preserved Ejection Fraction: Mediated by Circulating Cytokines? Circ. Heart Fail. 2016, 9, e003027. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J. Mol. Cell Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguere, V.; et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007, 6, 13–24. [Google Scholar] [CrossRef]
- Huang, Y.; Hickey, R.P.; Yeh, J.L.; Liu, D.; Dadak, A.; Young, L.H.; Johnson, R.S.; Giordano, F.J. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004, 18, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Seymour, E.M.; Bennink, M.R.; Bolling, S.F. Diet-relevant phytochemical intake affects the cardiac AhR and nrf2 transcriptome and reduces heart failure in hypertensive rats. J. Nutr. Biochem. 2013, 24, 1580–1586. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid. Redox Signal. 2011, 14, 425–437. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Kim, A.S.; Miller, E.J.; Wright, T.M.; Li, J.; Qi, D.; Atsina, K.; Zaha, V.; Sakamoto, K.; Young, L.H. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J. Mol. Cell Cardiol. 2011, 51, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, A.D.; Balteau, M.; Gelinas, R.; Renguet, E.; Ginion, A.; de Meester, C.; Sakamoto, K.; Balligand, J.L.; Bontemps, F.; Vanoverschelde, J.L.; et al. A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1619–H1630. [Google Scholar] [CrossRef]
- Ma, S.; Feng, J.; Zhang, R.; Chen, J.; Han, D.; Li, X.; Yang, B.; Li, X.; Fan, M.; Li, C.; et al. SIRT1 Activation by Resveratrol Alleviates Cardiac Dysfunction via Mitochondrial Regulation in Diabetic Cardiomyopathy Mice. Oxid. Med. Cell. Longev. 2017, 2017, 4602715. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Chen, E.; Pan, Z. The role of mitochondrial dysfunction in mesenchymal stem cell senescence. Cell Tissue Res. 2020, 382, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Hittelman, K.J.; Faulkner, J.A.; Beyer, R.E. Temperature, skeletal muscle mitochondrial functions, and oxygen debt. Am. J. Physiol. 1971, 220, 1053–1059. [Google Scholar] [CrossRef]
- Sommer, A.M.; Portner, H.O. Mitochondrial function in seasonal acclimatization versus latitudinal adaptation to cold in the lugworm Arenicola marina (L.). Physiol. Biochem. Zool. 2004, 77, 174–186. [Google Scholar] [CrossRef]
- Schafer, M.; Oeing, C.U.; Rohm, M.; Baysal-Temel, E.; Lehmann, L.H.; Bauer, R.; Volz, H.C.; Boutros, M.; Sohn, D.; Sticht, C.; et al. Ataxin-10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol. Metab. 2016, 5, 67–78. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. Fatty acid utilization in the hypertrophied and failing heart: Molecular regulatory mechanisms. Am. J. Med. Sci. 1999, 318, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Guven, B.; Wagg, C.S.; Almeida de Oliveira, A.; Silver, H.; Zhang, L.; Chen, B.; Wei, K.; Ketema, E.B.; Karwi, Q.G.; et al. Mitochondrial fatty acid oxidation is the major source of cardiac adenosine triphosphate production in heart failure with preserved ejection fraction. Cardiovasc. Res. 2024, 120, 360–371. [Google Scholar] [CrossRef]
- Yoshii, A.; McMillen, T.S.; Wang, Y.; Zhou, B.; Chen, H.; Banerjee, D.; Herrero, M.; Wang, P.; Muraoka, N.; Wang, W.; et al. Blunted Cardiac Mitophagy in Response to Metabolic Stress Contributes to HFpEF. Circ. Res. 2024, 135, 1004–1017. [Google Scholar] [CrossRef]
- Meddeb, M.; Koleini, N.; Binek, A.; Keykhaei, M.; Darehgazani, R.; Kwon, S.; Aboaf, C.; Margulies, K.B.; Bedi, K.C., Jr.; Lehar, M.; et al. Myocardial ultrastructure of human heart failure with preserved ejection fraction. Nat. Cardiovasc. Res. 2024, 3, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49 (Suppl. S1), 3–8. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Osko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczynska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed]
- Heart Outcomes Prevention Evaluation Study, I.; Yusuf, S.; Dagenais, G.; Pogue, J.; Bosch, J.; Sleight, P. Vitamin E supplementation and cardiovascular events in high-risk patients. N. Engl. J. Med. 2000, 342, 154–160. [Google Scholar] [CrossRef]
- Dresdale, A.R.; Barr, L.H.; Bonow, R.O.; Mathisen, D.J.; Myers, C.E.; Schwartz, D.E.; d’Angelo, T.; Rosenberg, S.A. Prospective randomized study of the role of N-acetyl cysteine in reversing doxorubicin-induced cardiomyopathy. Am. J. Clin. Oncol. 1982, 5, 657–663. [Google Scholar] [CrossRef]
- Unverferth, D.V.; Jagadeesh, J.M.; Unverferth, B.J.; Magorien, R.D.; Leier, C.V.; Balcerzak, S.P. Attempt to prevent doxorubicin-induced acute human myocardial morphologic damage with acetylcysteine. J. Natl. Cancer Inst. 1983, 71, 917–920. [Google Scholar] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Unger, R.H. Klotho-induced insulin resistance: A blessing in disguise? Nat. Med. 2006, 12, 56–57. [Google Scholar] [CrossRef]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef]
- Bell, E.L.; Guarente, L. The SirT3 divining rod points to oxidative stress. Mol. Cell 2011, 42, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Airhart, S.E.; Zhou, B.; Shireman, L.M.; Jiang, S.; Melendez Rodriguez, C.; Kirkpatrick, J.N.; Shen, D.D.; Tian, R.; O’Brien, K.D. Safety and Tolerability of Nicotinamide Riboside in Heart Failure With Reduced Ejection Fraction. JACC Basic. Transl. Sci. 2022, 7, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265. [Google Scholar] [CrossRef]
- Mei, Y.; Zhang, Y.; Yamamoto, K.; Xie, W.; Mak, T.W.; You, H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 5153–5158. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, M.J.; Huynh, K.; Qin, C.; Rosli, S.; Kiriazis, H.; Ayer, A.; Cemerlang, N.; Stocker, R.; Du, X.J.; McMullen, J.R.; et al. Therapeutic targeting of oxidative stress with coenzyme Q10 counteracts exaggerated diabetic cardiomyopathy in a mouse model of diabetes with diminished PI3K(p110alpha) signaling. Free Radic. Biol. Med. 2015, 87, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Fotino, A.D.; Thompson-Paul, A.M.; Bazzano, L.A. Effect of coenzyme Q(1)(0)supplementation on heart failure: A meta-analysis. Am. J. Clin. Nutr. 2013, 97, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Coleman, C.I.; Patel, A.A.; Kluger, J.; White, C.M. The impact of coenzyme Q10 on systolic function in patients with chronic heart failure. J. Card. Fail. 2006, 12, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Soja, A.M.; Mortensen, S.A. Treatment of congestive heart failure with coenzyme Q10 illuminated by meta-analyses of clinical trials. Mol. Asp. Med. 1997, 18, S159–S168. [Google Scholar] [CrossRef]
- Mlynarska, E.; Hajdys, J.; Czarnik, W.; Fularski, P.; Leszto, K.; Majchrowicz, G.; Lisinska, W.; Rysz, J.; Franczyk, B. The Role of Antioxidants in the Therapy of Cardiovascular Diseases-A Literature Review. Nutrients 2024, 16, 2587. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxid. Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef] [PubMed]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.H.; Gambardella, J.; Jankauskas, S.; Wang, X.; Santulli, G.; Gudas, L.J.; Levi, R. A Retinoic Acid Receptor beta (2) Agonist Improves Cardiac Function in a Heart Failure Model. J. Pharmacol. Exp. Ther. 2021, 379, 182–190. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Pulinilkunnil, T.; Nagendran, J.; Dyck, J.R. Myocardial triacylglycerol metabolism. J. Mol. Cell Cardiol. 2013, 55, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001, 2, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Baro, M.R.; Lewin, T.M.; Coleman, R.A. Regulation of Triglyceride Metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1195–G1199. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W. The structure and functions of human lysophosphatidic acid acyltransferases. Front. Biosci. 2001, 6, D944–D953. [Google Scholar] [CrossRef]
- Takeuchi, K.; Reue, K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1195–E1209. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Foryst-Ludwig, A.; Haemmerle, G.; Zechner, R. The Role of Adipose Triglyceride Lipase and Cytosolic Lipolysis in Cardiac Function and Heart Failure. Cell Rep. Med. 2020, 1, 100001. [Google Scholar] [CrossRef] [PubMed]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Munoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat. Commun. 2017, 8, 494. [Google Scholar] [CrossRef]
- Chen, X.; Ruiz-Velasco, A.; Zou, Z.; Hille, S.S.; Ross, C.; Fonseka, O.; Gare, S.R.; Alatawi, N.H.O.; Raja, R.; Zhang, J.; et al. PAK3 Exacerbates Cardiac Lipotoxicity via SREBP1c in Obesity Cardiomyopathy. Diabetes 2024, 73, 1805–1820. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.J.; Sartoni-D’Ambrosia, G.; Arbique, D.; Vongpatanasin, W.; Unger, R.; Victor, R.G. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 2003, 49, 417–423. [Google Scholar] [CrossRef]
- Zlobine, I.; Gopal, K.; Ussher, J.R. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim. Biophys. Acta 2016, 1861, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Lewin, T.M.; de Jong, H.; Schwerbrock, N.J.; Hammond, L.E.; Watkins, S.M.; Combs, T.P.; Coleman, R.A. Mice deficient in mitochondrial glycerol-3-phosphate acyltransferase-1 have diminished myocardial triacylglycerol accumulation during lipogenic diet and altered phospholipid fatty acid composition. Biochim. Biophys. Acta 2008, 1781, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Glenn, D.J.; Wang, F.; Nishimoto, M.; Cruz, M.C.; Uchida, Y.; Holleran, W.M.; Zhang, Y.; Yeghiazarians, Y.; Gardner, D.G. A murine model of isolated cardiac steatosis leads to cardiomyopathy. Hypertension 2011, 57, 216–222. [Google Scholar] [CrossRef]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Buttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Pulinilkunnil, T.; Nagendran, J.; Young, M.E.; Bogner-Strauss, J.G.; Hackl, H.; Khadour, R.; Heydari, E.; Haemmerle, G.; Zechner, R.; et al. Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation. Cardiovasc. Res. 2013, 99, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Jaeger, D.; Kolleritsch, S.; Zimmermann, R.; Zechner, R.; Lass, A.; Haemmerle, G. The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J. Biol. Chem. 2015, 290, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Schweiger, M.; Jaeger, D.; Kolb, D.; Kumari, M.; Schreiber, R.; Kolleritsch, S.; Markolin, P.; Grabner, G.F.; Heier, C.; et al. Cardiac-specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J. Lipid Res. 2013, 54, 1092–1102. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Gong, D.W.; O’Connell, K.A.; Dabkowski, E.R.; Hecker, P.A.; Ionica, N.; Konig, M.; Mahurkar, A.; Sun, Y.; et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J. Lipid Res. 2013, 54, 953–965. [Google Scholar] [CrossRef]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef]
- Dobbins, R.L.; Szczepaniak, L.S.; Bentley, B.; Esser, V.; Myhill, J.; McGarry, J.D. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes 2001, 50, 123–130. [Google Scholar] [CrossRef]
- Shao, D.; Kolwicz, S.C., Jr.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef]
- Kashiwagi, H.; Tomiyama, Y.; Kosugi, S.; Shiraga, M.; Lipsky, R.H.; Kanayama, Y.; Kurata, Y.; Matsuzawa, Y. Identification of molecular defects in a subject with type I CD36 deficiency. Blood 1994, 83, 3545–3552. [Google Scholar] [CrossRef]
- Watanabe, K.; Ohta, Y.; Toba, K.; Ogawa, Y.; Hanawa, H.; Hirokawa, Y.; Kodama, M.; Tanabe, N.; Hirono, S.; Ohkura, Y.; et al. Myocardial CD36 expression and fatty acid accumulation in patients with type I and II CD36 deficiency. Ann. Nucl. Med. 1998, 12, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Chiba, H.; Morimoto, M.; Abe, K.; Fujiwara, H.; Fuda, H.; Hui, S.P.; Takahashi, Y.; Akita, H.; Jamieson, G.A.; et al. Human CD36 deficiency is associated with elevation in low-density lipoprotein-cholesterol. Am. J. Med. Genet. 2000, 93, 299–304. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Arioglu, E.; De Almeida, S.; Akkoc, N.; Taylor, S.I.; Bowcock, A.M.; Barnes, R.I.; Garg, A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet. 2002, 31, 21–23. [Google Scholar] [CrossRef]
- Gandotra, S.; Le Dour, C.; Bottomley, W.; Cervera, P.; Giral, P.; Reznik, Y.; Charpentier, G.; Auclair, M.; Delepine, M.; Barroso, I.; et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med. 2011, 364, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Zietzer, A.; Dusing, P.; Reese, L.; Nickenig, G.; Jansen, F. Ceramide Metabolism in Cardiovascular Disease: A Network With High Therapeutic Potential. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2017, 2, e82922. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Hu, Y.; Noh, H.L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.C.; Abel, E.D.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J. Lipid Res. 2010, 51, 2202–2210. [Google Scholar] [CrossRef]
- Nakamura, M. Lipotoxicity as a therapeutic target in obesity and diabetic cardiomyopathy. J. Pharm. Pharm. Sci. 2024, 27, 12568. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 2016, 1861, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Fragasso, G.; Palloshi, A.; Puccetti, P.; Silipigni, C.; Rossodivita, A.; Pala, M.; Calori, G.; Alfieri, O.; Margonato, A. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J. Am. Coll. Cardiol. 2006, 48, 992–998. [Google Scholar] [CrossRef]
- Dyck, J.R.; Cheng, J.F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Battiprolu, P.K.; Fukushima, A.; Nguyen, K.; Milner, K.; Gupta, A.; Altamimi, T.; Byrne, N.; Mori, J.; et al. Malonyl CoA Decarboxylase Inhibition Improves Cardiac Function Post-Myocardial Infarction. JACC Basic Transl. Sci. 2019, 4, 385–400. [Google Scholar] [CrossRef]
- Stanley, W.C.; Morgan, E.E.; Huang, H.; McElfresh, T.A.; Sterk, J.P.; Okere, I.C.; Chandler, M.P.; Cheng, J.; Dyck, J.R.; Lopaschuk, G.D. Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2304–H2309. [Google Scholar] [CrossRef]
- Abdurrachim, D.; Luiken, J.J.; Nicolay, K.; Glatz, J.F.; Prompers, J.J.; Nabben, M. Good and bad consequences of altered fatty acid metabolism in heart failure: Evidence from mouse models. Cardiovasc. Res. 2015, 106, 194–205. [Google Scholar] [CrossRef]
- Zhang, C.; Syed, T.W.; Liu, R.; Yu, J. Role of Endoplasmic Reticulum Stress, Autophagy, and Inflammation in Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Liu, M.Q.; Chen, Z.; Chen, L.X. Endoplasmic reticulum stress: A novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol. Sin. 2016, 37, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dudley, S.C., Jr. Role for the Unfolded Protein Response in Heart Disease and Cardiac Arrhythmias. Int. J. Mol. Sci. 2015, 17, 52. [Google Scholar] [CrossRef]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef]
- Duan, Q.; Ni, L.; Wang, P.; Chen, C.; Yang, L.; Ma, B.; Gong, W.; Cai, Z.; Zou, M.H.; Wang, D.W. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell 2016, 15, 625–633. [Google Scholar] [CrossRef]
- Isodono, K.; Takahashi, T.; Imoto, H.; Nakanishi, N.; Ogata, T.; Asada, S.; Adachi, A.; Ueyama, T.; Oh, H.; Matsubara, H. PARM-1 is an endoplasmic reticulum molecule involved in endoplasmic reticulum stress-induced apoptosis in rat cardiac myocytes. PLoS ONE 2010, 5, e9746. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Ruiz-Velasco, A.; Raja, R.; Howell, G.; Miller, J.M.; Abouleisa, R.R.E.; Ou, Q.; Mace, K.; Hille, S.S.; Frey, N.; et al. Paracrine signal emanating from stressed cardiomyocytes aggravates inflammatory microenvironment in diabetic cardiomyopathy. iScience 2022, 25, 103973. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kwak, D.; Lu, Z.; Xu, X.; Fassett, J.; Wang, H.; Wei, Y.; Cavener, D.R.; Hu, X.; Hall, J.; et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension 2014, 64, 738–744. [Google Scholar] [CrossRef]
- Wang, J.; Lu, L.; Chen, S.; Xie, J.; Lu, S.; Zhou, Y.; Jiang, H. Up-regulation of PERK/Nrf2/HO-1 axis protects myocardial tissues of mice from damage triggered by ischemia-reperfusion through ameliorating endoplasmic reticulum stress. Cardiovasc. Diagn. Ther. 2020, 10, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Deng, Y.; Zhang, G.; Li, C.; Ding, G.; May, H.I.; Tran, D.H.; Luo, X.; Jiang, D.S.; Li, D.L.; et al. Spliced X-box Binding Protein 1 Stimulates Adaptive Growth Through Activation of mTOR. Circulation 2019, 140, 566–579. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef] [PubMed]
- Steiger, D.; Yokota, T.; Li, J.; Ren, S.; Minamisawa, S.; Wang, Y. The serine/threonine-protein kinase/endoribonuclease IRE1alpha protects the heart against pressure overload-induced heart failure. J. Biol. Chem. 2018, 293, 9652–9661. [Google Scholar] [CrossRef]
- Jin, J.K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Aghajani, M.; Alcock, C.D.; Blackwood, E.A.; Sandmann, C.; Herzog, N.; Gross, J.; Plate, L.; Wiseman, R.L.; Kaufman, R.J.; et al. ATF6 protects against protein misfolding during cardiac hypertrophy. J. Mol. Cell Cardiol. 2024, 189, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Binder, P.; Wang, S.; Radu, M.; Zin, M.; Collins, L.; Khan, S.; Li, Y.; Sekeres, K.; Humphreys, N.; Swanton, E.; et al. Pak2 as a Novel Therapeutic Target for Cardioprotective Endoplasmic Reticulum Stress Response. Circ. Res. 2019, 124, 696–711. [Google Scholar] [CrossRef]
- Turdi, S.; Hu, N.; Ren, J. Tauroursodeoxycholic acid mitigates high fat diet-induced cardiomyocyte contractile and intracellular Ca2+ anomalies. PLoS ONE 2013, 8, e63615. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Sreenivasaiah, P.K.; Kim, J.O.; Lee, M.Y.; Kang, W.S.; Kim, Y.S.; Ahn, Y.; Park, W.J.; Cho, C.; Kim, D.H. Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS ONE 2017, 12, e0176071. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H. Chemical chaperones: A pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Chen, B.; Wang, X. 4-PBA prevents pressure overload-induced myocardial hypertrophy and interstitial fibrosis by attenuating endoplasmic reticulum stress. Chem. Biol. Interact. 2015, 242, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Jian, L.; Lu, Y.; Lu, S.; Lu, C. Chemical Chaperone 4-Phenylbutyric Acid Reduces Cardiac Ischemia/Reperfusion Injury by Alleviating Endoplasmic Reticulum Stress and Oxidative Stress. Med. Sci. Monit. 2016, 22, 5218–5227. [Google Scholar] [CrossRef]
- Villani, S.; Dematteis, G.; Tapella, L.; Gagliardi, M.; Lim, D.; Corazzari, M.; Aprile, S.; Del Grosso, E. Quantification of the Chemical Chaperone 4-Phenylbutyric Acid (4-PBA) in Cell Culture Media via LC-HRMS: Applications in Fields of Neurodegeneration and Cancer. Pharmaceuticals 2023, 16, 298. [Google Scholar] [CrossRef] [PubMed]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 2018, 7, e37168. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, A.; Kok, B.P.; Rius, B.; Grandjean, J.M.D.; Alabi, A.; Albert, V.; Sukiasyan, A.; Powers, E.T.; Galmozzi, A.; Saez, E.; et al. Pharmacologic IRE1/XBP1s activation promotes systemic adaptive remodeling in obesity. Nat. Commun. 2022, 13, 608. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Rani, S.; Sreenivasaiah, P.K.; Cho, C.; Kim, D.H. Salubrinal Alleviates Pressure Overload-Induced Cardiac Hypertrophy by Inhibiting Endoplasmic Reticulum Stress Pathway. Mol. Cells 2017, 40, 66–72. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Qi, S.Y.; Ru, L.S.; Ding, C.; Wang, H.J.; Zhao, J.S.; Li, J.J.; Li, A.Y.; Wang, D.M. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and JNK pathways. Can. J. Cardiol. 2014, 30, 368–375. [Google Scholar] [CrossRef]
- Li, J.; Ahat, E.; Wang, Y. Golgi Structure and Function in Health, Stress, and Diseases. Results Probl. Cell Differ. 2019, 67, 441–485. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Fonseka, O.; Ganenthiran, H.; Andrea Ruiz, V.; Liu, W. The multifaceted roles of ER and Golgi in metabolic cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 999044. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Singal, P.K. Ultrastructure of failing myocardium due to induced chronic mitral insufficiency in dogs. Br. J. Exp. Pathol. 1977, 58, 289–300. [Google Scholar]
- Hatt, P.Y. Cellular changes and damage in mechanically overloaded hearts. Recent. Adv. Stud. Cardiac Struct. Metab. 1975, 6, 325–333. [Google Scholar] [PubMed]
- Muhammad, E.; Levitas, A.; Singh, S.R.; Braiman, A.; Ofir, R.; Etzion, S.; Sheffield, V.C.; Etzion, Y.; Carrier, L.; Parvari, R. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum. Mol. Genet. 2015, 24, 7227–7240. [Google Scholar] [CrossRef] [PubMed]
- Jungk, L.; Franke, H.; Salameh, A.; Dhein, S. Golgi Fragmentation in Human Patients with Chronic Atrial Fibrillation: A New Aspect of Remodeling. Thorac. Cardiovasc. Surg. 2019, 67, 98–106. [Google Scholar] [CrossRef]
- Filipeanu, C.M.; Zhou, F.; Wu, G. Analysis of Rab1 function in cardiomyocyte growth. Methods Enzymol. 2008, 438, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Filipeanu, C.M.; Zhou, F.; Claycomb, W.C.; Wu, G. Regulation of the cell surface expression and function of angiotensin II type 1 receptor by Rab1-mediated endoplasmic reticulum-to-Golgi transport in cardiac myocytes. J. Biol. Chem. 2004, 279, 41077–41084. [Google Scholar] [CrossRef]
- Nielsen, L.B.; Veniant, M.; Boren, J.; Raabe, M.; Wong, J.S.; Tam, C.; Flynn, L.; Vanni-Reyes, T.; Gunn, M.D.; Goldberg, I.J.; et al. Genes for apolipoprotein B and microsomal triglyceride transfer protein are expressed in the heart: Evidence that the heart has the capacity to synthesize and secrete lipoproteins. Circulation 1998, 98, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, Y.; Li, T.; Jiang, Z.; Zeng, L.; Hu, Z. The role of the Golgi apparatus in disease (Review). Int. J. Mol. Med. 2021, 47, 38. [Google Scholar] [CrossRef] [PubMed]
- Zappa, F.; Failli, M.; De Matteis, M.A. The Golgi complex in disease and therapy. Curr. Opin. Cell Biol. 2018, 50, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7–18. [Google Scholar] [CrossRef]
- Massey, A.C.; Zhang, C.; Cuervo, A.M. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006, 73, 205–235. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef]
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Tremel, S.; Ohashi, Y.; Morado, D.R.; Bertram, J.; Perisic, O.; Brandt, L.T.L.; von Wrisberg, M.K.; Chen, Z.A.; Maslen, S.L.; Kovtun, O.; et al. Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat. Commun. 2021, 12, 1564. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Velasco, A.; Raja, R.; Chen, X.; Ganenthiran, H.; Kaur, N.; Alatawi, N.H.O.; Miller, J.M.; Abouleisa, R.R.E.; Ou, Q.; Zhao, X.; et al. Restored autophagy is protective against PAK3-induced cardiac dysfunction. iScience 2023, 26, 106970. [Google Scholar] [CrossRef]
- Kassiotis, C.; Ballal, K.; Wellnitz, K.; Vela, D.; Gong, M.; Salazar, R.; Frazier, O.H.; Taegtmeyer, H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 2009, 120, S191–S197. [Google Scholar] [CrossRef] [PubMed]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Z.; Li, Q.; Trammell, S.A.; Schmidt, M.S.; Pires, K.M.; Cai, J.; Zhang, Y.; Kenny, H.; Boudina, S.; et al. Control of NAD(+) homeostasis by autophagic flux modulates mitochondrial and cardiac function. EMBO J. 2024, 43, 362–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef]
- Chi, C.; Riching, A.S.; Song, K. Lysosomal Abnormalities in Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 811. [Google Scholar] [CrossRef]
- Guan, J.; Mishra, S.; Qiu, Y.; Shi, J.; Trudeau, K.; Las, G.; Liesa, M.; Shirihai, O.S.; Connors, L.H.; Seldin, D.C.; et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol. Med. 2014, 6, 1493–1507. [Google Scholar] [CrossRef]
- Evans, S.; Ma, X.; Wang, X.; Chen, Y.; Zhao, C.; Weinheimer, C.J.; Kovacs, A.; Finck, B.; Diwan, A.; Mann, D.L. Targeting the Autophagy-Lysosome Pathway in a Pathophysiologically Relevant Murine Model of Reversible Heart Failure. JACC Basic. Transl. Sci. 2022, 7, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef]
- Alcalai, R.; Arad, M.; Wakimoto, H.; Yadin, D.; Gorham, J.; Wang, L.; Burns, E.; Maron, B.J.; Roberts, W.C.; Konno, T.; et al. LAMP2 Cardiomyopathy: Consequences of Impaired Autophagy in the Heart. J. Am. Heart Assoc. 2021, 10, e018829. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Meijer, A.J. Autophagy: A potential link between obesity and insulin resistance. Cell Metab. 2010, 11, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Pietzsch, S.; Stapel, B.; Ricke-Hoch, M.; Lee, C.W.; Bankstahl, J.P.; Scherr, M.; Heineke, J.; Scharf, G.; Haghikia, A.; et al. Insulin supplementation attenuates cancer-induced cardiomyopathy and slows tumor disease progression. JCI Insight 2017, 2, e93098. [Google Scholar] [CrossRef] [PubMed]
- Fidzianska, A.; Bilinska, Z.T.; Walczak, E.; Witkowski, A.; Chojnowska, L. Autophagy in transition from hypertrophic cardiomyopathy to heart failure. J. Electron. Microsc. 2010, 59, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Tang, M.; Zheng, Q.; Kumarapeli, A.R.; Horak, K.M.; Tian, Z.; Wang, X. Doxycycline attenuates protein aggregation in cardiomyocytes and improves survival of a mouse model of cardiac proteinopathy. J. Am. Coll. Cardiol. 2010, 56, 1418–1426. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef]
- Gao, G.; Chen, W.; Yan, M.; Liu, J.; Luo, H.; Wang, C.; Yang, P. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 2020, 45, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yao, X.; Zhang, Q.J.; Zhu, M.; Liu, Z.P.; Ci, B.; Xie, Y.; Carlson, D.; Rothermel, B.A.; Sun, Y.; et al. Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation 2018, 138, 2247–2262. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Meng, H.; Xiao, J.; Liu, F.; Du, J.; Zeng, H. Pretreatment of 3-MA prevents doxorubicin-induced cardiotoxicity through inhibition of autophagy initiation. Toxicology 2023, 490, 153512. [Google Scholar] [CrossRef] [PubMed]
- Aiad, N.; du Fay de Lavallaz, J.; Zhang, M.J.; Chaikijurajai, T.; Ye, B.; Nijjar, P.S.; Lahiri, J.A.; Martin, C.M.; Alexy, T.; Meyer, M. Cilostazol in patients with heart failure and preserved ejection fraction-The CLIP-HFpEF trial. ESC Heart Fail. 2024. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiang, X.; Xu, Z. Cilostazol protects against myocardial ischemia and reperfusion injury by activating transcription factor EB (TFEB). Biotechnol. Appl. Biochem. 2019, 66, 555–563. [Google Scholar] [CrossRef]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Bi, Y.; Ajoolabady, A.; You, F.; Sowers, J.; Wang, Q.; Ceylan, A.F.; Zhang, Y.; Ren, J. Parkin Insufficiency Accentuates High-Fat Diet-Induced Cardiac Remodeling and Contractile Dysfunction Through VDAC1-Mediated Mitochondrial Ca(2+) Overload. JACC Basic Transl. Sci. 2022, 7, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [PubMed]
- Abudureyimu, M.; Yu, W.; Cao, R.Y.; Zhang, Y.; Liu, H.; Zheng, H. Berberine Promotes Cardiac Function by Upregulating PINK1/Parkin-Mediated Mitophagy in Heart Failure. Front. Physiol. 2020, 11, 565751. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Chen, J.; Wang, L.; Hao, M.; Dong, X.; Luo, T.; Jiang, J.; Lin, Z.; Li, X.; Chen, P.; et al. Nuanxinkang prevents the development of myocardial infarction-induced chronic heart failure by promoting PINK1/Parkin-mediated mitophagy. Phytomedicine 2023, 108, 154494. [Google Scholar] [CrossRef]
- Hu, J.; Liu, T.; Fu, F.; Cui, Z.; Lai, Q.; Zhang, Y.; Yu, B.; Liu, F.; Kou, J.; Li, F. Omentin1 ameliorates myocardial ischemia-induced heart failure via SIRT3/FOXO3a-dependent mitochondrial dynamical homeostasis and mitophagy. J. Transl. Med. 2022, 20, 447. [Google Scholar] [CrossRef]
- Cai, W.; Chong, K.; Huang, Y.; Huang, C.; Yin, L. Empagliflozin improves mitochondrial dysfunction in diabetic cardiomyopathy by modulating ketone body metabolism and oxidative stress. Redox Biol. 2024, 69, 103010. [Google Scholar] [CrossRef]
- Nah, J.; Shirakabe, A.; Mukai, R.; Zhai, P.; Sung, E.A.; Ivessa, A.; Mizushima, W.; Nakada, Y.; Saito, T.; Hu, C.; et al. Ulk1-dependent alternative mitophagy plays a protective role during pressure overload in the heart. Cardiovasc. Res. 2022, 118, 2638–2651. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yan, Z.; Fan, Y.; Fan, X.; Li, A.; Qi, Z.; Zhang, J. Cardiac repair after myocardial infarction: A two-sided role of inflammation-mediated. Front. Cardiovasc. Med. 2023, 9, 1077290. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Miller, H. Role of cytokines in heart failure. Am. Heart J. 1998, 135, 181–186. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Hedayat, M.; Mahmoudi, M.J.; Rose, N.R.; Rezaei, N. Proinflammatory cytokines in heart failure: Double-edged swords. Heart Fail. Rev. 2010, 15, 543–562. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Gullestad, L.; Nymo, S.H.; Yndestad, A.; Aukrust, P.; Askevold, E.T. Inflammatory cytokines as biomarkers in heart failure. Clin. Chim. Acta 2015, 443, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Date, S.; Bhatt, L.K. Targeting high-mobility-group-box-1-mediated inflammation: A promising therapeutic approach for myocardial infarction. Inflammopharmacology 2024. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Wahid, A.; Wen, J.; Yang, Q.; Zhang, Z.; Zhao, X.; Tang, X. Serum HMGB1 is a biomarker for acute myocardial infarction with or without heart failure. Clin. Transl. Sci. 2023, 16, 2299–2309. [Google Scholar] [CrossRef]
- Yao, H.C.; Zhao, A.P.; Han, Q.F.; Wu, L.; Yao, D.K.; Wang, L.X. Correlation between serum high-mobility group box-1 levels and high-sensitivity C-reactive protein and troponin I in patients with coronary artery disease. Exp. Ther. Med. 2013, 6, 121–124. [Google Scholar] [CrossRef]
- Tao, A.; Song, J.; Lan, T.; Xu, X.; Kvietys, P.; Kao, R.; Martin, C.; Rui, T. Cardiomyocyte–fibroblast interaction contributes to diabetic cardiomyopathy in mice: Role of HMGB1/TLR4/IL-33 axis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Pan, W.; Li, F.; Sheikh, S.A.; Xie, Q.; Zhang, C. Berberine up-regulates miR-340-5p to protect myocardial ischaemia/reperfusion from HMGB1-mediated inflammatory injury. ESC Heart Fail. 2023, 10, 931–942. [Google Scholar] [CrossRef]
- Ago, T.; Sadoshima, J. GDF15, a Cardioprotective TGF-β Superfamily Protein. Circ. Res. 2006, 98, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Assadi, A.; Zahabi, A.; Hart, R.A. GDF15, an update of the physiological and pathological roles it plays: A review. Pflüg. Arch. Eur. J. Physiol. 2020, 472, 1535–1546. [Google Scholar] [CrossRef]
- Samarel, A.M. Focal adhesion signaling in heart failure. Pflug. Arch. 2014, 466, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Lok, S.I.; Winkens, B.; Goldschmeding, R.; van Geffen, A.J.; Nous, F.M.; van Kuik, J.; van der Weide, P.; Klöpping, C.; Kirkels, J.H.; Lahpor, J.R.; et al. Circulating growth differentiation factor-15 correlates with myocardial fibrosis in patients with non-ischaemic dilated cardiomyopathy and decreases rapidly after left ventricular assist device support. Eur. J. Heart Fail. 2012, 14, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Zeller, M.; Cottin, Y.; Vergely, C. GDF15 and Cardiac Cells: Current Concepts and New Insights. Int. J. Mol. Sci. 2021, 22, 8889. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M.; Tadross, J.A.; Al-Hadithi, A.B.A.K.; Zhao, X.; Villena-Gutiérrez, R.; Tromp, J.; Absar, S.; Au, M.; Harrison, J.; Coll, A.P.; et al. GDF15 antagonism limits severe heart failure and prevents cardiac cachexia. Cardiovasc. Res. 2024, 120, 2249–2260. [Google Scholar] [CrossRef]
- Ullah, N.; Wu, Y. Regulation of Conformational Changes in C-reactive Protein Alters its Bioactivity. Cell Biochem. Biophys. 2022, 80, 595–608. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Dutka, M.; Bobiński, R.; Ulman-Włodarz, I.; Hajduga, M.; Bujok, J.; Pająk, C.; Ćwiertnia, M. Various aspects of inflammation in heart failure. Heart Fail. Rev. 2020, 25, 537–548. [Google Scholar] [CrossRef]
- Burger, P.M.; Koudstaal, S.; Mosterd, A.; Fiolet, A.T.L.; Teraa, M.; van der Meer, M.G.; Cramer, M.J.; Visseren, F.L.J.; Ridker, P.M.; Dorresteijn, J.A.N.; et al. C-Reactive Protein and Risk of Incident Heart Failure in Patients With Cardiovascular Disease. J. Am. Coll. Cardiol. 2023, 82, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S. Interleukin-11 and its eminent role in tissue fibrosis: A possible therapeutic target. Clin. Exp. Immunol. 2023, 214, 154–161. [Google Scholar] [CrossRef]
- Cook, S.A. Understanding interleukin 11 as a disease gene and therapeutic target. Biochem. J. 2023, 480, 1987–2008. [Google Scholar] [CrossRef]
- Ye, J.; Wang, Z.; Ye, D.; Wang, Y.; Wang, M.; Ji, Q.; Huang, Y.; Liu, L.; Shi, Y.; Shi, L.; et al. Increased Interleukin-11 Levels Are Correlated with Cardiac Events in Patients with Chronic Heart Failure. Mediators Inflamm. 2019, 2019, 1575410. [Google Scholar] [CrossRef]
- Ng, B.; Widjaja, A.A.; Viswanathan, S.; Dong, J.; Chothani, S.P.; Lim, S.; Shekeran, S.G.; Tan, J.; McGregor, N.E.; Walker, E.C.; et al. Similarities and differences between IL11 and IL11RA1 knockout mice for lung fibro-inflammation, fertility and craniosynostosis. Sci. Rep. 2021, 11, 14088. [Google Scholar] [CrossRef] [PubMed]
- Jian, Y.; Zhou, X.; Shan, W.; Chen, C.; Ge, W.; Cui, J.; Yi, W.; Sun, Y. Crosstalk between macrophages and cardiac cells after myocardial infarction. Cell Commun. Signal. 2023, 21, 109. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Fu, S.; Yang, R.; Yang, K.; Lei, W.; Yang, Y.; Zhang, Q.; Zhao, Y.; Yu, J.; Yu, L.; et al. Advances in the study of macrophage polarization in inflammatory immune skin diseases. J. Inflamm. 2023, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, M.; Lu, S.; Dai, S.; Liu, J. Role of macrophage polarization in heart failure and traditional Chinese medicine treatment. Front. Pharmacol. 2024, 15, 1434654. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 Inhibits Inflammation and Attenuates Left Ventricular Remodeling After Myocardial Infarction via Activation of STAT3 and Suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Hovsepian, E.; Penas, F.; Siffo, S.; Mirkin, G.A.; Goren, N.B. IL-10 Inhibits the NF-κB and ERK/MAPK-Mediated Production of Pro-Inflammatory Mediators by Up-Regulation of SOCS-3 in Trypanosoma cruzi-Infected Cardiomyocytes. PLoS ONE 2013, 8, e79445. [Google Scholar] [CrossRef] [PubMed]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Fekete, A.; Szabó, A.J.; Vannay, Á. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediators Inflamm. 2015, 2015, 764641. [Google Scholar] [CrossRef]
- Stumpf, C.; Lehner, C.; Yilmaz, A.; Daniel, W.G.; Garlichs, C.D. Decrease of serum levels of the anti-inflammatory cytokine interleukin-10 in patients with advanced chronic heart failure. Clin. Sci. 2003, 105, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Sharma, A.K.; Singal, P.K. Significance of changes in TNF-alpha and IL-10 levels in the progression of heart failure subsequent to myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H106–H113. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; Anti, T.N.F.T.A.C.H.F.I. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, L.; Li, W.; Fang, J. Role of high-mobility group box-1 in myocardial ischemia/reperfusion injury and the effect of ethyl pyruvate. Exp. Ther. Med. 2015, 9, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Wu, F.; Ren, K.; Huo, J.L. Melatonin attenuates inflammation and cardiac dysfunction in myocardial infarction by regulating the miRNA-200b-3p/high mobility group box chromosomal protein 1 axis. J. Physiol. Pharmacol. 2023, 74, 377–387. [Google Scholar] [CrossRef]
- Halladin, N.L.; Busch, S.E.; Jensen, S.E.; Hansen, H.S.; Zaremba, T.; Aarøe, J.; Rosenberg, J.; Gögenur, I. Intracoronary and systemic melatonin to patients with acute myocardial infarction: Protocol for the IMPACT trial. Dan. Med. J. 2014, 61, A4773. [Google Scholar]
- Crossman, D.; Rothman, A. The Canakinumab Antiinflammatory Thrombosis Outcome Study trial—The starting gun has fired. J. Thorac. Dis. 2017, 9, 4922–4925. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Oddi Erdle, C.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e004373. [Google Scholar] [CrossRef]
- Garofalo, M.; Corso, R.; Tomasoni, D.; Adamo, M.; Lombardi, C.M.; Inciardi, R.M.; Gussago, C.; Di Mario, C.; Metra, M.; Pagnesi, M. Inflammation in acute heart failure. Front. Cardiovasc. Med. 2023, 10, 1235178. [Google Scholar] [CrossRef]
- Sansonetti, M.; Al Soodi, B.; Thum, T.; Jung, M. Macrophage-based therapeutic approaches for cardiovascular diseases. Basic. Res. Cardiol. 2024, 119, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Chernykh, E.R.; Kafanova, M.Y.; Shevela, E.Y.; Sirota, S.I.; Adonina, E.I.; Sakhno, L.V.; Ostanin, A.A.; Kozlov, V.V. Clinical experience with autologous M2 macrophages in children with severe cerebral palsy. Cell Transplant. 2014, 23 (Suppl. S1), S97–S104. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.N.; Henry, T.D.; Quyyumi, A.A.; Schaer, G.L.; Anderson, R.D.; Toma, C.; East, C.; Remmers, A.E.; Goodrich, J.; Desai, A.S.; et al. Ixmyelocel-T for patients with ischaemic heart failure: A prospective randomised double-blind trial. Lancet 2016, 387, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and cell death. Nat. Cell Biol. 2024, 26, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; Bonilha, V.L. Regulated cell death pathways in the sodium iodate model: Insights and implications for AMD. Exp. Eye Res. 2024, 238, 109728. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef]
- Narula, J.; Pandey, P.; Arbustini, E.; Haider, N.; Narula, N.; Kolodgie, F.D.; Dal Bello, B.; Semigran, M.J.; Bielsa-Masdeu, A.; Dec, G.W.; et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Natl. Acad. Sci. USA 1999, 96, 8144–8149. [Google Scholar] [CrossRef] [PubMed]
- Scheubel, R.J.; Bartling, B.; Simm, A.; Silber, R.E.; Drogaris, K.; Darmer, D.; Holtz, J. Apoptotic pathway activation from mitochondria and death receptors without caspase-3 cleavage in failing human myocardium: Fragile balance of myocyte survival? J. Am. Coll. Cardiol. 2002, 39, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Birks, E.J.; Latif, N.; Enesa, K.; Folkvang, T.; Luong, L.A.; Sarathchandra, P.; Khan, M.; Ovaa, H.; Terracciano, C.M.; Barton, P.J.; et al. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc. Res. 2008, 79, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Conte, J.V., Jr.; Foster, A.H.; McLaughlin, J.S.; Wei, C. Increased p53 protein expression in human failing myocardium. J. Heart Lung Transplant. 1999, 18, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Leri, A.; Liu, Y.; Malhotra, A.; Li, Q.; Stiegler, P.; Claudio, P.P.; Giordano, A.; Kajstura, J.; Hintze, T.H.; Anversa, P. Pacing-induced heart failure in dogs enhances the expression of p53 and p53-dependent genes in ventricular myocytes. Circulation 1998, 97, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liang, C.; Li, P.; Yang, L.; Hao, Z.; Kong, L.; Tian, X.; Guo, C.; Dong, J.; Zhang, Y.; et al. Runt-related transcription factor 1 (Runx1) aggravates pathological cardiac hypertrophy by promoting p53 expression. J. Cell Mol. Med. 2021, 25, 7867–7877. [Google Scholar] [CrossRef]
- Qi, P.; Zhai, Q.; Zhang, X. RUNX1 facilitates heart failure progression through regulating TGF-beta-induced cardiac remodeling. PeerJ 2023, 11, e16202. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Yang, J.; Zhang, M.; Tian, T.; Jiang, Y.; Liu, X.; Xue, G.; Li, X.; Zhang, X.; et al. Interdependent Nuclear Co-Trafficking of ASPP1 and p53 Aggravates Cardiac Ischemia/Reperfusion Injury. Circ. Res. 2023, 132, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Huby, A.C.; Turdi, S.; James, J.; Towbin, J.A.; Purevjav, E. FasL expression in cardiomyocytes activates the ERK1/2 pathway, leading to dilated cardiomyopathy and advanced heart failure. Clin. Sci. 2016, 130, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Borne, Y.; Mattisson, I.Y.; Wigren, M.; Melander, O.; Ohro-Melander, M.; Bengtsson, E.; Fredrikson, G.N.; Nilsson, J.; Engstrom, G. FADD, Caspase-3, and Caspase-8 and Incidence of Coronary Events. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 983–989. [Google Scholar] [CrossRef]
- Yin, H.; Guo, X.; Chen, Y.; Zeng, Y.; Mo, X.; Hong, S.; He, H.; Li, J.; Steinmetz, R.; Liu, Q. TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and necroptosis. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Steenbergen, C.; Afshari, C.A.; Petranka, J.G.; Collins, J.; Martin, K.; Bennett, L.; Haugen, A.; Bushel, P.; Murphy, E. Alterations in apoptotic signaling in human idiopathic cardiomyopathic hearts in failure. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H268–H276. [Google Scholar] [CrossRef]
- Li, Y.Z.; Wu, H.; Liu, D.; Yang, J.; Yang, J.; Ding, J.W.; Zhou, G.; Zhang, J.; Zhang, D. cFLIP(L) Alleviates Myocardial Ischemia-Reperfusion Injury by Inhibiting Endoplasmic Reticulum Stress. Cardiovasc. Drugs Ther. 2023, 37, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, B.; Cavalieri, T.A.; Hock, C.E. Pifithrin-alpha attenuates p53-mediated apoptosis and improves cardiac function in response to myocardial ischemia/reperfusion in aged rats. Shock 2006, 26, 608–614. [Google Scholar] [CrossRef]
- Liu, X.; Chua, C.C.; Gao, J.; Chen, Z.; Landy, C.L.; Hamdy, R.; Chua, B.H. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H933–H939. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200. [Google Scholar] [CrossRef]
- Jia, K.; Dai, Y.; Liu, A.; Li, X.; Wu, L.; Lu, L.; Bao, Y.; Jin, Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J. Cardiovasc. Pharmacol. 2020, 76, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Lerida-Viso, A.; Estepa-Fernandez, A.; Morella-Aucejo, A.; Lozano-Torres, B.; Alfonso, M.; Blandez, J.F.; Bisbal, V.; Sepulveda, P.; Garcia-Fernandez, A.; Orzaez, M.; et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 2022, 183, 106356. [Google Scholar] [CrossRef] [PubMed]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 2024, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, S.; Zhao, C.; Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 2019, 516, 37–43. [Google Scholar] [CrossRef]
- Gawargi, F.I.; Mishra, P.K. Regulation of cardiac ferroptosis in diabetic human heart failure: Uncovering molecular pathways and key targets. Cell Death Discov. 2024, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhao, C.; Li, H.; Chen, X.; Ding, Y.; Xu, S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 2018, 497, 233–240. [Google Scholar] [CrossRef]
- Ma, S.; He, L.L.; Zhang, G.R.; Zuo, Q.J.; Wang, Z.L.; Zhai, J.L.; Zhang, T.T.; Wang, Y.; Ma, H.J.; Guo, Y.F. Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn Schmiedebergs Arch. Pharmacol. 2022, 395, 945–962. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Omiya, S.; Rusu, M.C.; Ueda, H.; Murakawa, T.; Tanada, Y.; Abe, H.; Nakahara, K.; Asahi, M.; Taneike, M.; et al. Iron derived from autophagy-mediated ferritin degradation induces cardiomyocyte death and heart failure in mice. Elife 2021, 10, e62174. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, C.; Gao, Z.; Chen, H.; Li, K.; Wang, L.; Zheng, Y.; Li, C.; Zhang, H.; Gong, M.; et al. SLC7A11/xCT Prevents Cardiac Hypertrophy by Inhibiting Ferroptosis. Cardiovasc. Drugs Ther. 2022, 36, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.Y.; Zhao, Y.C.; Zhao, C.X.; Gu, Z.C.; Lu, X.Y.; Jiang, W.L.; Gao, L.C.; Li, W.L.; Qin, Z.H.; Ge, H.; et al. The Role of CD147 in Pathological Cardiac Hypertrophy Is Regulated by Glycosylation. Oxid. Med. Cell. Longev. 2022, 2022, 6603296. [Google Scholar] [CrossRef] [PubMed]
- Gawargi, F.I.; Mishra, P.K. MMP9 drives ferroptosis by regulating GPX4 and iron signaling. iScience 2024, 27, 110622. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Zhou, Y.J.; Tang, L.J.; Xiong, X.M.; Zhang, X.J.; Ali Sheikh, M.S.; Zhang, J.J.; Luo, X.J.; Yuan, C.; Peng, J. Combination of ponatinib with deferoxamine synergistically mitigates ischemic heart injury via simultaneous prevention of necroptosis and ferroptosis. Eur. J. Pharmacol. 2021, 898, 173999. [Google Scholar] [CrossRef]
- Ishimaru, K.; Ikeda, M.; Miyamoto, H.D.; Furusawa, S.; Abe, K.; Watanabe, M.; Kanamura, T.; Fujita, S.; Nishimura, R.; Toyohara, T.; et al. Deferasirox Targeting Ferroptosis Synergistically Ameliorates Myocardial Ischemia Reperfusion Injury in Conjunction With Cyclosporine A. J. Am. Heart Assoc. 2024, 13, e031219. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, X.; Jiang, L.; Hao, T.; Wang, Y.; Li, T. Inhibition of ferroptosis reverses heart failure with preserved ejection fraction in mice. J. Transl. Med. 2024, 22, 199. [Google Scholar] [CrossRef]
- Yan, L. Ferrostatin-1-loaded liposome treatment for coronary in-stent restenosis. J. Am. Coll. Cardiol. 2024, 83, 902. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, R.; Ouyang, Y.; Gu, W.; Xiao, T.; Yang, H.; Tang, L.; Wang, H.; Xiang, B.; Chen, P. Pyroptosis in health and disease: Mechanisms, regulation and clinical perspective. Signal Transduct. Target. Ther. 2024, 9, 245. [Google Scholar] [CrossRef]
- Merkle, S.; Frantz, S.; Schon, M.P.; Bauersachs, J.; Buitrago, M.; Frost, R.J.; Schmitteckert, E.M.; Lohse, M.J.; Engelhardt, S. A role for caspase-1 in heart failure. Circ. Res. 2007, 100, 645–653. [Google Scholar] [CrossRef]
- Segiet, O.A.; Piecuch, A.; Mielanczyk, L.; Michalski, M.; Nowalany-Kozielska, E. Role of interleukins in heart failure with reduced ejection fraction. Anatol. J. Cardiol. 2019, 22, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhao, H.; Wen, X.; Li, G.; Guo, S.; Zhang, D. NLRP3-inflammasome inhibition by MCC950 attenuates cardiac and pulmonary artery remodelling in heart failure with preserved ejection fraction. Life Sci. 2023, 333, 122185. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Du, N.; Zhang, Q.; Li, J.; Chen, X.; Liu, X.; Hu, Y.; Qin, W.; Shen, N.; Xu, C.; et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014, 5, e1479. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Zheng, Z.; Luo, Y.; Wang, X.; Lv, M.; Qin, D.; Tan, Q.; Zhang, Y.; Wang, T.; Hu, H. NLRP3-mediated pyroptosis aggravates pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction in mice: Cardioprotective role of irisin. Cell Death Discov. 2021, 7, 50. [Google Scholar] [CrossRef]
- Wang, J.; Deng, B.; Liu, Q.; Huang, Y.; Chen, W.; Li, J.; Zhou, Z.; Zhang, L.; Liang, B.; He, J.; et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020, 11, 574. [Google Scholar] [CrossRef] [PubMed]
- Onodi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Toth, V.E.; Koncsos, G.; Novak, J.; Brenner, G.B.; Makkos, A.; Baranyai, T.; et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 2021, 117, 2639–2651. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.P.; Wu, C.; Ma, Y.J.; Zhu, J.B.; Ma, L.L.; Kong, F.J. AIM2 Deficiency Alleviates Cardiac Inflammation and Hypertrophy in HFD/STZ-Induced Diabetic Mice by Inhibiting the NLRC4/IRF1 Signaling Pathway. J. Cardiovasc. Transl. Res. 2024. [Google Scholar] [CrossRef]
- Durga Devi, T.; Babu, M.; Makinen, P.; Kaikkonen, M.U.; Heinaniemi, M.; Laakso, H.; Yla-Herttuala, E.; Rieppo, L.; Liimatainen, T.; Naumenko, N.; et al. Aggravated Postinfarct Heart Failure in Type 2 Diabetes Is Associated with Impaired Mitophagy and Exaggerated Inflammasome Activation. Am. J. Pathol. 2017, 187, 2659–2673. [Google Scholar] [CrossRef]
- Wang, X.; Pan, J.; Liu, H.; Zhang, M.; Liu, D.; Lu, L.; Tian, J.; Liu, M.; Jin, T.; An, F. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019, 221, 249–258. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Figal, D.; Nunez, J.; Perez-Martinez, M.T.; Gonzalez-Juanatey, J.R.; Taibo-Urquia, M.; Llacer Iborra, P.; Delgado, J.; Villar, S.; Mirabet, S.; Aimo, A.; et al. Colchicine in acutely decompensated heart failure: The COLICA trial. Eur. Heart J. 2024, 45, 4826–4836. [Google Scholar] [CrossRef]
- Sun, X.; Duan, J.; Gong, C.; Feng, Y.; Hu, J.; Gu, R.; Xu, B. Colchicine Ameliorates Dilated Cardiomyopathy Via SIRT2-Mediated Suppression of NLRP3 Inflammasome Activation. J. Am. Heart Assoc. 2022, 11, e025266. [Google Scholar] [CrossRef] [PubMed]
- Wohlford, G.F.; Van Tassell, B.W.; Billingsley, H.E.; Kadariya, D.; Canada, J.M.; Carbone, S.; Mihalick, V.L.; Bonaventura, A.; Vecchie, A.; Chiabrando, J.G.; et al. Phase 1B, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects With NYHA II-III Systolic Heart Failure. J. Cardiovasc. Pharmacol. 2020, 77, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Abellan, A.; Angosto-Bazarra, D.; Martinez-Banaclocha, H.; de Torre-Minguela, C.; Ceron-Carrasco, J.P.; Perez-Sanchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhao, L.; Wang, J.; Liu, S.; Zhang, Y.; Qin, Q. The selective NLRP3 inflammasome inhibitor MCC950 improves isoproterenol-induced cardiac dysfunction by inhibiting cardiomyocyte senescence. Eur. J. Pharmacol. 2022, 937, 175364. [Google Scholar] [CrossRef]
- Audia, J.P.; Yang, X.M.; Crockett, E.S.; Housley, N.; Haq, E.U.; O’Donnell, K.; Cohen, M.V.; Downey, J.M.; Alvarez, D.F. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y(12) receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic. Res. Cardiol. 2018, 113, 32. [Google Scholar] [CrossRef]
- Su, X.L.; Wang, S.H.; Komal, S.; Cui, L.G.; Ni, R.C.; Zhang, L.R.; Han, S.N. The caspase-1 inhibitor VX765 upregulates connexin 43 expression and improves cell-cell communication after myocardial infarction via suppressing the IL-1beta/p38 MAPK pathway. Acta Pharmacol. Sin. 2022, 43, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Baldus, S.; von Kodolitsch, Y.; Rudolph, V.; Meinertz, T. Systemic endothelial dysfunction as an early predictor of adverse outcome in heart failure. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Zuchi, C.; Ambrosio, G.; Luscher, T.F.; Landmesser, U. Nutraceuticals in cardiovascular prevention: Lessons from studies on endothelial function. Cardiovasc. Ther. 2010, 28, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C., Jr. Coronary endothelial function in health and disease. Drugs 1997, 53 (Suppl. S1), 20–29. [Google Scholar] [CrossRef] [PubMed]
- Paolocci, N.; Biondi, R.; Bettini, M.; Lee, C.I.; Berlowitz, C.O.; Rossi, R.; Xia, Y.; Ambrosio, G.; L’Abbate, A.; Kass, D.A.; et al. Oxygen radical-mediated reduction in basal and agonist-evoked NO release in isolated rat heart. J. Mol. Cell Cardiol. 2001, 33, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Dikalov, S.; Tatge, H.; Wilke, R.; Kohler, C.; Harrison, D.G.; Hornig, B.; Drexler, H. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: Role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 2002, 106, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Stamler, J.S. NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Investig. 2005, 115, 509–517. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Tang, E.H.; Feletou, M. Endothelial dysfunction and vascular disease. Acta. Physiol. 2009, 196, 193–222. [Google Scholar] [CrossRef]
- Gebhart, V.; Reiss, K.; Kollau, A.; Mayer, B.; Gorren, A.C.F. Site and mechanism of uncoupling of nitric-oxide synthase: Uncoupling by monomerization and other misconceptions. Nitric Oxide 2019, 89, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Parodi, O.; De Maria, R.; Roubina, E. Redox state, oxidative stress and endothelial dysfunction in heart failure: The puzzle of nitrate-thiol interaction. J. Cardiovasc. Med. 2007, 8, 765–774. [Google Scholar] [CrossRef]
- Tousoulis, D.; Charakida, M.; Stefanadis, C. Endothelial function and inflammation in coronary artery disease. Heart 2006, 92, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.H.; Rector, T.S.; Bank, A.J.; Williams, R.E.; Heifetz, S.M. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation 1991, 84, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef]
- Tromp, J.; Lim, S.L.; Tay, W.T.; Teng, T.K.; Chandramouli, C.; Ouwerkerk, W.; Wander, G.S.; Sawhney, J.P.S.; Yap, J.; MacDonald, M.R.; et al. Microvascular Disease in Patients With Diabetes With Heart Failure and Reduced Ejection Versus Preserved Ejection Fraction. Diabetes Care 2019, 42, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Rossa, S.; Landmesser, U.; Spiekermann, S.; Engberding, N.; Hornig, B.; Drexler, H. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur. Heart J. 2005, 26, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.P.; Arnott, C.; Beale, A.L.; Chandramouli, C.; Hilfiker-Kleiner, D.; Kaye, D.M.; Ky, B.; Santema, B.T.; Sliwa, K.; Voors, A.A. Sex differences in heart failure. Eur. Heart J. 2019, 40, 3859–3868c. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Blenck, C.L.; Harvey, P.A.; Reckelhoff, J.F.; Leinwand, L.A. The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circ. Res. 2016, 118, 1294–1312. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Oliviero, P.; Damy, T.; Robidel, E.; Marotte, F.; Heymes, C.; Samuel, J.L. Effects of sex differences on constitutive nitric oxide synthase expression and activity in response to pressure overload in rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2650–H2658. [Google Scholar] [CrossRef]
- Loyer, X.; Damy, T.; Chvojkova, Z.; Robidel, E.; Marotte, F.; Oliviero, P.; Heymes, C.; Samuel, J.L. 17beta-estradiol regulates constitutive nitric oxide synthase expression differentially in the myocardium in response to pressure overload. Endocrinology 2007, 148, 4579–4584. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. Molecular and cellular basis of cardiovascular gender differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef]
- Ambrosio, G. The elusive difference between hibernation and stunning in patients. Basic. Res. Cardiol. 1995, 90, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, C.; Biondi, R.; Xia, Y.; Cardounel, A.J.; Druhan, L.J.; Ambrosio, G.; Zweier, J.L. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc. Natl. Acad. Sci. USA 2007, 104, 15081–15086. [Google Scholar] [CrossRef]
- Reyes, L.A.; Boslett, J.; Varadharaj, S.; De Pascali, F.; Hemann, C.; Druhan, L.J.; Ambrosio, G.; El-Mahdy, M.; Zweier, J.L. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. USA 2015, 112, 11648–11653. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.P.; Greer, J.J.; Kakkar, A.K.; Ware, P.D.; Turnage, R.H.; Hicks, M.; van Haperen, R.; de Crom, R.; Kawashima, S.; Yokoyama, M.; et al. Endothelial nitric oxide synthase overexpression attenuates myocardial reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H276–H282. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Rao, R.; Berman, J.W.; Schwarz, M.; Demopoulos, L.; Bijou, R.; LeJemtel, T.H. Pathophysiological correlates of increased serum tumor necrosis factor in patients with congestive heart failure. Relation to nitric oxide-dependent vasodilation in the forearm circulation. Circulation 1994, 90, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Agnoletti, L.; Curello, S.; Bachetti, T.; Malacarne, F.; Gaia, G.; Comini, L.; Volterrani, M.; Bonetti, P.; Parrinello, G.; Cadei, M.; et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: Role of tumor necrosis factor-alpha. Circulation 1999, 100, 1983–1991. [Google Scholar] [CrossRef]
- Paulus, W.J.; Bronzwaer, J.G. Myocardial contractile effects of nitric oxide. Heart Fail. Rev. 2002, 7, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Calderone, A.; Thaik, C.M.; Takahashi, N.; Chang, D.L.; Colucci, W.S. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Investig. 1998, 101, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Radauceanu, A. Effect of MR blockade on collagen formation and cardiovascular disease with a specific emphasis on heart failure. Heart Fail. Rev. 2005, 10, 71–78. [Google Scholar] [CrossRef]
- Singh, A.; Bhatt, K.S.; Nguyen, H.C.; Frisbee, J.C.; Singh, K.K. Endothelial-to-Mesenchymal Transition in Cardiovascular Pathophysiology. Int. J. Mol. Sci. 2024, 25, 6180. [Google Scholar] [CrossRef]
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550. [Google Scholar] [CrossRef] [PubMed]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model. Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Songia, P.; Branchetti, E.; Parolari, A.; Myasoedova, V.; Ferrari, G.; Alamanni, F.; Tremoli, E.; Poggio, P. Mitral valve endothelial cells secrete osteoprotegerin during endothelial mesenchymal transition. J. Mol. Cell Cardiol. 2016, 98, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- van de Wouw, J.; Sorop, O.; van Drie, R.W.A.; van Duin, R.W.B.; Nguyen, I.T.N.; Joles, J.A.; Verhaar, M.C.; Merkus, D.; Duncker, D.J. Perturbations in myocardial perfusion and oxygen balance in swine with multiple risk factors: A novel model of ischemia and no obstructive coronary artery disease. Basic Res. Cardiol. 2020, 115, 21. [Google Scholar] [CrossRef] [PubMed]
- Van Empel, V.P.; Mariani, J.; Borlaug, B.A.; Kaye, D.M. Impaired myocardial oxygen availability contributes to abnormal exercise hemodynamics in heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2014, 3, e001293. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Shah, S.J.; Guazzi, M. Inflammation in Heart Failure With Preserved Ejection Fraction: Time to Put Out the Fire. JACC Heart Fail. 2016, 4, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Saito, N.; Kirigaya, H.; Gyotoku, D.; Iinuma, N.; Kusakawa, Y.; Iguchi, K.; Nakachi, T.; Fukui, K.; Futaki, M.; et al. Impairment of Coronary Flow Reserve Evaluated by Phase Contrast Cine-Magnetic Resonance Imaging in Patients With Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2016, 5, e002649. [Google Scholar] [CrossRef]
- Srivaratharajah, K.; Coutinho, T.; deKemp, R.; Liu, P.; Haddad, H.; Stadnick, E.; Davies, R.A.; Chih, S.; Dwivedi, G.; Guo, A.; et al. Reduced Myocardial Flow in Heart Failure Patients With Preserved Ejection Fraction. Circ. Heart Fail. 2016, 9, e002562. [Google Scholar] [CrossRef]
- Dryer, K.; Gajjar, M.; Narang, N.; Lee, M.; Paul, J.; Shah, A.P.; Nathan, S.; Butler, J.; Davidson, C.J.; Fearon, W.F.; et al. Coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1033–H1042. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Wright, J.W.; Mizutani, S.; Harding, J.W. Pathways involved in the transition from hypertension to hypertrophy to heart failure. Treatment strategies. Heart Fail. Rev. 2008, 13, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, M.; Skarda, V.; Leenen, F.H. Effects of ACE inhibitors on circulating versus cardiac angiotensin II in volume overload-induced cardiac hypertrophy in rats. Circulation 1995, 92, 3568–3573. [Google Scholar] [CrossRef]
- Schunkert, H.; Ingelfinger, J.R.; Hirsch, A.T.; Tang, S.S.; Litwin, S.E.; Talsness, C.E.; Dzau, V.J. Evidence for tissue-specific activation of renal angiotensinogen mRNA expression in chronic stable experimental heart failure. J. Clin. Investig. 1992, 90, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Mulatero, P.; Schiavone, D.; Mengozzi, G.; Tesio, L.; Chiandussi, L.; Veglio, F. Vasoactive hormones induce nitric oxide synthase mRNA expression and nitric oxide production in human endothelial cells and monocytes. Am. J. Hypertens. 1999, 12, 388–397. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Lips, D.J.; deWindt, L.J.; van Kraaij, D.J.; Doevendans, P.A. Molecular determinants of myocardial hypertrophy and failure: Alternative pathways for beneficial and maladaptive hypertrophy. Eur. Heart J. 2003, 24, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Suzuki, Y.; Mezzano, S.; Plaza, J.J.; Egido, J. Role of the renin-angiotensin system in vascular diseases: Expanding the field. Hypertension 2001, 38, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.Y.; Blann, A.D.; Patel, J.; Freestone, B.; Hughes, E.; Lip, G.Y. Endothelial dysfunction and damage in congestive heart failure: Relation of flow-mediated dilation to circulating endothelial cells, plasma indexes of endothelial damage, and brain natriuretic peptide. Circulation 2004, 110, 1794–1798. [Google Scholar] [CrossRef]
- Constans, J.; Conri, C. Circulating markers of endothelial function in cardiovascular disease. Clin. Chim. Acta 2006, 368, 33–47. [Google Scholar] [CrossRef]
- Meigs, J.B.; Hu, F.B.; Rifai, N.; Manson, J.E. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004, 291, 1978–1986. [Google Scholar] [CrossRef]
- Anderssohn, M.; Schwedhelm, E.; Luneburg, N.; Vasan, R.S.; Boger, R.H. Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: An intriguing interaction with diabetes mellitus. Diab Vasc. Dis. Res. 2010, 7, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Eid, H.M.; Arnesen, H.; Hjerkinn, E.M.; Lyberg, T.; Seljeflot, I. Relationship between obesity, smoking, and the endogenous nitric oxide synthase inhibitor, asymmetric dimethylarginine. Metabolism 2004, 53, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, M.T.; Creager, S.J.; Scales, K.M.; Cusco, J.A.; Lee, B.K.; Creager, M.A. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation 1993, 88, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, A.D.; Cross, J.; Kharbanda, R.K.; Mullen, M.J.; Bhagat, K.; Taylor, M.; Donald, A.E.; Palacios, M.; Griffin, G.E.; Deanfield, J.E.; et al. Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation 2000, 102, 994–999. [Google Scholar] [CrossRef]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The Role of Asymmetric Dimethylarginine (ADMA) in Endothelial Dysfunction and Cardiovascular Disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Kocher, A.A.; Schuster, M.D.; Szabolcs, M.J.; Takuma, S.; Burkhoff, D.; Wang, J.; Homma, S.; Edwards, N.M.; Itescu, S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 2001, 7, 430–436. [Google Scholar] [CrossRef]
- Taguchi, K.; Hida, M.; Narimatsu, H.; Matsumoto, T.; Kobayashi, T. Glucose and angiotensin II-derived endothelial extracellular vesicles regulate endothelial dysfunction via ERK1/2 activation. Pflug. Arch. 2017, 469, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Wang, A.; Wang, Q.; Han, W.; Rong, R.; Wang, L.; Liu, S.; Zhang, Y.; Dong, C.; Li, Y. Plasma endothelial cells-derived extracellular vesicles promote wound healing in diabetes through YAP and the PI3K/Akt/mTOR pathway. Aging 2020, 12, 12002–12018. [Google Scholar] [CrossRef] [PubMed]
- Abid Hussein, M.N.; Boing, A.N.; Biro, E.; Hoek, F.J.; Vogel, G.M.; Meuleman, D.G.; Sturk, A.; Nieuwland, R. Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties. Thromb. Res. 2008, 121, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.Q. MicroRNA targets and biomarker validation for diabetes-associated cardiac fibrosis. Pharmacol. Res. 2021, 174, 105941. [Google Scholar] [CrossRef] [PubMed]
- Amabile, N.; Cheng, S.; Renard, J.M.; Larson, M.G.; Ghorbani, A.; McCabe, E.; Griffin, G.; Guerin, C.; Ho, J.E.; Shaw, S.Y.; et al. Association of circulating endothelial microparticles with cardiometabolic risk factors in the Framingham Heart Study. Eur. Heart J. 2014, 35, 2972–2979. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, L.; Jy, W.; Fierro, C.; Macdonough, R.; Velazques, H.A.; Purow, J.; Jimenez, J.J.; Horstman, L.L.; Ferreira, A.; de Marchena, E.; et al. Endothelial microparticles correlate with high-risk angiographic lesions in acute coronary syndromes. Int. J. Cardiol. 2004, 97, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Gokce, N.; Keaney, J.F., Jr.; Hunter, L.M.; Watkins, M.T.; Nedeljkovic, Z.S.; Menzoian, J.O.; Vita, J.A. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J. Am. Coll. Cardiol. 2003, 41, 1769–1775. [Google Scholar] [CrossRef]
- Yeboah, J.; Folsom, A.R.; Burke, G.L.; Johnson, C.; Polak, J.F.; Post, W.; Lima, J.A.; Crouse, J.R.; Herrington, D.M. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: The multi-ethnic study of atherosclerosis. Circulation 2009, 120, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Boerma, E.C.; Cecconi, M.; De Backer, D.; Shapiro, N.I.; Duranteau, J.; Pinsky, M.R.; Artigas, A.; Teboul, J.L.; Reiss, I.K.M.; et al. Second consensus on the assessment of sublingual microcirculation in critically ill patients: Results from a task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2018, 44, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Chia, P.Y.; Teo, A.; Yeo, T.W. Overview of the Assessment of Endothelial Function in Humans. Front. Med. 2020, 7, 542567. [Google Scholar] [CrossRef] [PubMed]
- Aykut, G.; Veenstra, G.; Scorcella, C.; Ince, C.; Boerma, C. Cytocam-IDF (incident dark field illumination) imaging for bedside monitoring of the microcirculation. Intensive Care Med. Exp. 2015, 3, 40. [Google Scholar] [CrossRef]
- Lee, D.H.; Dane, M.J.; van den Berg, B.M.; Boels, M.G.; van Teeffelen, J.W.; de Mutsert, R.; den Heijer, M.; Rosendaal, F.R.; van der Vlag, J.; van Zonneveld, A.J.; et al. Deeper penetration of erythrocytes into the endothelial glycocalyx is associated with impaired microvascular perfusion. PLoS ONE 2014, 9, e96477. [Google Scholar] [CrossRef]
- Taqueti, V.R.; Di Carli, M.F. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2625–2641. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Taqueti, V.R.; van de Hoef, T.P.; Bajaj, N.S.; Bravo, P.E.; Murthy, V.L.; Osborne, M.T.; Seidelmann, S.B.; Vita, T.; Bibbo, C.F.; et al. Integrated Noninvasive Physiological Assessment of Coronary Circulatory Function and Impact on Cardiovascular Mortality in Patients With Stable Coronary Artery Disease. Circulation 2017, 136, 2325–2336. [Google Scholar] [CrossRef]
- Murthy, V.L.; Naya, M.; Foster, C.R.; Gaber, M.; Hainer, J.; Klein, J.; Dorbala, S.; Blankstein, R.; Di Carli, M.F. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012, 126, 1858–1868. [Google Scholar] [CrossRef]
- Taqueti, V.R.; Solomon, S.D.; Shah, A.M.; Desai, A.S.; Groarke, J.D.; Osborne, M.T.; Hainer, J.; Bibbo, C.F.; Dorbala, S.; Blankstein, R.; et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur. Heart J. 2018, 39, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Mrakic-Sposta, S.; Di Santo, S.G.; Franchini, F.; Arlati, S.; Zangiacomi, A.; Greci, L.; Moretti, S.; Jesuthasan, N.; Marzorati, M.; Rizzo, G.; et al. Effects of Combined Physical and Cognitive Virtual Reality-Based Training on Cognitive Impairment and Oxidative Stress in MCI Patients: A Pilot Study. Front. Aging Neurosci. 2018, 10, 282. [Google Scholar] [CrossRef]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Ploska, A.; Wieronska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: Molecular mechanisms as potential therapeutic targets. Cell Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Hong, H.J.; Hsiao, G.; Cheng, T.H.; Yen, M.H. Supplemention with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension 2001, 38, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Nishio, Y.; Okamura, T.; Yoshida, Y.; Maegawa, H.; Kojima, H.; Masada, M.; Toda, N.; Kikkawa, R.; Kashiwagi, A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ. Res. 2000, 87, 566–573. [Google Scholar] [CrossRef]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Fukuda, Y.; Matsuura, H.; Oshima, T.; Chayama, K. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 2002, 15, 326–332. [Google Scholar] [CrossRef]
- Maier, W.; Cosentino, F.; Lutolf, R.B.; Fleisch, M.; Seiler, C.; Hess, O.M.; Meier, B.; Luscher, T.F. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J. Cardiovasc. Pharmacol. 2000, 35, 173–178. [Google Scholar] [CrossRef]
- Setoguchi, S.; Hirooka, Y.; Eshima, K.; Shimokawa, H.; Takeshita, A. Tetrahydrobiopterin improves impaired endothelium-dependent forearm vasodilation in patients with heart failure. J. Cardiovasc. Pharmacol. 2002, 39, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Joannides, R.; Bizet-Nafeh, C.; Costentin, A.; Iacob, M.; Derumeaux, G.; Cribier, A.; Thuillez, C. Chronic ACE inhibition enhances the endothelial control of arterial mechanics and flow-dependent vasodilatation in heart failure. Hypertension 2001, 38, 1446–1450. [Google Scholar] [CrossRef] [PubMed]
- Hornig, B.; Landmesser, U.; Kohler, C.; Ahlersmann, D.; Spiekermann, S.; Christoph, A.; Tatge, H.; Drexler, H. Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: Role of superoxide dismutase. Circulation 2001, 103, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kusano, K.; Nakamura, Y.; Kakishita, M.; Ohta, K.; Nagase, S.; Yamamoto, M.; Miyaji, K.; Saito, H.; Morita, H.; et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002, 105, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Rossig, L.; Haendeler, J.; Mallat, Z.; Hugel, B.; Freyssinet, J.M.; Tedgui, A.; Dimmeler, S.; Zeiher, A.M. Congestive heart failure induces endothelial cell apoptosis: Protective role of carvedilol. J. Am. Coll. Cardiol. 2000, 36, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Balidemaj, K.; Homma, S.; Wu, H.; Wang, J.; Maybaum, S. Acute type 5 phosphodiesterase inhibition with sildenafil enhances flow-mediated vasodilation in patients with chronic heart failure. J. Am. Coll. Cardiol. 2000, 36, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Costello-Boerrigter, L.C.; Cataliotti, A.; Tsuruda, T.; Harty, G.J.; Lapp, H.; Stasch, J.P.; Burnett, J.C., Jr. Cardiorenal and humoral properties of a novel direct soluble guanylate cyclase stimulator BAY 41-2272 in experimental congestive heart failure. Circulation 2003, 107, 686–689. [Google Scholar] [CrossRef]
- Lapp, H.; Mitrovic, V.; Franz, N.; Heuer, H.; Buerke, M.; Wolfertz, J.; Mueck, W.; Unger, S.; Wensing, G.; Frey, R. Cinaciguat (BAY 58-2667) improves cardiopulmonary hemodynamics in patients with acute decompensated heart failure. Circulation 2009, 119, 2781–2788. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Anderson, A.S. The sympathetic nervous system and heart failure. Cardiol. Clin. 2014, 32, 33–45. [Google Scholar] [CrossRef]
- Schwartz, P.J. Vagal stimulation for heart diseases: From animals to men.—An example of translational cardiology. Circ. J. Off. J. Jpn. Circ. Soc. 2011, 75, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.Y.; Xiao, R.P. β-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Iwase, M.; Bishop, S.P.; Uechi, M.; Vatner, D.E.; Shannon, R.P.; Kudej, R.K.; Wight, D.C.; Wagner, T.E.; Ishikawa, Y.; Homcy, C.J.; et al. Adverse effects of chronic endogenous sympathetic drive induced by cardiac GS alpha overexpression. Circ. Res. 1996, 78, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Parichatikanond, W.; Duangrat, R.; Kurose, H.; Mangmool, S. Regulation of β-Adrenergic Receptors in the Heart: A Review on Emerging Therapeutic Strategies for Heart Failure. Cells 2024, 13, 1674. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Janssen, P.M.L. Myocardial relaxation in human heart failure: Why sarcomere kinetics should be center-stage. Arch. Biochem. Biophys. 2019, 661, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Santulli, G.; Bahlouli, L.; Miotto, M.C.; Weninger, G.; Marks, A.R. Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart. Biomolecules 2023, 13, 1409. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.D.; Capogrossi, M.C.; Lakatta, E.G. Spontaneous calcium release from the sarcoplasmic reticulum in myocardial cells: Mechanisms and consequences. Cell Calcium 1988, 9, 247–256. [Google Scholar] [CrossRef]
- Shen, X.; van den Brink, J.; Bergan-Dahl, A.; Kolstad, T.R.; Norden, E.S.; Hou, Y.; Laasmaa, M.; Aguilar-Sanchez, Y.; Quick, A.P.; Espe, E.K.S.; et al. Prolonged β-adrenergic stimulation disperses ryanodine receptor clusters in cardiomyocytes and has implications for heart failure. eLife 2022, 11, e77725. [Google Scholar] [CrossRef]
- Kim, J.C.; Pérez-Hernández, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)(i) Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Van Opbergen, C.J.M.; Noorman, M.; Pfenniger, A.; Copier, J.S.; Vermij, S.H.; Li, Z.; van der Nagel, R.; Zhang, M.; de Bakker, J.M.T.; Glass, A.M.; et al. Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress. Int. J. Mol. Sci. 2019, 20, 4076. [Google Scholar] [CrossRef]
- Fujii, S.; Kobayashi, S.; Chang, Y.; Nawata, J.; Yoshitomi, R.; Tanaka, S.; Kohno, M.; Nakamura, Y.; Ishiguchi, H.; Suetomi, T.; et al. RyR2-targeting therapy prevents left ventricular remodeling and ventricular tachycardia in post-infarction heart failure. J. Mol. Cell. Cardiol. 2023, 178, 36–50. [Google Scholar] [CrossRef]
- Kansakar, U.; Varzideh, F.; Jankauskas, S.S.; Gambardella, J.; Trimarco, B.; Santulli, G. Advances in the understanding of excitation-contraction coupling: The pulsing quest for drugs against heart failure and arrhythmias. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, e91–e93. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef]
- Hesketh, G.G.; Van Eyk, J.E.; Tomaselli, G.F. Mechanisms of gap junction traffic in health and disease. J. Cardiovasc. Pharmacol. 2009, 54, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef]
- Michela, P.; Velia, V.; Aldo, P.; Ada, P. Role of connexin 43 in cardiovascular diseases. Eur. J. Pharmacol. 2015, 768, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gerdes, A.M. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: III. Intercalated disc remodeling. J. Mol. Cell. Cardiol. 1999, 31, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.M.; Goodenough, D.A.; Paul, D.L. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr. Biol. CB 1998, 8, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Kreuzberg, M.M.; Willecke, K.; Bukauskas, F.F. Connexin-mediated cardiac impulse propagation: Connexin 30.2 slows atrioventricular conduction in mouse heart. Trends Cardiovasc. Med. 2006, 16, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; van der Heyden, M.A.; van Veen, T.A.; Cox, M.G.; Hauer, R.N.; de Bakker, J.M.; van Rijen, H.V. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J. Mol. Cell. Cardiol. 2009, 47, 23–31. [Google Scholar] [CrossRef]
- Harrison, O.J.; Brasch, J.; Lasso, G.; Katsamba, P.S.; Ahlsen, G.; Honig, B.; Shapiro, L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc. Natl. Acad. Sci. USA 2016, 113, 7160–7165. [Google Scholar] [CrossRef]
- Al-Jassar, C.; Bikker, H.; Overduin, M.; Chidgey, M. Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol. 2013, 425, 4006–4022. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwaag, P.A.; Jongbloed, J.D.; van den Berg, M.P.; van der Smagt, J.J.; Jongbloed, R.; Bikker, H.; Hofstra, R.M.; van Tintelen, J.P. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum. Mutat. 2009, 30, 1278–1283. [Google Scholar] [CrossRef]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef]
- Kant, S.; Krull, P.; Eisner, S.; Leube, R.E.; Krusche, C.A. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012, 348, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef]
- Ferreira-Cornwell, M.C.; Luo, Y.; Narula, N.; Lenox, J.M.; Lieberman, M.; Radice, G.L. Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J. Cell Sci. 2002, 115, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Tepass, U.; Truong, K.; Godt, D.; Ikura, M.; Peifer, M. Cadherins in embryonic and neural morphogenesis. Nat. Rev. Mol. Cell Biol. 2000, 1, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, D.O.; Blefari, V.; Prado, F.P.; Silva, C.A.; Fazan, R., Jr.; Salgado, H.C.; Ramos, S.G.; Prado, C.M. Reduced expression of adherens and gap junction proteins can have a fundamental role in the development of heart failure following cardiac hypertrophy in rats. Exp. Mol. Pathol. 2016, 100, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kostetskii, I.; Li, J.; Xiong, Y.; Zhou, R.; Ferrari, V.A.; Patel, V.V.; Molkentin, J.D.; Radice, G.L. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 2005, 96, 346–354. [Google Scholar] [CrossRef]
- Matsushita, T.; Oyamada, M.; Fujimoto, K.; Yasuda, Y.; Masuda, S.; Wada, Y.; Oka, T.; Takamatsu, T. Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ. Res. 1999, 85, 1046–1055. [Google Scholar] [CrossRef]
- Mahmoodzadeh, S.; Eder, S.; Nordmeyer, J.; Ehler, E.; Huber, O.; Martus, P.; Weiske, J.; Pregla, R.; Hetzer, R.; Regitz-Zagrosek, V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.; Lechat, P.; Verkenne, P.; Wiemann, H. Results from post-hoc analyses of the CIBIS II trial: Effect of bisoprolol in high-risk patient groups with chronic heart failure. Eur. J. Heart Fail. 2001, 3, 469–479. [Google Scholar] [CrossRef]
- Gómez, A.; Sánchez-Roman, I.; Gomez, J.; Cruces, J.; Mate, I.; Lopez-Torres, M.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; De la Fuente, M.; et al. Lifelong treatment with atenolol decreases membrane fatty acid unsaturation and oxidative stress in heart and skeletal muscle mitochondria and improves immunity and behavior, without changing mice longevity. Aging Cell 2014, 13, 551–560. [Google Scholar] [CrossRef]
- Sanchez-Roman, I.; Gomez, J.; Naudi, A.; Ayala, V.; Portero-Otín, M.; Lopez-Torres, M.; Pamplona, R.; Barja, G. The β-blocker atenolol lowers the longevity-related degree of fatty acid unsaturation, decreases protein oxidative damage, and increases extracellular signal-regulated kinase signaling in the heart of C57BL/6 mice. Rejuvenation Res. 2010, 13, 683–693. [Google Scholar] [CrossRef] [PubMed]
- McKeever, R.G.; Patel, P.; Hamilton, R.J. Calcium Channel Blockers. In StatPearls; StatPearls Publishing, Copyright © 2025; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Ferrandi, M.; Barassi, P.; Tadini-Buoninsegni, F.; Bartolommei, G.; Molinari, I.; Tripodi, M.G.; Reina, C.; Moncelli, M.R.; Bianchi, G.; Ferrari, P. Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br. J. Pharmacol. 2013, 169, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Carubelli, V.; Zhang, Y.; Metra, M.; Lombardi, C.; Felker, G.M.; Filippatos, G.; O’Connor, C.M.; Teerlink, J.R.; Simmons, P.; Segal, R.; et al. Treatment with 24 hour istaroxime infusion in patients hospitalised for acute heart failure: A randomised, placebo-controlled trial. Eur. J. Heart Fail. 2020, 22, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Abuelazm, M.; Ali, S.; AlBarakat, M.M.; Mahmoud, A.; Tanashat, M.; Suilik, H.A.; Abdelazeem, B.; Brasic, J.R. Istaroxime for Patients with Acute Heart Failure: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Diseases 2023, 11, 183. [Google Scholar] [CrossRef] [PubMed]
- Forzano, I.; Mone, P.; Mottola, G.; Kansakar, U.; Salemme, L.; De Luca, A.; Tesorio, T.; Varzideh, F.; Santulli, G. Efficacy of the New Inotropic Agent Istaroxime in Acute Heart Failure. J. Clin. Med. 2022, 11, 7503. [Google Scholar] [CrossRef]
- Yano, M.; Kobayashi, S.; Kohno, M.; Doi, M.; Tokuhisa, T.; Okuda, S.; Suetsugu, M.; Hisaoka, T.; Obayashi, M.; Ohkusa, T.; et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 2003, 107, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sorensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Gong, B.; Li, Z.; Yat Wong, P.C. Levosimendan Treatment for Heart Failure: A Systematic Review and Meta-Analysis. J. Cardiothorac. Vasc. Anesth. 2015, 29, 1415–1425. [Google Scholar] [CrossRef]
- Masarone, D.; Kittleson, M.M.; Pollesello, P.; Marini, M.; Iacoviello, M.; Oliva, F.; Caiazzo, A.; Petraio, A.; Pacileo, G. Use of Levosimendan in Patients with Advanced Heart Failure: An Update. J. Clin. Med. 2022, 11, 6408. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.S.; Fruhwald, S.; Heunks, L.M.; Suominen, P.K.; Gordon, A.C.; Kivikko, M.; Pollesello, P. Levosimendan: Current data, clinical use and future development. Heart Lung Vessel 2013, 5, 227–245. [Google Scholar] [PubMed]
- Leybaert, L.; De Smet, M.A.; Lissoni, A.; Allewaert, R.; Roderick, H.L.; Bultynck, G.; Delmar, M.; Sipido, K.R.; Witschas, K. Connexin hemichannels as candidate targets for cardioprotective and anti-arrhythmic treatments. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Falck, A.T.; Lund, B.A.; Johansen, D.; Lund, T.; Ytrehus, K. The Ambivalence of Connexin43 Gap Peptides in Cardioprotection of the Isolated Heart against Ischemic Injury. Int. J. Mol. Sci. 2022, 23, 10197. [Google Scholar] [CrossRef] [PubMed]
- Freitas-Andrade, M.; Wang, N.; Bechberger, J.F.; De Bock, M.; Lampe, P.D.; Leybaert, L.; Naus, C.C. Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 2019, 216, 916–935. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hoagland, D.; Palatinus, J.A.; He, H.; Iyyathurai, J.; Jourdan, L.J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of α Carboxyl Terminus 1 Peptide With the Connexin 43 Carboxyl Terminus Preserves Left Ventricular Function After Ischemia-Reperfusion Injury. J. Am. Heart Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Higo, S.; Kondo, T.; Li, J.; Liu, L.; Ikeda, Y.; Kohama, Y.; Kameda, S.; Tabata, T.; Inoue, H.; et al. Phenotypic recapitulation and correction of desmoglein-2-deficient cardiomyopathy using human-induced pluripotent stem cell-derived cardiomyocytes. Hum. Mol. Genet. 2021, 30, 1384–1397. [Google Scholar] [CrossRef]
- Devemy, E.; Blaschuk, O.W. Identification of a novel dual E- and N-cadherin antagonist. Peptides 2009, 30, 1539–1547. [Google Scholar] [CrossRef] [PubMed]

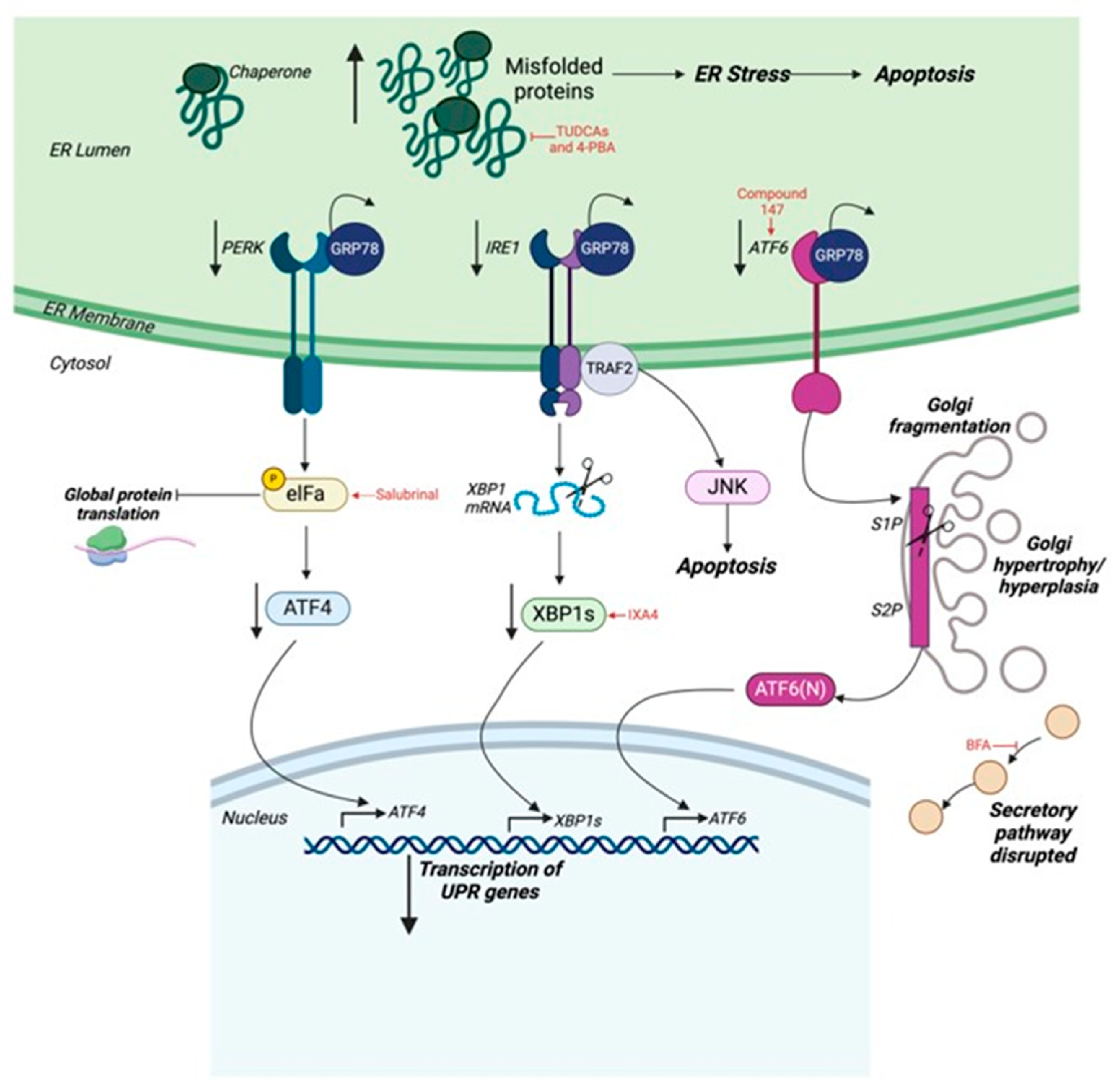
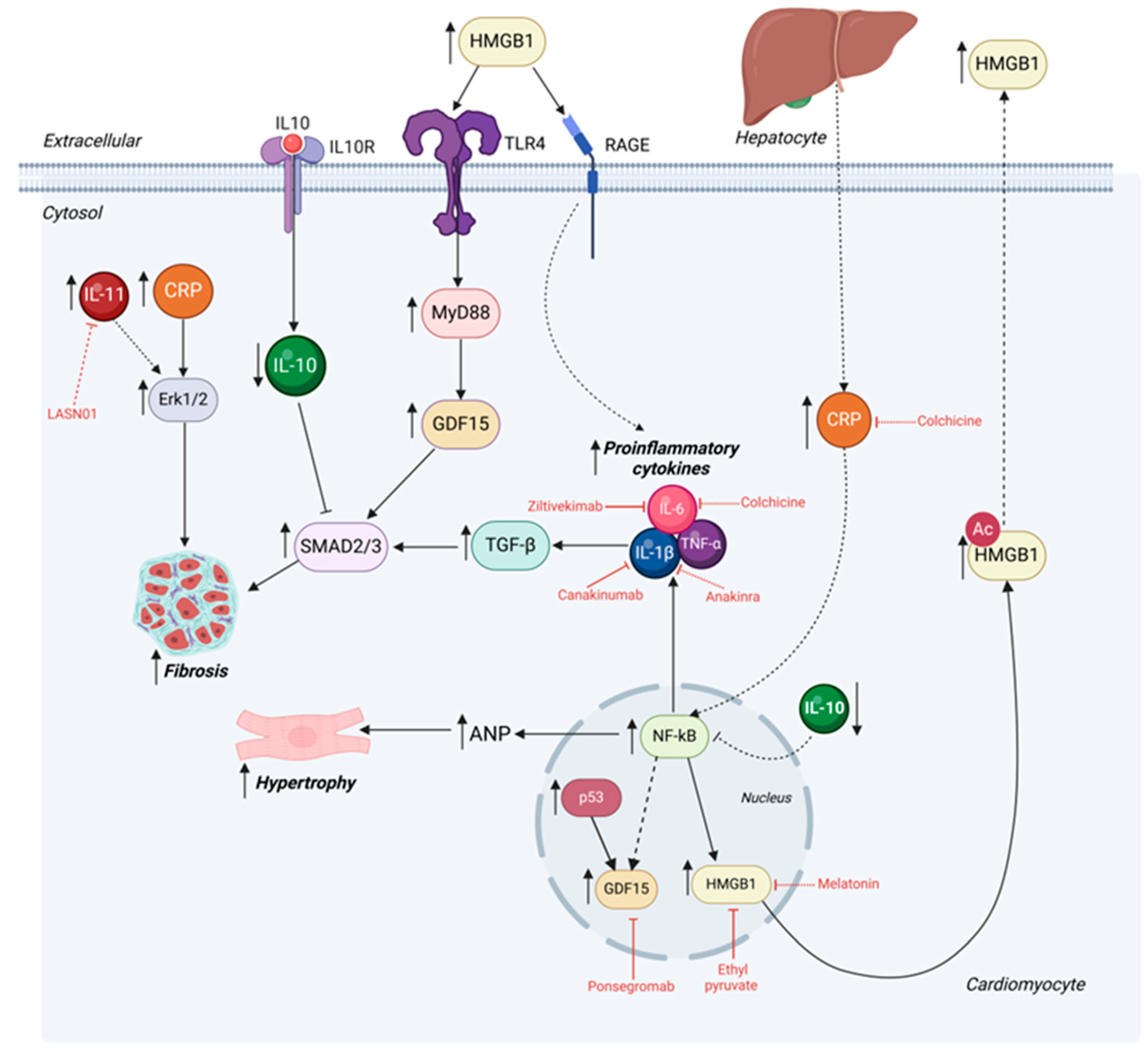
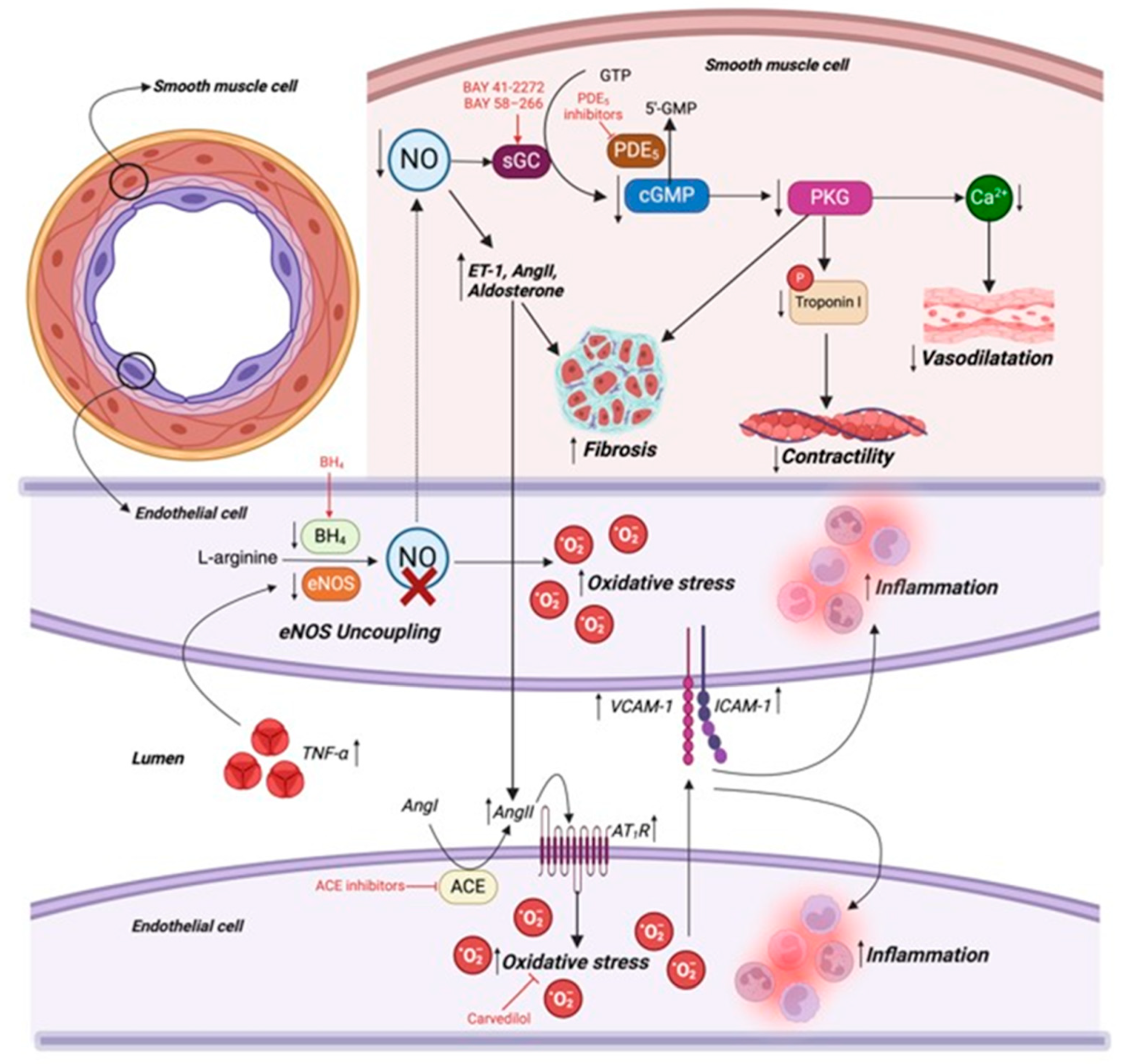
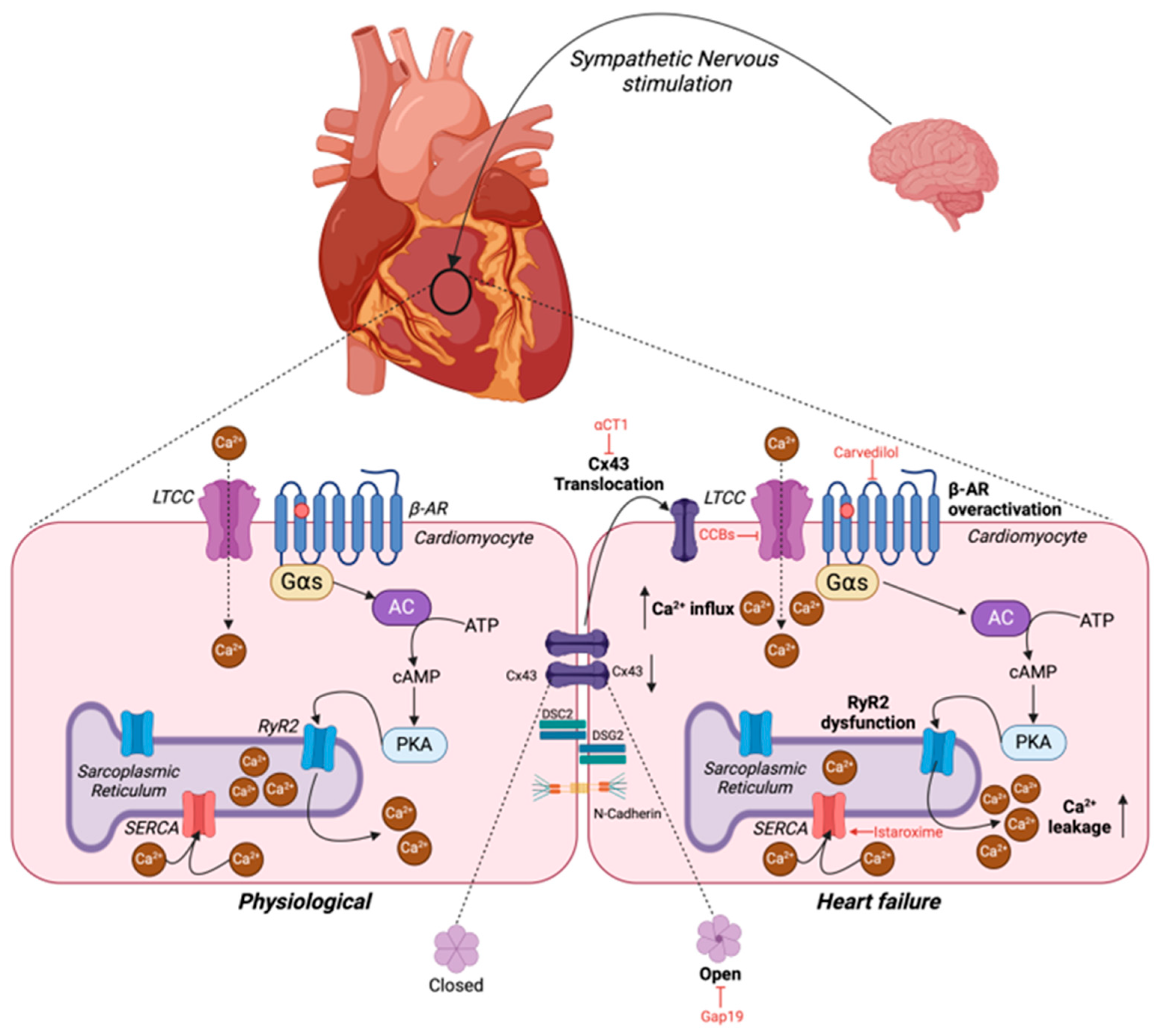
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseka, O.; Gare, S.R.; Chen, X.; Zhang, J.; Alatawi, N.H.; Ross, C.; Liu, W. Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential. Cells 2025, 14, 324. https://doi.org/10.3390/cells14050324
Fonseka O, Gare SR, Chen X, Zhang J, Alatawi NH, Ross C, Liu W. Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential. Cells. 2025; 14(5):324. https://doi.org/10.3390/cells14050324
Chicago/Turabian StyleFonseka, Oveena, Sanskruti Ravindra Gare, Xinyi Chen, Jiayan Zhang, Nasser Hawimel Alatawi, Claire Ross, and Wei Liu. 2025. "Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential" Cells 14, no. 5: 324. https://doi.org/10.3390/cells14050324
APA StyleFonseka, O., Gare, S. R., Chen, X., Zhang, J., Alatawi, N. H., Ross, C., & Liu, W. (2025). Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential. Cells, 14(5), 324. https://doi.org/10.3390/cells14050324