
PPAR-γ in Melanoma and Immune Cells: Insights into Disease Pathogenesis and Therapeutic Implications

 , , and
, , and
Abstract
:
1. Hypo- and Hyperpigmentation of the Skin, Nevi and Melanoma
2. The Expression and Regulation of PPAR-γ
2.1. The Ligand-Dependent Activation of PPAR-γ
2.1.1. Thiazolidinediones
2.1.2. Ciglitazone
2.1.3. Troglitazone
2.1.4. Pioglitazone
2.1.5. Rosiglitazone
2.1.6. Efatutazone
2.1.7. Endogenous and Natural Agonists of PPAR-γ
2.2. Selective Antagonists of PPAR-γ
2.2.1. MM902
2.2.2. T0070907
2.2.3. GW9662
2.3. PPAR-γ Ligands as a Therapeutic Strategy
2.4. Ligand-Independent Changes in the Transcriptional Activity of PPAR-γ
2.5. Coactivators of PPAR-γ
2.6. Corepressors of PPAR-γ
3. Activation of PPAR-γ in Melanoma Cells and Its Biological Effects
3.1. Activation of PPAR-γ in Melanoma Cells by Stimulation of Melanocortin 1 Receptor
3.2. The Role of PPAR-γ in Pigmentation
3.3. Effect of PPAR-γ on Proliferation
3.4. Role of PPAR-γ in Initiation of Apoptosis
3.5. Influence of PPAR-γ on Terminal Differentiation of Melanoma Cells
3.6. Role of PPAR-γ in Modulating Tumor Microenvironment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 15d-PGJ2 | 15-deoxy-Δ12,14-prostaglandin J2 |
| 5-FU | fluorouracil |
| AF | activation function |
| AgRC | agonist–receptor complex |
| AnRC | antagonist–receptor complex |
| AIF | apoptosis-inducing factor |
| ALT | alanine aminotransferase |
| ALPI | alkaline phosphatase |
| AMPK | AMP-activated protein kinase |
| AP1 | activator protein 1 |
| BAX | BCL2-associated X protein |
| Bcl | B-cell lymphoma proteins (Bcl-2 and Bcl-xL) |
| BRAF | B-rapidly accelerated fibrosarcoma |
| BRCA1 | breast cancer gene 1 |
| cAMP | cyclic adenosine monophosphate |
| CAF | cancer-associated fibroblasts |
| CASP1 | caspase 1 |
| CAT | catalase |
| CBP | CREB-binding protein |
| CCL2 | C-C motif chemokine ligand 2 |
| CCND1 | cyclin D1 |
| CD | complex of differentiation |
| CDK5 | cyclin-dependent kinase 5 |
| c-FLIP | flice-inhibitory protein |
| CHK2 | checkpoint kinase 2 |
| CGZ | ciglitazone |
| CNS | central nervous system |
| CCND1 | cyclin D1 |
| COX2 | cyclooxygenase 2 |
| CTL | cytotoxic lymphocyte |
| CTLA | cytotoxic T-lymphocyte antigen |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| DBD | DNA-binding domain |
| DC | dendritic cell |
| DHA | docosahexanoic acid |
| DOPA | 3,4-dihydroxyphenylalanine |
| DOPA enzyme | tyrosine hydroxylase |
| DR | death receptor |
| DTIC | dacarbazine |
| ECM | extracellular matrix |
| EFZ | multidrug resistance 1 |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| eIF2 | eukaryotic initiation factor 2 |
| EPA | eicosopentanoic acid |
| ERK1/2 | extracellular signal-regulated kinases ½ |
| FABP4 | fatty acid-binding protein 4 |
| FAK | focal adhesion kinase |
| FDA | Food and Drug Administration |
| FOS | this protein name originates from Finkel–Biskis–Jinkins (FBJ) murine osteosarcoma virus |
| GPX | glutathione peroxidase |
| GSK-3β | glycogen synthase kinase-3β |
| HCC | hepatocellular carcinoma |
| HDAC3 | histone deacetylase 3 |
| HIF1 α | hypoxia-inducible factor 1α |
| HMOX1 | heme oxygenase-1 |
| IFNG | interferon γ |
| IGF1 | insulin-like growth factor 1 |
| IGFBP3 | insulin growth factor-binding protein 3 |
| IGF1 | insulin growth factor 1 |
| IKK-α | inhibitor of nuclear factor κB kinase subunit α |
| JNK | c-Jun N-terminal kinase |
| JUN | this protein name originates from “ju-nana”, the Japanese word for “17”,じゅうなな, (e.g., a viral oncogene v-jun means the seventeenth viral oncogene in English) |
| KLRG1 | killer cell lectin-like receptor subfamily G member 1 |
| LAG3 | lymphocyte-activation gene 3 |
| LBD | ligand-binding domain |
| LC-PUFA | long-chain polyunsaturated fatty acid |
| LXR | liver X receptor |
| MAPK | mitogen-activated protein kinase |
| MC1R | melanocortin-1 receptor |
| MCAD1 | medium-chain acyl-CoA dehydrogenase |
| MDR1 | multidrug resistance 1 |
| MDSC | myeloid-derived suppressor cell |
| MHC1 | major histocompatibility complex class I |
| MITF | microphthalmia-associated transcription factor |
| α-MSH | α-melanocyte-stimulating hormone |
| mTOR | mechanistic target of rapamycin |
| NCoA | nuclear receptor coactivator |
| NCoR | nuclear receptor corepressor |
| NFκB | nuclear factor κB |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| NSCLC | non-small cell lung cancer |
| NYHA | New York Heart Association |
| Octa | 2,4,6-octatrienoic acid |
| O-GlcNAc | β-O-linked N-acetylglucosamine |
| p70S6K | p70 ribosomal S6 kinase |
| PDGF | platelet-derived growth factor |
| PDK4 | pyruvate dehydrogenase kinase 4 |
| PD-L1 | programmed death ligand 1 |
| PERK | protein kinase RNA-like endoplasmic reticulum kinase |
| PGC-1α | PPAR-γ coactivator-1α |
| PGC-1β | PPAR-γ coactivator-1β |
| PGZ | pioglitazone |
| PI3K | phosphoinositide 3-kinase |
| PKA | protein kinase A |
| PKB | protein kinase B |
| PKC | protein kinase C |
| PKR | protein kinase RNA-activated |
| PLA2 | phospholipase A2 |
| PLC | phospholipase C |
| PPAR-α | peroxisome proliferator-activated receptor α |
| PPAR-β/δ | peroxisome proliferator-activated receptor β/δ |
| PPAR-γ | peroxisome proliferator-activated receptor γ |
| PPRE | peroxisome proliferator response element |
| PTEN | phosphatase and tensin homolog deleted on chromosome 10 |
| PUFA | polyunsaturated fatty acid |
| RB1 | retinoblastoma 1 |
| RGZ | rosiglitazone |
| ROS | reactive oxygen species |
| RXR- α | retinoid X receptor α |
| SHC | Src homology and collagen |
| SMAD2 | mothers against decapentaplegic homolog 2 |
| SMRT | silencing mediator of retinoid and thyroid hormone receptors |
| SNP | single-nucleotide polymorphism |
| SOD | superoxide dismutase |
| SRC | sarcoma family member |
| STAT3 | signal transducer and activator of transcription 3 |
| T2D | type 2 diabetes |
| TAM | tumor-associated macrophage |
| TetO | tetracycline operator |
| TGF-β | transforming growth factor-β |
| TIM3 | T-cell immunoglobulin and mucin domain-3 |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor-α |
| TP53 | tumor protein p53 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Treg | regulatory T cell |
| TRPM1 | transient receptor potential melastatin 1 |
| TRP1 | tyrosinase-related protein-1 |
| TRP2 | tyrosinase-related protein-2 |
| TZD | thiazolidinedione |
| TYR | tyrosinase |
| VCAM1 | vascular cell adhesion molecule 1 |
| VDR | vitamin D receptor |
| VEGF | vascular endothelial growth factor |
| VILL1 | villin 1 |
| WNT | wingless-related integration site |
References
- Praetorius, C.; Sturm, R.A.; Steingrimsson, E. Sun-induced freckling: Ephelides and solar lentigines. Pigment. Cell Melanoma Res. 2014, 27, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kanlayavattanakul, M.; Lourith, N. Skin hyperpigmentation treatment using herbs: A review of clinical evidences. J. Cosmet. Laser Ther. 2018, 20, 123–131. [Google Scholar] [CrossRef]
- Khamaysi, Z.; Jiryis, B. Prevention and Treatment of Skin Pigmentation Disorders. J. Clin. Med. 2024, 13, 4312. [Google Scholar] [CrossRef]
- Mosca, S.; Morrone, A. Human Skin Pigmentation: From a Biological Feature to a Social Determinant. Healthcare 2023, 11, 2091. [Google Scholar] [CrossRef]
- Balu, M.; Kelly, K.M.; Zachary, C.B.; Harris, R.M.; Krasieva, T.B.; König, K.; Durkin, A.J.; Tromberg, B.J. Distinguishing between benign and malignant melanocytic nevi by in vivo multiphoton microscopy. Cancer Res. 2014, 74, 2688–2697. [Google Scholar] [CrossRef]
- Kiszner, G.; Wichmann, B.; Nemeth, I.B.; Varga, E.; Meggyeshazi, N.; Teleki, I.; Balla, P.; Maros, M.E.; Penksza, K.; Krenacs, T. Cell cycle analysis can differentiate thin melanomas from dysplastic nevi and reveals accelerated replication in thick melanomas. Virchows Arch. 2014, 464, 603–612. [Google Scholar] [CrossRef]
- Reed, J.A.; Finnerty, B.; Albino, A.P. Divergent cellular differentiation pathways during the invasive stage of cutaneous malignant melanoma progression. Am. J. Pathol. 1999, 155, 549–555. [Google Scholar] [CrossRef]
- Tabassum, A.; Iqbal, M.S. Benign Nevi Mimicking Melanoma: A Diagnostic Dilemma. Cureus 2024, 16, e74821. [Google Scholar] [CrossRef]
- Zhang, Y.; Ostrowski, S.M.; Fisher, D.E. Nevi and Melanoma. Hematol. Oncol. Clin. N. Am. 2024, 38, 939–952. [Google Scholar] [CrossRef]
- Baigrie, D.; Tanner, L.S. Dysplastic Nevi. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Goldstein, A.M.; Tucker, M.A. Dysplastic nevi and melanoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 528–532. [Google Scholar] [CrossRef]
- Conforti, C.; Zalaudek, I. Epidemiology and Risk Factors of Melanoma: A Review. Dermatol. Pract. Concept. 2021, 11, e2021161S. [Google Scholar] [CrossRef]
- Liu, W.; Dowling, J.P.; Murray, W.K.; McArthur, G.A.; Thompson, J.F.; Wolfe, R.; Kelly, J.W. Rate of growth in melanomas: Characteristics and associations of rapidly growing melanomas. Arch. Dermatol. 2006, 142, 1551–1558. [Google Scholar] [CrossRef]
- Urso, C. Are growth phases exclusive to cutaneous melanoma? J. Clin. Pathol. 2004, 57, 560. [Google Scholar] [CrossRef]
- Betti, R.; Agape, E.; Vergani, R.; Moneghini, L.; Cerri, A. An observational study regarding the rate of growth in vertical and radial growth phase superficial spreading melanomas. Oncol. Lett. 2016, 12, 2099–2102. [Google Scholar] [CrossRef]
- Guerry, D.t.; Synnestvedt, M.; Elder, D.E.; Schultz, D. Lessons from tumor progression: The invasive radial growth phase of melanoma is common, incapable of metastasis, and indolent. J. Investig. Dermatol. 1993, 100, 342s–345s. [Google Scholar] [CrossRef]
- Crowson, A.N.; Magro, C.M.; Mihm, M.C. Prognosticators of melanoma, the melanoma report, and the sentinel lymph node. Mod. Pathol. 2006, 19 (Suppl. S2), S71–S87. [Google Scholar] [CrossRef]
- Oliveira Filho, R.S.; Ferreira, L.M.; Biasi, L.J.; Enokihara, M.M.; Paiva, G.R.; Wagner, J. Vertical growth phase and positive sentinel node in thin melanoma. Braz. J. Med. Biol. Res. 2003, 36, 347–350. [Google Scholar] [CrossRef]
- Xuan, J.; Gao, Z.; Wei, C.; Gu, J. Insights for the immunotherapy in malignant melanoma: A new revolution. Clin. Cancer Bull. 2024, 3, 21. [Google Scholar] [CrossRef]
- Scolyer, R.A.; Judge, M.J.; Evans, A.; Frishberg, D.P.; Prieto, V.G.; Thompson, J.F.; Trotter, M.J.; Walsh, M.Y.; Walsh, N.M.; Ellis, D.W. Data set for pathology reporting of cutaneous invasive melanoma: Recommendations from the international collaboration on cancer reporting (ICCR). Am. J. Surg. Pathol. 2013, 37, 1797–1814. [Google Scholar] [CrossRef]
- Atkins, M.B.; Curiel-Lewandrowski, C.; Fisher, D.E.; Swetter, S.M.; Tsao, H.; Aguirre-Ghiso, J.A.; Soengas, M.S.; Weeraratna, A.T.; Flaherty, K.T.; Herlyn, M.; et al. The State of Melanoma: Emergent Challenges and Opportunities. Clin. Cancer Res. 2021, 27, 2678–2697. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Burri, L.; Thoresen, G.H.; Berge, R.K. The Role of PPARα Activation in Liver and Muscle. PPAR Res. 2010, 2010, 542359. [Google Scholar] [CrossRef]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Peña, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vázquez-Carrera, M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef]
- Sobolev, V.V.; Tchepourina, E.; Korsunskaya, I.M.; Geppe, N.A.; Chebysheva, S.N.; Soboleva, A.G.; Mezentsev, A. The Role of Transcription Factor PPAR-γ in the Pathogenesis of Psoriasis, Skin Cells, and Immune Cells. Int. J. Mol. Sci. 2022, 23, 9708. [Google Scholar] [CrossRef]
- Drew, P.D.; Xu, J.; Racke, M.K. PPAR-gamma: Therapeutic Potential for Multiple Sclerosis. PPAR Res. 2008, 2008, 627463. [Google Scholar] [CrossRef]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [PubMed]
- Scirpo, R.; Fiorotto, R.; Villani, A.; Amenduni, M.; Spirli, C.; Strazzabosco, M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-γ limits NF-κB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology 2015, 62, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Moreau, F.; Chadee, K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papavassiliou, A.G. NF-kappaB/PPAR gamma and/or AP-1/PPAR gamma ‘on/off’ switches and induction of CBP in colon adenocarcinomas: Correlation with COX-2 expression. Int. J. Colorectal Dis. 2007, 22, 57–68. [Google Scholar] [CrossRef]
- Sobolev, V.V.; Khashukoeva, A.Z.; Evina, O.E.; Geppe, N.A.; Chebysheva, S.N.; Korsunskaya, I.M.; Tchepourina, E.; Mezentsev, A. Role of the Transcription Factor FOSL1 in Organ Development and Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 1521. [Google Scholar] [CrossRef]
- Kotla, S.; Singh, N.K.; Rao, G.N. Corrigendum to “ROS via BTK-p300-STAT1-PPARγ signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation” [Redox Biol. 11 (2017) 350-364/513]. Redox Biol. 2022, 50, 102254. [Google Scholar] [CrossRef]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, B.M.; Ahn, Y.H.; Choi, J.H.; Choi, Y.H.; Kang, J.L. STAT6 Signaling Mediates PPARγ Activation and Resolution of Acute Sterile Inflammation in Mice. Cells 2021, 10, 501. [Google Scholar] [CrossRef]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef]
- Adams, M.; Reginato, M.J.; Shao, D.; Lazar, M.A.; Chatterjee, V.K. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem. 1997, 272, 5128–5132. [Google Scholar] [CrossRef]
- Konger, R.L.; Martel, K.C.; Jernigan, D.; Zhang, Q.; Travers, J.B. The peroxisome proliferator-activated receptor gamma system regulates ultraviolet B-induced prostaglandin e(2) production in human epidermal keratinocytes. PPAR Res. 2010, 2010, 467053. [Google Scholar] [CrossRef] [PubMed]
- Flori, E.; Mastrofrancesco, A.; Kovacs, D.; Ramot, Y.; Briganti, S.; Bellei, B.; Paus, R.; Picardo, M. 2,4,6-Octatrienoic acid is a novel promoter of melanogenesis and antioxidant defence in normal human melanocytes via PPAR-γ activation. Pigment. Cell Melanoma Res. 2011, 24, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Mastrofrancesco, A.; Ottaviani, M.; Cardinali, G.; Flori, E.; Briganti, S.; Ludovici, M.; Zouboulis, C.C.; Lora, V.; Camera, E.; Picardo, M. Pharmacological PPARγ modulation regulates sebogenesis and inflammation in SZ95 human sebocytes. Biochem. Pharmacol. 2017, 138, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Milam, J.E.; Keshamouni, V.G.; Phan, S.H.; Hu, B.; Gangireddy, S.R.; Hogaboam, C.M.; Standiford, T.J.; Thannickal, V.J.; Reddy, R.C. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L891–L901. [Google Scholar] [CrossRef]
- Mössner, R.; Schulz, U.; Krüger, U.; Middel, P.; Schinner, S.; Füzesi, L.; Neumann, C.; Reich, K. Agonists of peroxisome proliferator-activated receptor gamma inhibit cell growth in malignant melanoma. J. Investig. Dermatol. 2002, 119, 576–582. [Google Scholar] [CrossRef]
- Kang, H.Y.; Chung, E.; Lee, M.; Cho, Y.; Kang, W.H. Expression and function of peroxisome proliferator-activated receptors in human melanocytes. Br. J. Dermatol. 2004, 150, 462–468. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications--a review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Liu, C.; Feng, T.; Zhu, N.; Liu, P.; Han, X.; Chen, M.; Wang, X.; Li, N.; Li, Y.; Xu, Y.; et al. Corrigendum: Identification of a novel selective agonist of PPARγ with no promotion of adipogenesis and less inhibition of osteoblastogenesis. Sci. Rep. 2015, 5, 12185. [Google Scholar] [CrossRef]
- Yi, W.; Shi, J.; Zhao, G.; Zhou, X.E.; Suino-Powell, K.; Melcher, K.; Xu, H.E. Identification of a novel selective PPARγ ligand with a unique binding mode and improved therapeutic profile in vitro. Sci. Rep. 2017, 7, 41487. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, M.; Yuan, B.; Zhang, K.; Zhong, M.; Yi, W.; Xu, X.; Duan, X. VSP-17, a New PPARγ Agonist, Suppresses the Metastasis of Triple-Negative Breast Cancer via Upregulating the Expression of E-Cadherin. Molecules 2018, 23, 121. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T. Mechanisms by which thiazolidinediones induce anti-cancer effects in cancers in digestive organs. J. Gastroenterol. 2010, 45, 1097–1102. [Google Scholar] [CrossRef]
- Wei, S.; Yang, J.; Lee, S.L.; Kulp, S.K.; Chen, C.S. PPARgamma-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009, 276, 119–124. [Google Scholar] [CrossRef]
- Read, W.L.; Baggstrom, M.Q.; Fracasso, P.M.; Govindan, R. A phase I study of bexarotene and rosiglitazone in patients with refractory cancers. Chemotherapy 2008, 54, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Bross, K.; Vogt, T.; Bataille, F.; Wild, P.; Berand, A.; Krause, S.W.; Andreesen, R. Pioglitazone and rofecoxib combined with angiostatically scheduled trofosfamide in the treatment of far-advanced melanoma and soft tissue sarcoma. Cancer 2004, 101, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.B.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Rhodes, L.E.; Al-Aasswad, N.M.; Massey, K.A.; Nicolaou, A. Impact of EPA ingestion on COX- and LOX-mediated eicosanoid synthesis in skin with and without a pro-inflammatory UVR challenge--report of a randomised controlled study in humans. Mol. Nutr. Food Res. 2014, 58, 580–590. [Google Scholar] [CrossRef]
- Brown, V.A.; Patel, K.R.; Viskaduraki, M.; Crowell, J.A.; Perloff, M.; Booth, T.D.; Vasilinin, G.; Sen, A.; Schinas, A.M.; Piccirilli, G.; et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: Safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res. 2010, 70, 9003–9011. [Google Scholar] [CrossRef]
- Greil, R.; Greil-Ressler, S.; Weiss, L.; Schönlieb, C.; Magnes, T.; Radl, B.; Bolger, G.T.; Vcelar, B.; Sordillo, P.P. A phase 1 dose-escalation study on the safety, tolerability and activity of liposomal curcumin (Lipocurc(™)) in patients with locally advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 695–706. [Google Scholar] [CrossRef]
- Grillier-Vuissoz, I.; Mazerbourg, S.; Boisbrun, M.; Kuntz, S.; Chapleur, Y.; Flament, S. PPARγ-independent Activity of Thiazolidinediones: A Promising Mechanism of Action for New Anticancer Drugs? J. Carcinog. Mutagen. 2012, 12, S8:002. [Google Scholar] [CrossRef]
- Palakurthi, S.S.; Aktas, H.; Grubissich, L.M.; Mortensen, R.M.; Halperin, J.A. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001, 61, 6213–6218. [Google Scholar] [PubMed]
- Chi, T.; Wang, M.; Wang, X.; Yang, K.; Xie, F.; Liao, Z.; Wei, P. PPAR-γ Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 737776. [Google Scholar] [CrossRef]
- Botton, T.; Puissant, A.; Bahadoran, P.; Annicotte, J.S.; Fajas, L.; Ortonne, J.P.; Gozzerino, G.; Zamoum, T.; Tartare-Deckert, S.; Bertolotto, C.; et al. In vitro and in vivo anti-melanoma effects of ciglitazone. J. Investig. Dermatol. 2009, 129, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Wahl, R. Chemotherapy and chemoprevention by thiazolidinediones. Biomed. Res. Int. 2015, 2015, 845340. [Google Scholar] [CrossRef]
- Mrówka, P.; Glodkowska, E.; Nowis, D.; Legat, M.; Issat, T.; Makowski, M.; Szokalska, A.; Janowska, S.; Stoklosa, T.; Jakóbisiak, M.; et al. Ciglitazone, an agonist of peroxisome proliferator-activated receptor gamma, exerts potentiated cytostatic/cytotoxic effects against tumor cells when combined with lovastatin. Int. J. Oncol. 2008, 32, 249–255. [Google Scholar]
- Keen, H. Insulin resistance and the prevention of diabetes mellitus. N. Engl. J. Med. 1994, 331, 1226–1227. [Google Scholar] [CrossRef]
- Henry, R.R. Effects of troglitazone on insulin sensitivity. Diabet. Med. 1996, 13, S148–S150. [Google Scholar]
- Kores, K.; Konc, J.; Bren, U. Mechanistic Insights into Side Effects of Troglitazone and Rosiglitazone Using a Novel Inverse Molecular Docking Protocol. Pharmaceutics 2021, 13, 315. [Google Scholar] [CrossRef]
- Roman, J. Moving away from PPARs—EGFR signaling and the anti-cancer effects of thiazolinedinediones. Cell Res. 2009, 19, 669–671. [Google Scholar] [CrossRef]
- Li, X.; Yang, X.; Xu, Y.; Jiang, X.; Li, X.; Nan, F.; Tang, H. Troglitazone inhibits cell proliferation by attenuation of epidermal growth factor receptor signaling independent of peroxisome proliferator-activated receptor gamma. Cell Res. 2009, 19, 720–732. [Google Scholar] [CrossRef]
- Tsujiya, Y.; Hasegawa, A.; Yamamori, M.; Okamura, N. Troglitazone-Induced Autophagic Cytotoxicity in Lung Adenocarcinoma Cell Lines. Biol. Pharm. Bull. 2022, 45, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Shi, W.; Shao, B.; Shi, J.; Shen, A.; Ma, Y.; Chen, J.; Lan, Q. Peroxisome proliferator-activated receptor γ agonist pioglitazone inhibits β-catenin-mediated glioma cell growth and invasion. Mol. Cell Biochem. 2011, 349, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.X.; Lin, S.Y.; Lian, S.X.; Qiu, Y.R.; Li, Z.H.; Chen, Z.H.; Lu, W.Q.; Zhang, Y.; Deng, L.; Jiang, Y.; et al. The inhibition of the breast cancer by PPARγ agonist pioglitazone through JAK2/STAT3 pathway. Neoplasma 2020, 67, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Keith, R.L.; Blatchford, P.J.; Merrick, D.T.; Bunn, P.A., Jr.; Bagwell, B.; Dwyer-Nield, L.D.; Jackson, M.K.; Geraci, M.W.; Miller, Y.E. A Randomized Phase II Trial of Pioglitazone for Lung Cancer Chemoprevention in High-Risk Current and Former Smokers. Cancer Prev. Res. 2019, 12, 721–730. [Google Scholar] [CrossRef]
- Moghareabed, R.; Hemati, S.; Akhavan, A.; Emami, H.; Farghadani, M.; Roayaei, M.; Tavajoh, S.; Feizi, A. Randomized phase II clinical trial of pioglitazone plus chemotherapy versus chemotherapy alone in patients with metastatic breast cancer. J. Glob. Oncol. 2019, 5, 83. [Google Scholar] [CrossRef]
- Yeh, J.S.; Sarpatwari, A.; Kesselheim, A.S. Ethical and Practical Considerations in Removing Black Box Warnings from Drug Labels. Drug Saf. 2016, 39, 709–714. [Google Scholar] [CrossRef]
- Bo, Q.F.; Sun, X.M.; Liu, J.; Sui, X.M.; Li, G.X. Antitumor action of the peroxisome proliferator-activated receptor-γ agonist rosiglitazone in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 1979–1984. [Google Scholar] [CrossRef]
- Wu, K.; Yang, Y.; Liu, D.; Qi, Y.; Zhang, C.; Zhao, J.; Zhao, S. Activation of PPARγ suppresses proliferation and induces apoptosis of esophageal cancer cells by inhibiting TLR4-dependent MAPK pathway. Oncotarget 2016, 7, 44572–44582. [Google Scholar] [CrossRef]
- Cao, L.Q.; Wang, X.L.; Wang, Q.; Xue, P.; Jiao, X.Y.; Peng, H.P.; Lu, H.W.; Zheng, Q.; Chen, X.L.; Huang, X.H.; et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARgamma signaling pathway. Acta Pharmacol. Sin. 2009, 30, 1316–1322. [Google Scholar] [CrossRef]
- Han, S.; Roman, J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol. Cancer Ther. 2006, 5, 430–437. [Google Scholar] [CrossRef]
- Konger, R.L.; Derr-Yellin, E.; Ermatov, N.; Ren, L.; Sahu, R.P. The PPARγ Agonist Rosiglitazone Suppresses Syngeneic Mouse SCC (Squamous Cell Carcinoma) Tumor Growth through an Immune-Mediated Mechanism. Molecules 2019, 24, 2192. [Google Scholar] [CrossRef] [PubMed]
- Luconi, M.; Mangoni, M.; Gelmini, S.; Poli, G.; Nesi, G.; Francalanci, M.; Pratesi, N.; Cantini, G.; Lombardi, A.; Pepi, M.; et al. Rosiglitazone impairs proliferation of human adrenocortical cancer: Preclinical study in a xenograft mouse model. Endocr. Relat. Cancer 2010, 17, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Rui, M.; Huang, Z.; Liu, Y.; Wang, Z.; Liu, R.; Fu, J.; Huang, H. Rosiglitazone suppresses angiogenesis in multiple myeloma via downregulation of hypoxia-inducible factor-1α and insulin-like growth factor-1 mRNA expression. Mol. Med. Rep. 2014, 10, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Teresi, R.E.; Waite, K.A. PPARgamma, PTEN, and the Fight against Cancer. PPAR Res. 2008, 2008, 932632. [Google Scholar] [CrossRef]
- Pignatelli, M.; Cocca, C.; Santos, A.; Perez-Castillo, A. Enhancement of BRCA1 gene expression by the peroxisome proliferator-activated receptor gamma in the MCF-7 breast cancer cell line. Oncogene 2003, 22, 5446–5450. [Google Scholar] [CrossRef]
- Apostoli, A.J.; Roche, J.M.; Schneider, M.M.; SenGupta, S.K.; Di Lena, M.A.; Rubino, R.E.; Peterson, N.T.; Nicol, C.J. Opposing roles for mammary epithelial-specific PPARγ signaling and activation during breast tumour progression. Mol. Cancer 2015, 14, 85. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, W.; Li, X.; Feng, Y.; Qian, K.; Wang, G.; Gao, Y.; Xu, X.; Zhang, S.; Yue, L.; et al. The PPARγ Agonist Rosiglitazone Enhances the Radiosensitivity of Human Pancreatic Cancer Cells. Drug Des. Dev. Ther. 2020, 14, 3099–3110. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, Y.; Li, H.; Li, J.; Fu, J.; Liu, Y.; Chen, X.G. Growth inhibition and differentiation induced by peroxisome proliferator activated receptor gamma ligand rosiglitazone in human melanoma cell line A375. Med. Oncol. 2006, 23, 393–402. [Google Scholar] [CrossRef]
- Smith, A.G.; Beaumont, K.A.; Smit, D.J.; Thurber, A.E.; Cook, A.L.; Boyle, G.M.; Parsons, P.G.; Sturm, R.A.; Muscat, G.E. PPARgamma agonists attenuate proliferation and modulate Wnt/beta-catenin signalling in melanoma cells. Int. J. Biochem. Cell Biol. 2009, 41, 844–852. [Google Scholar] [CrossRef]
- Pich, C.; Meylan, P.; Mastelic-Gavillet, B.; Nguyen, T.N.; Loyon, R.; Trang, B.K.; Moser, H.; Moret, C.; Goepfert, C.; Hafner, J.; et al. Induction of Paracrine Signaling in Metastatic Melanoma Cells by PPARγ Agonist Rosiglitazone Activates Stromal Cells and Enhances Tumor Growth. Cancer Res. 2018, 78, 6447–6461. [Google Scholar] [CrossRef]
- Esteva, F.J.; Moulder, S.L.; Gonzalez-Angulo, A.M.; Ensor, J.; Murray, J.L.; Green, M.C.; Koenig, K.B.; Lee, M.H.; Hortobagyi, G.N.; Yeung, S.C. Phase I trial of exemestane in combination with metformin and rosiglitazone in nondiabetic obese postmenopausal women with hormone receptor-positive metastatic breast cancer. Cancer Chemother. Pharmacol. 2013, 71, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.D.; Williams, N.; Wen, P.; Young, D.C.; Lester, J.; Johnson, M.V.; Farrar, W.B.; Walker, M.J.; Povoski, S.P.; Suster, S.; et al. Pilot study of rosiglitazone therapy in women with breast cancer: Effects of short-term therapy on tumor tissue and serum markers. Clin. Cancer Res. 2007, 13, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Peng, M.; Reiff, E.; Treseler, P.; Woeber, K.A.; Clark, O.H.; Greenspan, F.S.; Lindsay, S.; Duh, Q.Y.; Morita, E. A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery 2006, 140, 960–966, discussion 966–967. [Google Scholar] [CrossRef]
- Debrock, G.; Vanhentenrijk, V.; Sciot, R.; Debiec-Rychter, M.; Oyen, R.; Van Oosterom, A. A phase II trial with rosiglitazone in liposarcoma patients. Br. J. Cancer 2003, 89, 1409–1412. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef]
- Serizawa, M.; Murakami, H.; Watanabe, M.; Takahashi, T.; Yamamoto, N.; Koh, Y. Peroxisome proliferator-activated receptor γ agonist efatutazone impairs transforming growth factor β2-induced motility of epidermal growth factor receptor tyrosine kinase inhibitor-resistant lung cancer cells. Cancer Sci. 2014, 105, 683–689. [Google Scholar] [CrossRef]
- Ni, J.; Zhou, L.L.; Ding, L.; Zhang, X.Q.; Zhao, X.; Li, H.; Cao, H.; Liu, S.; Wang, Z.; Ma, R.; et al. Efatutazone and T0901317 exert synergistically therapeutic effects in acquired gefitinib-resistant lung adenocarcinoma cells. Cancer Med. 2018, 7, 1955–1966. [Google Scholar] [CrossRef]
- Ory, V.; Kietzman, W.B.; Boeckelman, J.; Kallakury, B.V.; Wellstein, A.; Furth, P.A.; Riegel, A.T. The PPARγ agonist efatutazone delays invasive progression and induces differentiation of ductal carcinoma in situ. Breast Cancer Res. Treat. 2018, 169, 47–57. [Google Scholar] [CrossRef]
- Murakami, H.; Ono, A.; Takahashi, T.; Onozawa, Y.; Tsushima, T.; Yamazaki, K.; Jikoh, T.; Boku, N.; Yamamoto, N. Phase I study of Efatutazone, an oral PPARγ agonist, in patients with metastatic solid tumors. Anticancer. Res. 2014, 34, 5133–5141. [Google Scholar]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an oral PPAR-γ agonist, in combination with paclitaxel in anaplastic thyroid cancer: Results of a multicenter phase 1 trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400. [Google Scholar] [CrossRef]
- Williams, R. Discontinued in 2013: Oncology drugs. Expert. Opin. Investig. Drugs 2015, 24, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Ballav, S.; Biswas, B.; Sahu, V.K.; Ranjan, A.; Basu, S. PPAR-γ Partial Agonists in Disease-Fate Decision with Special Reference to Cancer. Cells 2022, 11, 3215. [Google Scholar] [CrossRef]
- Lathion, C.; Michalik, L.; Wahli, W. Physiological ligands of PPARs in inflammation and lipid homeostasis. Future Lipidol. 2006, 1, 191–201. [Google Scholar] [CrossRef]
- Straus, D.S.; Pascual, G.; Li, M.; Welch, J.S.; Ricote, M.; Hsiang, C.H.; Sengchanthalangsy, L.L.; Ghosh, G.; Glass, C.K. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4844–4849. [Google Scholar] [CrossRef]
- Xu, M.; Yang, H.; Zhang, Q.; Lu, P.; Feng, Y.; Geng, X.; Zhang, L.; Jia, X. Alpha-Tocopherol prevents esophageal squamous cell carcinoma by modulating PPARγ-Akt signaling pathway at the early stage of carcinogenesis. Oncotarget 2017, 8, 95914–95930. [Google Scholar] [CrossRef]
- Ge, L.N.; Yan, L.; Li, C.; Cheng, K. Bavachinin exhibits antitumor activity against non-small cell lung cancer by targeting PPARγ. Mol. Med. Rep. 2019, 20, 2805–2811. [Google Scholar] [CrossRef]
- Mineda, A.; Nishimura, M.; Kagawa, T.; Takiguchi, E.; Kawakita, T.; Abe, A.; Irahara, M. Resveratrol suppresses proliferation and induces apoptosis of uterine sarcoma cells by inhibiting the Wnt signaling pathway. Exp. Ther. Med. 2019, 17, 2242–2246. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Q.; Yang, C.; Yang, H.; Rao, J.; Zhang, X. Curcumin exerts anti-tumor effects on diffuse large B cell lymphoma via regulating PPARγ expression. Biochem. Biophys. Res. Commun. 2020, 524, 70–76. [Google Scholar] [CrossRef]
- Flori, E.; Mastrofrancesco, A.; Kovacs, D.; Bellei, B.; Briganti, S.; Maresca, V.; Cardinali, G.; Picardo, M. The activation of PPARγ by 2,4,6-Octatrienoic acid protects human keratinocytes from UVR-induced damages. Sci. Rep. 2017, 7, 9241. [Google Scholar] [CrossRef]
- Wang, Z.; Coleman, D.J.; Bajaj, G.; Liang, X.; Ganguli-Indra, G.; Indra, A.K. RXRα ablation in epidermal keratinocytes enhances UVR-induced DNA damage, apoptosis, and proliferation of keratinocytes and melanocytes. J. Investig. Dermatol. 2011, 131, 177–187. [Google Scholar] [CrossRef]
- Ruggiero, F.; Pfäffle, M.; von der Mark, K.; Garrone, R. Retention of carboxypropeptides in type-II collagen fibrils in chick embryo chondrocyte cultures. Cell Tissue Res. 1988, 252, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Liu, X.K.; Tian, X.T.; Liu, F.; Yao, X.J.; Dong, J.F. PPARG: A Promising Therapeutic Target in Breast Cancer and Regulation by Natural Drugs. PPAR Res. 2023, 2023, 4481354. [Google Scholar] [CrossRef] [PubMed]
- Augimeri, G.; Giordano, C.; Gelsomino, L.; Plastina, P.; Barone, I.; Catalano, S.; Andò, S.; Bonofiglio, D. The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies. Cancers 2020, 12, 2623. [Google Scholar] [CrossRef]
- Hatton, J.L.; Yee, L.D. Clinical Use of PPARgamma Ligands in Cancer. PPAR Res. 2008, 2008, 159415. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.V.; Gonçalves-de-Albuquerque, C.F.; Silva, A.R. PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. Int. J. Mol. Sci. 2021, 22, 805. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Q.; Zielinski, R.M.; Howells, R.D.; Welsh, W.J. Identification of an irreversible PPARγ antagonist with potent anticancer activity. Pharmacol. Res. Perspect. 2020, 8, e00693. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Demchenko, Y.N.; Kuehl, W.M. A critical role for the NFkB pathway in multiple myeloma. Oncotarget 2010, 1, 59–68. [Google Scholar] [CrossRef]
- Brust, R.; Shang, J.; Fuhrmann, J.; Mosure, S.A.; Bass, J.; Cano, A.; Heidari, Z.; Chrisman, I.M.; Nemetchek, M.D.; Blayo, A.L.; et al. A structural mechanism for directing corepressor-selective inverse agonism of PPARγ. Nat. Commun. 2018, 9, 4687. [Google Scholar] [CrossRef]
- Lee, G.; Elwood, F.; McNally, J.; Weiszmann, J.; Lindstrom, M.; Amaral, K.; Nakamura, M.; Miao, S.; Cao, P.; Learned, R.M.; et al. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J. Biol. Chem. 2002, 277, 19649–19657. [Google Scholar] [CrossRef]
- An, Z.; Muthusami, S.; Yu, J.R.; Park, W.Y. T0070907, a PPAR γ inhibitor, induced G2/M arrest enhances the effect of radiation in human cervical cancer cells through mitotic catastrophe. Reprod. Sci. 2014, 21, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Wallis, N.K.; Southard, R.C.; Kilgore, M.W. The PPARgamma antagonist T0070907 suppresses breast cancer cell proliferation and motility via both PPARgamma-dependent and -independent mechanisms. Anticancer. Res. 2011, 31, 813–823. [Google Scholar] [PubMed]
- Brust, R.; Lin, H.; Fuhrmann, J.; Asteian, A.; Kamenecka, T.M.; Kojetin, D.J. Modification of the Orthosteric PPARγ Covalent Antagonist Scaffold Yields an Improved Dual-Site Allosteric Inhibitor. ACS Chem. Biol. 2017, 12, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Burger, K.; Brandt, A.; Staltner, R.; Jung, F.; Rajcic, D.; Lorenzo Pisarello, M.J.; Bergheim, I. GW9662, a peroxisome proliferator-activated receptor gamma antagonist, attenuates the development of non-alcoholic fatty liver disease. Metabolism 2022, 133, 155233. [Google Scholar] [CrossRef]
- Wu, B.; Sun, X.; Yuan, B.; Ge, F.; Gupta, H.B.; Chiang, H.C.; Li, J.; Hu, Y.; Curiel, T.J.; Li, R. PPARγ inhibition boosts efficacy of PD-L1 Checkpoint Blockade Immunotherapy against Murine Melanoma in a sexually dimorphic manner. Int. J. Biol. Sci. 2020, 16, 1526–1535. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Schumacher, U.; Reinert, S.; Hoffmann, J. The Critical Role of PPARgamma in Human Malignant Melanoma. PPAR Res. 2008, 2008, 503797. [Google Scholar] [CrossRef]
- Sohda, T.; Momose, Y.; Meguro, K.; Kawamatsu, Y.; Sugiyama, Y.; Ikeda, H. Studies on antidiabetic agents. Synthesis and hypoglycemic activity of 5-[4-(pyridylalkoxy)benzyl]-2,4-thiazolidinediones. Arzneimittelforschung 1990, 40, 37–42. [Google Scholar] [CrossRef]
- Shiau, C.W.; Yang, C.C.; Kulp, S.K.; Chen, K.F.; Chen, C.S.; Huang, J.W.; Chen, C.S. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARgamma. Cancer Res. 2005, 65, 1561–1569. [Google Scholar] [CrossRef]
- Wei, S.; Yang, H.C.; Chuang, H.C.; Yang, J.; Kulp, S.K.; Lu, P.J.; Lai, M.D.; Chen, C.S. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J. Biol. Chem. 2008, 283, 26759–26770. [Google Scholar] [CrossRef]
- Chang, A.Y.; Wyse, B.M.; Gilchrist, B.J.; Peterson, T.; Diani, A.R. Ciglitazone, a new hypoglycemic agent. I. Studies in ob/ob and db/db mice, diabetic Chinese hamsters, and normal and streptozotocin-diabetic rats. Diabetes 1983, 32, 830–838. [Google Scholar] [CrossRef]
- Topal, M.A.D.; Şahin, E.; Alver, A. The Effects of Ciglitazone on Enzyme Activities of Carbonic Anhydrase II and Glucose-6-Phosphate Dehydrogenase. Gümüşhane Üniversitesi Sağlık Bilim. Derg. 2022, 11, 105–111. [Google Scholar] [CrossRef]
- Wieczfinska, J.; Pawliczak, R. Anti-fibrotic effect of ciglitazone in HRV-induced airway remodelling cell model. J. Cell Mol. Med. 2023, 27, 1867–1879. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, K.; Murakami, K.; Todo, M.; Aoki, T.; Asaki, T.; Murai, M.; Yano, J. A novel selective peroxisome proliferator-activated receptor alpha agonist, 2-methyl-c-5-[4-[5-methyl-2-(4-methylphenyl)-4-oxazolyl]butyl]-1,3-dioxane-r-2-carboxylic acid (NS-220), potently decreases plasma triglyceride and glucose levels and modifies lipoprotein profiles in KK-Ay mice. J. Pharmacol. Exp. Ther. 2004, 309, 970–977. [Google Scholar] [CrossRef]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. The effect of pioglitazone on peroxisome proliferator-activated receptor-gamma target genes related to lipid storage in vivo. Diabetes Care 2004, 27, 1660–1667. [Google Scholar] [CrossRef]
- Griggs, R.B.; Donahue, R.R.; Morgenweck, J.; Grace, P.M.; Sutton, A.; Watkins, L.R.; Taylor, B.K. Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPARγ mechanisms. Pain 2015, 156, 469–482. [Google Scholar] [CrossRef]
- Orasanu, G.; Ziouzenkova, O.; Devchand, P.R.; Nehra, V.; Hamdy, O.; Horton, E.S.; Plutzky, J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J. Am. Coll. Cardiol. 2008, 52, 869–881. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Tomonari, Y.; Kawashima, K.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Pioglitazone inhibits cancer cell growth through STAT3 inhibition and enhanced AIF expression via a PPARγ-independent pathway. J. Cell Physiol. 2018, 233, 3638–3647. [Google Scholar] [CrossRef]
- Osman, I.; Segar, L. Pioglitazone, a PPARγ agonist, attenuates PDGF-induced vascular smooth muscle cell proliferation through AMPK-dependent and AMPK-independent inhibition of mTOR/p70S6K and ERK signaling. Biochem. Pharmacol. 2016, 101, 54–70. [Google Scholar] [CrossRef]
- Yang, H.C.; Deleuze, S.; Zuo, Y.; Potthoff, S.A.; Ma, L.J.; Fogo, A.B. The PPARgamma agonist pioglitazone ameliorates aging-related progressive renal injury. J. Am. Soc. Nephrol. 2009, 20, 2380–2388. [Google Scholar] [CrossRef]
- Chen, H.Y.; Zhao, H.; Yang, J.J.; Zhang, Q.; Yan, M.M.; Qiu, X.Y. Reassessment of pioglitazone and bladder cancer based on FAERS database. Expert. Opin. Drug Saf. 2024, 23, 1–8. [Google Scholar] [CrossRef]
- Baddam, S.; Banka, A.V.; Divity, S.; Sandesara, M.; Vityala, Y. Association between pioglitazone use and bladder cancer: A systematic review. Bladder 2024, 11, e21200023. [Google Scholar] [CrossRef] [PubMed]
- Viscoli, C.M.; Inzucchi, S.E.; Young, L.H.; Insogna, K.L.; Conwit, R.; Furie, K.L.; Gorman, M.; Kelly, M.A.; Lovejoy, A.M.; Kernan, W.N. Pioglitazone and Risk for Bone Fracture: Safety Data From a Randomized Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 914–922. [Google Scholar] [CrossRef]
- Shah, P.; Mudaliar, S. Pioglitazone: Side effect and safety profile. Expert. Opin. Drug Saf. 2010, 9, 347–354. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef]
- Willson, T.M.; Cobb, J.E.; Cowan, D.J.; Wiethe, R.W.; Correa, I.D.; Prakash, S.R.; Beck, K.D.; Moore, L.B.; Kliewer, S.A.; Lehmann, J.M. The structure-activity relationship between peroxisome proliferator-activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones. J. Med. Chem. 1996, 39, 665–668. [Google Scholar] [CrossRef]
- Vandewalle, B.; Moerman, E.; Lefebvre, B.; Defrance, F.; Gmyr, V.; Lukowiak, B.; Kerr Conte, J.; Pattou, F. PPARgamma-dependent and -independent effects of rosiglitazone on lipotoxic human pancreatic islets. Biochem. Biophys. Res. Commun. 2008, 366, 1096–1101. [Google Scholar] [CrossRef]
- Riess, M.L.; Elorbany, R.; Weihrauch, D.; Stowe, D.F.; Camara, A.K.S. PPARγ-Independent Side Effects of Thiazolidinediones on Mitochondrial Redox State in Rat Isolated Hearts. Cells 2020, 9, 252. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arter. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef]
- Matthews, L.; Berry, A.; Tersigni, M.; D’Acquisto, F.; Ianaro, A.; Ray, D. Thiazolidinediones are partial agonists for the glucocorticoid receptor. Endocrinology 2009, 150, 75–86. [Google Scholar] [CrossRef]
- Nuwormegbe, S.A.; Sohn, J.H.; Kim, S.W. A PPAR-Gamma Agonist Rosiglitazone Suppresses Fibrotic Response in Human Pterygium Fibroblasts by Modulating the p38 MAPK Pathway. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5217–5226. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Wallach, J.D.; Wang, K.; Zhang, A.D.; Cheng, D.; Grossetta Nardini, H.K.; Lin, H.; Bracken, M.B.; Desai, M.; Krumholz, H.M.; Ross, J.S. Updating insights into rosiglitazone and cardiovascular risk through shared data: Individual patient and summary level meta-analyses. BMJ 2020, 368, l7078. [Google Scholar] [CrossRef]
- Lu, Y.; Ma, D.; Xu, W.; Shao, S.; Yu, X. Effect and cardiovascular safety of adding rosiglitazone to insulin therapy in type 2 diabetes: A meta-analysis. J. Diabetes Investig. 2015, 6, 78–86. [Google Scholar] [CrossRef]
- Ma, Y.; Du, X.; Zhao, D.; Tang, K.; Wang, X.; Guo, S.; Li, X.; Mei, S.; Sun, N.; Liu, J.; et al. 18:0 Lyso PC, a natural product with potential PPAR-γ agonistic activity, plays hypoglycemic effect with lower liver toxicity and cardiotoxicity in db/db mice. Biochem. Biophys. Res. Commun. 2021, 579, 168–174. [Google Scholar] [CrossRef]
- Davidson, M.A.; Mattison, D.R.; Azoulay, L.; Krewski, D. Thiazolidinedione drugs in the treatment of type 2 diabetes mellitus: Past, present and future. Crit. Rev. Toxicol. 2018, 48, 52–108. [Google Scholar] [CrossRef]
- Yokoi, T. Troglitazone. Handb. Exp. Pharmacol. 2010, 196, 419–435. [Google Scholar] [CrossRef]
- Zhou, Y.M.; Wen, Y.H.; Kang, X.Y.; Qian, H.H.; Yang, J.M.; Yin, Z.F. Troglitazone, a peroxisome proliferator-activated receptor gamma ligand, induces growth inhibition and apoptosis of HepG2 human liver cancer cells. World J. Gastroenterol. 2008, 14, 2168–2173. [Google Scholar] [CrossRef]
- Weng, J.R.; Chen, C.Y.; Pinzone, J.J.; Ringel, M.D.; Chen, C.S. Beyond peroxisome proliferator-activated receptor gamma signaling: The multi-facets of the antitumor effect of thiazolidinediones. Endocr. Relat. Cancer 2006, 13, 401–413. [Google Scholar] [CrossRef]
- Welbourne, T.; Friday, E.; Fowler, R.; Turturro, F.; Nissim, I. Troglitazone acts by PPARgamma and PPARgamma-independent pathways on LLC-PK1-F+ acid-base metabolism. Am. J. Physiol. Renal Physiol. 2004, 286, F100–F110. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Clem, B.F. Troglitazone suppresses glutamine metabolism through a PPAR-independent mechanism. Biol. Chem. 2015, 396, 937–947. [Google Scholar] [CrossRef]
- Salomone, S. Pleiotropic effects of glitazones: A double edge sword? Front. Pharmacol. 2011, 2, 14. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Kosaka, K.; Kuzuya, T.; Akanuma, Y.; Shigeta, Y.; Kaneko, T. Effects of troglitazone: A new hypoglycemic agent in patients with NIDDM poorly controlled by diet therapy. Diabetes Care 1996, 19, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, I.; Wozniak, R.; Hawkins, M.; Shamoon, H. Troglitazone amplifies counterregulatory responses to hypoglycemia in nondiabetic subjects. J. Clin. Endocrinol. Metab. 2001, 86, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Yoshino, T.; Yamazaki, K.; Yuki, S.; Machida, N.; Sasaki, T.; Hyodo, I.; Yachi, Y.; Onuma, H.; Ohtsu, A. Phase 1 study of efatutazone, a novel oral peroxisome proliferator-activated receptor gamma agonist, in combination with FOLFIRI as second-line therapy in patients with metastatic colorectal cancer. Investig. New Drugs 2014, 32, 473–480. [Google Scholar] [CrossRef]
- Marshall, J.; Shuster, D.E.; Goldberg, T.R.; Copigneaux, C.; Chen, S.; Zahir, H.; Dutta, D.; Saleh, M.N.; Pishvaian, M.J.; Varela, M.S.; et al. A randomized, open-label phase II study of efatutazone in combination with FOLFIRI as second-line therapy for metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2014, 32, 535. [Google Scholar]
- Kawahara, A.; Haraguchi, N.; Tsuchiya, H.; Ikeda, Y.; Hama, S.; Kogure, K. Peroxisome proliferator-activated receptor γ (PPARγ)-independent specific cytotoxicity against immature adipocytes induced by PPARγ antagonist T0070907. Biol. Pharm. Bull. 2013, 36, 1428–1434. [Google Scholar] [CrossRef]
- Leesnitzer, L.M.; Parks, D.J.; Bledsoe, R.K.; Cobb, J.E.; Collins, J.L.; Consler, T.G.; Davis, R.G.; Hull-Ryde, E.A.; Lenhard, J.M.; Patel, L.; et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 2002, 41, 6640–6650. [Google Scholar] [CrossRef]
- Seargent, J.M.; Yates, E.A.; Gill, J.H. GW9662, a potent antagonist of PPARgamma, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARgamma agonist rosiglitazone, independently of PPARgamma activation. Br. J. Pharmacol. 2004, 143, 933–937. [Google Scholar] [CrossRef]
- Schubert, M.; Becher, S.; Wallert, M.; Maeß, M.B.; Abhari, M.; Rennert, K.; Mosig, A.S.; Große, S.; Heller, R.; Grün, M.; et al. The Peroxisome Proliferator-Activated Receptor (PPAR)-γ Antagonist 2-Chloro-5-Nitro-N-Phenylbenzamide (GW9662) Triggers Perilipin 2 Expression via PPARδ and Induces Lipogenesis and Triglyceride Accumulation in Human THP-1 Macrophages. Mol. Pharmacol. 2020, 97, 212–225. [Google Scholar] [CrossRef]
- Jaudszus, A.; Lorkowski, S.; Gruen, M.; Roth, A.; Jahreis, G. Limited Applicability of GW9662 to Elucidate PPARγ-Mediated Fatty Acid Effects in Primary Human T-Helper Cells. Int. J. Inflam. 2014, 2014, 149628. [Google Scholar] [CrossRef]
- Werman, A.; Hollenberg, A.; Solanes, G.; Bjorbaek, C.; Vidal-Puig, A.J.; Flier, J.S. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor gamma (PPARgamma). Differential activity of PPARgamma1 and -2 isoforms and influence of insulin. J. Biol. Chem. 1997, 272, 20230–20235. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef]
- Tian, L.; Wang, C.; Hagen, F.K.; Gormley, M.; Addya, S.; Soccio, R.; Casimiro, M.C.; Zhou, J.; Powell, M.J.; Xu, P.; et al. Acetylation-defective mutant of Pparγ is associated with decreased lipid synthesis in breast cancer cells. Oncotarget 2014, 5, 7303–7315. [Google Scholar] [CrossRef] [PubMed]
- Aaron, N.; Zahr, T.; He, Y.; Yu, L.; Mayfield, B.; Pajvani, U.B.; Qiang, L. Acetylation of PPARγ in macrophages promotes visceral fat degeneration in obesity. Life Metab. 2022, 1, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Chana, R.S.; Baines, R.J.; Willars, G.B.; Brunskill, N.J. Ligand-independent activation of peroxisome proliferator-activated receptor-gamma by insulin and C-peptide in kidney proximal tubular cells: Dependent on phosphatidylinositol 3-kinase activity. J. Biol. Chem. 2004, 279, 49747–49754. [Google Scholar] [CrossRef]
- Dias, M.M.G.; Batista, F.A.H.; Tittanegro, T.H.; de Oliveira, A.G.; Le Maire, A.; Torres, F.R.; Filho, H.V.R.; Silveira, L.R.; Figueira, A.C.M. PPARγ S273 Phosphorylation Modifies the Dynamics of Coregulator Proteins Recruitment. Front. Endocrinol. 2020, 11, 561256. [Google Scholar] [CrossRef]
- Bae, H.; Jang, J.Y.; Choi, S.S.; Lee, J.J.; Kim, H.; Jo, A.; Lee, K.J.; Choi, J.H.; Suh, S.W.; Park, S.B. Mechanistic elucidation guided by covalent inhibitors for the development of anti-diabetic PPARγ ligands. Chem. Sci. 2016, 7, 5523–5529. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Banks, A.S.; Laznik-Bogoslavski, D.; White, J.P.; Choi, J.H.; Kazak, L.; Lo, J.C.; Cohen, P.; Wong, K.K.; Kamenecka, T.M.; et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2018, 115, 561–566. [Google Scholar] [CrossRef]
- Norris, A.W.; Sigmund, C.D. A second chance for a PPARγ targeted therapy? Circ. Res. 2012, 110, 8–11. [Google Scholar] [CrossRef]
- Camp, H.S.; Tafuri, S.R. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 10811–10816. [Google Scholar] [CrossRef]
- Shao, D.; Rangwala, S.M.; Bailey, S.T.; Krakow, S.L.; Reginato, M.J.; Lazar, M.A. Interdomain communication regulating ligand binding by PPAR-gamma. Nature 1998, 396, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Iankova, I.; Petersen, R.K.; Annicotte, J.S.; Chavey, C.; Hansen, J.B.; Kratchmarova, I.; Sarruf, D.; Benkirane, M.; Kristiansen, K.; Fajas, L. Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol. 2006, 20, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Yamashita, D.; Yamaguchi, T.; Hirose, F.; Osumi, T. Aspects of the regulatory mechanisms of PPAR functions: Analysis of a bidirectional response element and regulation by sumoylation. Mol. Cell. Biochem. 2006, 286, 33–42. [Google Scholar] [CrossRef]
- Chung, S.S.; Ahn, B.Y.; Kim, M.; Kho, J.H.; Jung, H.S.; Park, K.S. SUMO modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor γ in C2C12 myotubes. Biochem. J. 2011, 433, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Park, S.Y.; Roth, J.; Kim, H.S.; Cho, J.W. O-GlcNAc modification of PPARγ reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 2012, 417, 1158–1163. [Google Scholar] [CrossRef]
- Shoag, J.; Haq, R.; Zhang, M.; Liu, L.; Rowe, G.C.; Jiang, A.; Koulisis, N.; Farrel, C.; Amos, C.I.; Wei, Q.; et al. PGC-1 coactivators regulate MITF and the tanning response. Mol. Cell 2013, 49, 145–157. [Google Scholar] [CrossRef]
- Miller, A.J.; Du, J.; Rowan, S.; Hershey, C.L.; Widlund, H.R.; Fisher, D.E. Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res. 2004, 64, 509–516. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Nan, H.; Kraft, P.; Qureshi, A.A.; Guo, Q.; Chen, C.; Hankinson, S.E.; Hu, F.B.; Thomas, G.; Hoover, R.N.; Chanock, S.; et al. Genome-wide association study of tanning phenotype in a population of European ancestry. J. Investig. Dermatol. 2009, 129, 2250–2257. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Choi, H.I.; Kim, H.J.; Park, J.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1α attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3β inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef] [PubMed]
- Gelato, K.A.; Schöckel, L.; Klingbeil, O.; Rückert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Héroult, M.; Lejeune, P.; Mönning, U.; et al. Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Korzus, E.; Rosenfeld, M.G.; Mayford, M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 2004, 42, 961–972. [Google Scholar] [CrossRef]
- Ding, L.; Wen, Y.; Zhang, X.; Zhao, F.; Lv, K.; Shi, J.H.; Shen, S.; Pan, X. Transcriptional network constituted of CBP, Ku70, NOX2, and BAX prevents the cell death of necrosis, paraptosis, and apoptosis in human melanoma. Cell Death Discov. 2021, 7, 40. [Google Scholar] [CrossRef]
- Kim, E.; Zucconi, B.E.; Wu, M.; Nocco, S.E.; Meyers, D.J.; McGee, J.S.; Venkatesh, S.; Cohen, D.L.; Gonzalez, E.C.; Ryu, B.; et al. MITF Expression Predicts Therapeutic Vulnerability to p300 Inhibition in Human Melanoma. Cancer Res. 2019, 79, 2649–2661. [Google Scholar] [CrossRef]
- Deblacam, C.; Byrne, C.; Hughes, E.; McIlroy, M.; Bane, F.; Hill, A.D.; Young, L.S. HOXC11-SRC-1 regulation of S100beta in cutaneous melanoma: New targets for the kinase inhibitor dasatinib. Br. J. Cancer 2011, 105, 118–123. [Google Scholar] [CrossRef]
- Bezrookove, V.; Kashani-Sabet, M. NCOA3, a new player in melanoma susceptibility and a therapeutic target. Cancer Gene Ther. 2022, 29, 399–401. [Google Scholar] [CrossRef]
- Dutto, I.; Scalera, C.; Prosperi, E. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell. Mol. Life Sci. 2018, 75, 1325–1338. [Google Scholar] [CrossRef]
- Rangel, J.; Torabian, S.; Shaikh, L.; Nosrati, M.; Baehner, F.L.; Haqq, C.; Leong, S.P.; Miller, J.R., 3rd; Sagebiel, R.W.; Kashani-Sabet, M. Prognostic significance of nuclear receptor coactivator-3 overexpression in primary cutaneous melanoma. J. Clin. Oncol. 2006, 24, 4565–4569. [Google Scholar] [CrossRef]
- Rotte, A.; Bhandaru, M.; Cheng, Y.; Sjoestroem, C.; Martinka, M.; Li, G. Decreased expression of nuclear p300 is associated with disease progression and worse prognosis of melanoma patients. PLoS ONE 2013, 8, e75405. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Markan, K.; Temple, K.A.; Deplewski, D.; Brady, M.J.; Cohen, R.N. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J. Biol. Chem. 2005, 280, 13600–13605. [Google Scholar] [CrossRef] [PubMed]
- Emont, M.P.; Mantis, S.; Kahn, J.H.; Landeche, M.; Han, X.; Sargis, R.M.; Cohen, R.N. Silencing Mediator of Retinoid and Thyroid Hormone Receptors (SMRT) regulates glucocorticoid action in adipocytes. Mol. Cell. Endocrinol. 2015, 407, 52–56. [Google Scholar] [CrossRef]
- Shang, J.; Mosure, S.A.; Zheng, J.; Brust, R.; Bass, J.; Nichols, A.; Solt, L.A.; Griffin, P.R.; Kojetin, D.J. A molecular switch regulating transcriptional repression and activation of PPARγ. Nat. Commun. 2020, 11, 956. [Google Scholar] [CrossRef]
- Guan, H.P.; Ishizuka, T.; Chui, P.C.; Lehrke, M.; Lazar, M.A. Corepressors selectively control the transcriptional activity of PPARgamma in adipocytes. Genes Dev. 2005, 19, 453–461. [Google Scholar] [CrossRef]
- Cohen, R.N. Nuclear receptor corepressors and PPARgamma. Nucl. Recept. Signal 2006, 4, e003. [Google Scholar] [CrossRef]
- Umemoto, T.; Fujiki, Y. Ligand-dependent nucleo-cytoplasmic shuttling of peroxisome proliferator-activated receptors, PPARα and PPARγ. Genes Cells 2012, 17, 576–596. [Google Scholar] [CrossRef]
- Gallardo, F.; Padrón, A.; Garcia-Carbonell, R.; Rius, C.; González-Perez, A.; Arumí-Uria, M.; Iglesias, M.; Nonell, L.; Bellosillo, B.; Segura, S.; et al. Cytoplasmic accumulation of NCoR in malignant melanoma: Consequences of altered gene repression and prognostic significance. Oncotarget 2015, 6, 9284–9294. [Google Scholar] [CrossRef]
- Eastham, L.L.; Mills, C.N.; Niles, R.M. PPARalpha/gamma expression and activity in mouse and human melanocytes and melanoma cells. Pharm. Res. 2008, 25, 1327–1333. [Google Scholar] [CrossRef]
- Wolf Horrell, E.M.; Boulanger, M.C.; D’Orazio, J.A. Melanocortin 1 Receptor: Structure, Function, and Regulation. Front. Genet. 2016, 7, 95. [Google Scholar] [CrossRef]
- Swope, V.B.; Abdel-Malek, Z.A. Significance of the Melanocortin 1 and Endothelin B Receptors in Melanocyte Homeostasis and Prevention of Sun-Induced Genotoxicity. Front. Genet. 2016, 7, 146. [Google Scholar] [CrossRef]
- Herraiz, C.; Martínez-Vicente, I.; Maresca, V. The α-melanocyte-stimulating hormone/melanocortin-1 receptor interaction: A driver of pleiotropic effects beyond pigmentation. Pigment. Cell Melanoma Res. 2021, 34, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Gelmi, M.C.; Houtzagers, L.E.; Strub, T.; Krossa, I.; Jager, M.J. MITF in Normal Melanocytes, Cutaneous and Uveal Melanoma: A Delicate Balance. Int. J. Mol. Sci. 2022, 23, 6001. [Google Scholar] [CrossRef]
- Maresca, V.; Flori, E.; Camera, E.; Bellei, B.; Aspite, N.; Ludovici, M.; Catricalà, C.; Cardinali, G.; Picardo, M. Linking αMSH with PPARγ in B16-F10 melanoma. Pigment. Cell Melanoma Res. 2013, 26, 113–127. [Google Scholar] [CrossRef]
- Flori, E.; Rosati, E.; Cardinali, G.; Kovacs, D.; Bellei, B.; Picardo, M.; Maresca, V. The α-melanocyte stimulating hormone/peroxisome proliferator activated receptor-γ pathway down-regulates proliferation in melanoma cell lines. J. Exp. Clin. Cancer Res. 2017, 36, 142. [Google Scholar] [CrossRef]
- Motiani, R.K.; Tanwar, J.; Raja, D.A.; Vashisht, A.; Khanna, S.; Sharma, S.; Srivastava, S.; Sivasubbu, S.; Natarajan, V.T.; Gokhale, R.S. STIM1 activation of adenylyl cyclase 6 connects Ca2+ and cAMP signaling during melanogenesis. EMBO J. 2018, 37, e97597. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Moll, I.; Schumacher, U.; Thies, A. Anti-proliferative effect of peroxisome proliferator-activated receptor gamma agonists on human malignant melanoma cells in vitro. Anticancer Drugs 2006, 17, 325–332. [Google Scholar] [CrossRef]
- Yang, L.K.; Tao, Y.X. Biased signaling at neural melanocortin receptors in regulation of energy homeostasis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2486–2495. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, X.; Zhou, R.; Gong, R. Melanocortin 5 receptor signaling pathway in health and disease. Cell. Mol. Life Sci. 2020, 77, 3831–3840. [Google Scholar] [CrossRef]
- Slominski, R.M.; Kim, T.K.; Janjetovic, Z.; Brożyna, A.A.; Podgorska, E.; Dixon, K.M.; Mason, R.S.; Tuckey, R.C.; Sharma, R.; Crossman, D.K.; et al. Malignant Melanoma: An Overview, New Perspectives, and Vitamin D Signaling. Cancers 2024, 16, 2262. [Google Scholar] [CrossRef]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The role of melanin pigment in melanoma. Exp. Dermatol. 2015, 24, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Fateeva, A.; Eddy, K.; Chen, S. Overview of current melanoma therapies. Pigment. Cell Melanoma Res. 2024, 37, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Valencia, J.C.; Lai, B.; Zhang, G.; Paterson, J.K.; Rouzaud, F.; Berens, W.; Wincovitch, S.M.; Garfield, S.H.; Leapman, R.D.; et al. Melanosomal sequestration of cytotoxic drugs contributes to the intractability of malignant melanomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9903–9907. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.P.; Lindgren, S.; Powell, W.; Green, H. Melanin inhibits cytotoxic effects of doxorubicin and daunorubicin in MOLT 4 cells. Pigment. Cell Res. 2003, 16, 351–354. [Google Scholar] [CrossRef]
- Xie, T.; Nguyen, T.; Hupe, M.; Wei, M.L. Multidrug resistance decreases with mutations of melanosomal regulatory genes. Cancer Res. 2009, 69, 992–999. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jóźwicki, W.; Carlson, J.A.; Slominski, A.T. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum. Pathol. 2013, 44, 2071–2074. [Google Scholar] [CrossRef]
- Schoonjans, K.; Auwerx, J. Thiazolidinediones: An update. Lancet 2000, 355, 1008–1010. [Google Scholar] [CrossRef]
- Wiechers, J.W.; Rawlings, A.V.; Garcia, C.; Chesné, C.; Balaguer, P.; Nicolas, J.C.; Corre, S.; Galibert, M.D. A new mechanism of action for skin whitening agents: Binding to the peroxisome proliferator-activated receptor. Int. J. Cosmet. Sci. 2005, 27, 123–132. [Google Scholar] [CrossRef]
- Grabacka, M.; Wieczorek, J.; Michalczyk-Wetula, D.; Malinowski, M.; Wolan, N.; Wojcik, K.; Plonka, P.M. Peroxisome proliferator-activated receptor α (PPARα) contributes to control of melanogenesis in B16 F10 melanoma cells. Arch. Dermatol. Res. 2017, 309, 141–157. [Google Scholar] [CrossRef]
- Placha, W.; Gil, D.; Dembińska-Kieć, A.; Laidler, P. The effect of PPARgamma ligands on the proliferation and apoptosis of human melanoma cells. Melanoma Res. 2003, 13, 447–456. [Google Scholar] [CrossRef]
- Becker, A.L.; Carpenter, E.L.; Slominski, A.T.; Indra, A.K. The Role of the Vitamin D Receptor in the Pathogenesis, Prognosis, and Treatment of Cutaneous Melanoma. Front. Oncol. 2021, 11, 743667. [Google Scholar] [CrossRef]
- Sertznig, P.; Dunlop, T.; Seifert, M.; Tilgen, W.; Reichrath, J. Cross-talk between vitamin D receptor (VDR)- and peroxisome proliferator-activated receptor (PPAR)-signaling in melanoma cells. Anticancer. Res. 2009, 29, 3647–3658. [Google Scholar] [PubMed]
- Lee, C.S.; Park, M.; Han, J.; Lee, J.H.; Bae, I.H.; Choi, H.; Son, E.D.; Park, Y.H.; Lim, K.M. Liver X receptor activation inhibits melanogenesis through the acceleration of ERK-mediated MITF degradation. J. Investig. Dermatol. 2013, 133, 1063–1071. [Google Scholar] [CrossRef]
- Klopper, J.P.; Sharma, V.; Berenz, A.; Hays, W.R.; Loi, M.; Pugazhenthi, U.; Said, S.; Haugen, B.R. Retinoid and thiazolidinedione therapies in melanoma: An analysis of differential response based on nuclear hormone receptor expression. Mol. Cancer 2009, 8, 16. [Google Scholar] [CrossRef]
- Dana, N.; Vaseghi, G.; Haghjooy Javanmard, S. Crosstalk between Peroxisome Proliferator-Activated Receptors and Toll-Like Receptors: A Systematic Review. Adv. Pharm. Bull. 2019, 9, 12–21. [Google Scholar] [CrossRef]
- Amiri, K.I.; Richmond, A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005, 24, 301–313. [Google Scholar] [CrossRef]
- Elrod, H.A.; Sun, S.Y. PPARgamma and Apoptosis in Cancer. PPAR Res. 2008, 2008, 704165. [Google Scholar] [CrossRef]
- Wang, C.; Fu, M.; D’Amico, M.; Albanese, C.; Zhou, J.N.; Brownlee, M.; Lisanti, M.P.; Chatterjee, V.K.; Lazar, M.A.; Pestell, R.G. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol. Cell. Biol. 2001, 21, 3057–3070. [Google Scholar] [CrossRef]
- Qin, C.; Burghardt, R.; Smith, R.; Wormke, M.; Stewart, J.; Safe, S. Peroxisome proliferator-activated receptor gamma agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer Res. 2003, 63, 958–964. [Google Scholar]
- Plissonnier, M.L.; Fauconnet, S.; Bittard, H.; Mougin, C.; Rommelaere, J.; Lascombe, I. Cell death and restoration of TRAIL-sensitivity by ciglitazone in resistant cervical cancer cells. Oncotarget 2017, 8, 107744–107762. [Google Scholar] [CrossRef]
- Kim, Y.H.; Jung, E.M.; Lee, T.J.; Kim, S.H.; Choi, Y.H.; Park, J.W.; Park, J.W.; Choi, K.S.; Kwon, T.K. Rosiglitazone promotes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic. Biol. Med. 2008, 44, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Yoshida, T.; Shiraishi, T.; Horinaka, M.; Kouhara, J.; Wakada, M.; Sakai, T. 15-Deoxy-Delta12,14-prostaglandin J(2) induces death receptor 5 expression through mRNA stabilization independently of PPARgamma and potentiates TRAIL-induced apoptosis. Mol. Cancer Ther. 2006, 5, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Koshizuka, K.; Williamson, E.A.; Asou, H.; Said, J.W.; Holden, S.; Miyoshi, I.; Koeffler, H.P. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998, 58, 3344–3352. [Google Scholar]
- Demetri, G.D.; Fletcher, C.D.; Mueller, E.; Sarraf, P.; Naujoks, R.; Campbell, N.; Spiegelman, B.M.; Singer, S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. USA 1999, 96, 3951–3956. [Google Scholar] [CrossRef]
- Rubin, G.L.; Zhao, Y.; Kalus, A.M.; Simpson, E.R. Peroxisome proliferator-activated receptor gamma ligands inhibit estrogen biosynthesis in human breast adipose tissue: Possible implications for breast cancer therapy. Cancer Res. 2000, 60, 1604–1608. [Google Scholar]
- Senior, J.R. Evolution of the Food and Drug Administration approach to liver safety assessment for new drugs: Current status and challenges. Drug Saf. 2014, 37 (Suppl. S1), S9–S17. [Google Scholar] [CrossRef]
- Katoch, S.; Sharma, V.; Patial, V. Peroxisome proliferator-activated receptor gamma as a therapeutic target for hepatocellular carcinoma: Experimental and clinical scenarios. World J. Gastroenterol. 2022, 28, 3535–3554. [Google Scholar] [CrossRef]
- Mueller, E.; Sarraf, P.; Tontonoz, P.; Evans, R.M.; Martin, K.J.; Zhang, M.; Fletcher, C.; Singer, S.; Spiegelman, B.M. Terminal differentiation of human breast cancer through PPAR gamma. Mol. Cell 1998, 1, 465–470. [Google Scholar] [CrossRef]
- Kitamura, S.; Miyazaki, Y.; Shinomura, Y.; Kondo, S.; Kanayama, S.; Matsuzawa, Y. Peroxisome proliferator-activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. Jpn. J. Cancer Res. 1999, 90, 75–80. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, W.H. Peroxisome proliferator-activated receptor γ and colorectal cancer. World J. Gastrointest. Oncol. 2010, 2, 159–164. [Google Scholar] [CrossRef]
- Sun, J.; Yu, L.; Qu, X.; Huang, T. The role of peroxisome proliferator-activated receptors in the tumor microenvironment, tumor cell metabolism, and anticancer therapy. Front. Pharmacol. 2023, 14, 1184794. [Google Scholar] [CrossRef] [PubMed]
- Bren-Mattison, Y.; Van Putten, V.; Chan, D.; Winn, R.; Geraci, M.W.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene 2005, 24, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. PPARγ as a Novel Therapeutic Target in Lung Cancer. PPAR Res. 2016, 2016, 8972570. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Kim, H.; Kim, H.G.; Cho, Y.M.; Jung, W.Y.; Han, H.S.; Hwang, T.S.; Kwon, G.Y.; Lim, S.D. Expression of Peroxisome Proliferator Activated Receptor gamma in Prostatic Adenocarcinoma. J. Korean Med. Sci. 2015, 30, 533–541. [Google Scholar] [CrossRef]
- Rapuano, R.; Riccio, A.; Mercuri, A.; Madera, J.R.; Dallavalle, S.; Moricca, S.; Lupo, A. Proliferation and migration of PC-3 prostate cancer cells is counteracted by PPARγ-cladosporol binding-mediated apoptosis and a decreased lipid biosynthesis and accumulation. Biochem. Pharmacol. 2024, 222, 116097. [Google Scholar] [CrossRef]
- Paulitschke, V.; Gruber, S.; Hofstätter, E.; Haudek-Prinz, V.; Klepeisz, P.; Schicher, N.; Jonak, C.; Petzelbauer, P.; Pehamberger, H.; Gerner, C.; et al. Proteome analysis identified the PPARγ ligand 15d-PGJ2 as a novel drug inhibiting melanoma progression and interfering with tumor-stroma interaction. PLoS ONE 2012, 7, e46103. [Google Scholar] [CrossRef]
- Briganti, S.; Mosca, S.; Di Nardo, A.; Flori, E.; Ottaviani, M. New Insights into the Role of PPARγ in Skin Physiopathology. Biomolecules 2024, 14, 728. [Google Scholar] [CrossRef]
- Ferrara, A.; Lewis, J.D.; Quesenberry, C.P., Jr.; Peng, T.; Strom, B.L.; Van Den Eeden, S.K.; Ehrlich, S.F.; Habel, L.A. Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diabetes Care 2011, 34, 923–929. [Google Scholar] [CrossRef]
- Zhang, S.; Lv, K.; Liu, Z.; Zhao, R.; Li, F. Fatty acid metabolism of immune cells: A new target of tumour immunotherapy. Cell Death Discov. 2024, 10, 39. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Mi, S.; Ye, J.; Lou, G. Aberrant lipid metabolism in cancer cells and tumor microenvironment: The player rather than bystander in cancer progression and metastasis. J. Cancer 2021, 12, 7498–7506. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e1005. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 2021, 54, 1561–1577.e1567. [Google Scholar] [CrossRef]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, M.; Su, J.; Li, Y.; Long, J.; Chu, J.; Wan, X.; Cao, Y.; Li, Q. Lipid Metabolism and Cancer. Life 2022, 12, 784. [Google Scholar] [CrossRef]
- Xin, X.; Yang, S.; Kowalski, J.; Gerritsen, M.E. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J. Biol. Chem. 1999, 274, 9116–9121. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Z.; Zuo, Q.; Kang, Y. Regulation of CD8+ T cells by lipid metabolism in cancer progression. Cell. Mol. Immunol. 2024, 21, 1215–1230. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8+ T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Tsai, Y.S.; Lin, S.C.; Liao, N.S.; Jan, M.S.; Liang, C.T.; Hsu, S.W.; Chen, W.C.; Sung, J.M.; Maeda, N.; et al. Quantitative PPARγ expression affects the balance between tolerance and immunity. Sci. Rep. 2016, 6, 26646. [Google Scholar] [CrossRef] [PubMed]
- Bertschi, N.L.; Steck, O.; Luther, F.; Bazzini, C.; von Meyenn, L.; Schärli, S.; Vallone, A.; Felser, A.; Keller, I.; Friedli, O.; et al. PPAR-γ regulates the effector function of human T helper 9 cells by promoting glycolysis. Nat. Commun. 2023, 14, 2471. [Google Scholar] [CrossRef]
- Shah, R.; Katopodi, X.L.; Christofides, A.; Pal, R.; Vlachos, I.; Patsoukis, N.; Boussiotis, V.A. Loss of the Nuclear Receptor Pparγ Primes T Effector (TEFF) Differentiation of Antigen-Specific CD8+ T Cells. Blood 2022, 140, 2637–2638. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Kondo, M.; Kumagai, S.; Nishikawa, H. Metabolic advantages of regulatory T cells dictated by cancer cells. Int. Immunol. 2024, 36, 75–86. [Google Scholar] [CrossRef]
- Xu, C.; Fu, Y.; Liu, S.; Trittipo, J.; Lu, X.; Qi, R.; Du, H.; Yan, C.; Zhang, C.; Wan, J.; et al. BATF Regulates T Regulatory Cell Functional Specification and Fitness of Triglyceride Metabolism in Restraining Allergic Responses. J. Immunol. 2021, 206, 2088–2100. [Google Scholar] [CrossRef]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12, 702726. [Google Scholar] [CrossRef]
- Qiu, Y.; Ke, S.; Chen, J.; Qin, Z.; Zhang, W.; Yuan, Y.; Meng, D.; Zhao, G.; Wu, K.; Li, B.; et al. FOXP3+ regulatory T cells and the immune escape in solid tumours. Front. Immunol. 2022, 13, 982986. [Google Scholar] [CrossRef]
- Hontecillas, R.; Bassaganya-Riera, J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J. Immunol. 2007, 178, 2940–2949. [Google Scholar] [CrossRef]
- Guri, A.J.; Mohapatra, S.K.; Horne, W.T., 2nd; Hontecillas, R.; Bassaganya-Riera, J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. 2010, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Wohlfert, E.A.; Nichols, F.C.; Nevius, E.; Clark, R.B. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: Enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J. Immunol. 2007, 178, 4129–4135. [Google Scholar] [CrossRef] [PubMed]
- Nobs, S.P.; Natali, S.; Pohlmeier, L.; Okreglicka, K.; Schneider, C.; Kurrer, M.; Sallusto, F.; Kopf, M. PPARγ in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J. Exp. Med. 2017, 214, 3015–3035. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, Q.; Peng, H. Tumor-associated macrophages: New insights on their metabolic regulation and their influence in cancer immunotherapy. Front. Immunol. 2023, 14, 1157291. [Google Scholar] [CrossRef]
- De Simone, G.; Soldani, C.; Morabito, A.; Franceschini, B.; Ferlan, F.; Costa, G.; Pastorelli, R.; Donadon, M.; Brunelli, L. Implication of metabolism in the polarization of tumor-associated-macrophages: The mass spectrometry-based point of view. Front. Immunol. 2023, 14, 1193235. [Google Scholar] [CrossRef]
- Szatmari, I.; Gogolak, P.; Im, J.S.; Dezso, B.; Rajnavolgyi, E.; Nagy, L. Activation of PPARgamma specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity 2004, 21, 95–106. [Google Scholar] [CrossRef]
- Maher, M.; Diesch, J.; Casquero, R.; Buschbeck, M. Epigenetic-Transcriptional Regulation of Fatty Acid Metabolism and Its Alterations in Leukaemia. Front. Genet. 2018, 9, 405. [Google Scholar] [CrossRef]
- Johnson, A.R.; Qin, Y.; Cozzo, A.J.; Freemerman, A.J.; Huang, M.J.; Zhao, L.; Sampey, B.P.; Milner, J.J.; Beck, M.A.; Damania, B.; et al. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol. Metab. 2016, 5, 506–526. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2019, 10, 2993. [Google Scholar] [CrossRef]
- Balestrieri, B.; Di Costanzo, D.; Dwyer, D.F. Macrophage-Mediated Immune Responses: From Fatty Acids to Oxylipins. Molecules 2021, 27, 152. [Google Scholar] [CrossRef]
- Conos, S.A.; Lawlor, K.E.; Vaux, D.L.; Vince, J.E.; Lindqvist, L.M. Cell death is not essential for caspase-1-mediated interleukin-1β activation and secretion. Cell Death Differ. 2016, 23, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Tyurin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; Celis, E.; et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Hemmert, K.C.; Ochi, A.; Jamal, M.; Henning, J.R.; Barilla, R.; Quesada, J.P.; Zambirinis, C.P.; Tang, K.; Ego-Osuala, M.; et al. Role of fatty-acid synthesis in dendritic cell generation and function. J. Immunol. 2013, 190, 4640–4649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef]
- Szatmari, I.; Töröcsik, D.; Agostini, M.; Nagy, T.; Gurnell, M.; Barta, E.; Chatterjee, K.; Nagy, L. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood 2007, 110, 3271–3280. [Google Scholar] [CrossRef]
- Ye, J.H.; Chen, Y.L.; Ogg, G. CD1a and skin T cells: A pathway for therapeutic intervention. Clin. Exp. Dermatol. 2024, 49, 450–458. [Google Scholar] [CrossRef]
- Vandoros, G.P.; Konstantinopoulos, P.A.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papachristou, G.I.; Karamouzis, M.V.; Gkermpesi, M.; Varakis, I.; Papavassiliou, A.G. PPAR-gamma is expressed and NF-kB pathway is activated and correlates positively with COX-2 expression in stromal myofibroblasts surrounding colon adenocarcinomas. J. Cancer Res. Clin. Oncol. 2006, 132, 76–84. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Sng, M.K.; Teo, Z.Q.; Chong, H.C.; Twang, J.S.; Tan, N.S. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 2018, 37, 160–173. [Google Scholar] [CrossRef]
- Avena, P.; Anselmo, W.; Whitaker-Menezes, D.; Wang, C.; Pestell, R.G.; Lamb, R.S.; Hulit, J.; Casaburi, I.; Andò, S.; Martinez-Outschoorn, U.E.; et al. Compartment-specific activation of PPARγ governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle 2013, 12, 1360–1370. [Google Scholar] [CrossRef]
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal-Epithelial Crosstalk and Carcinogenesis. Cancers 2021, 13, 2153. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Hu, X.; Wang, D.; Tang, D. Metabolic Reprogramming of Cancer-Associated Fibroblast in the Tumor Microenvironment: From Basics to Clinic. Clin. Med. Insights Oncol. 2024, 18, 11795549241287058. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.; Ahmad, I. The role of PPARγ in prostate cancer development and progression. Br. J. Cancer 2023, 128, 940–945. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef]
- Asgharzadeh, F.; Memarzia, A.; Alikhani, V.; Beigoli, S.; Boskabady, M.H. Peroxisome proliferator-activated receptors: Key regulators of tumor progression and growth. Transl. Oncol. 2024, 47, 102039. [Google Scholar] [CrossRef]








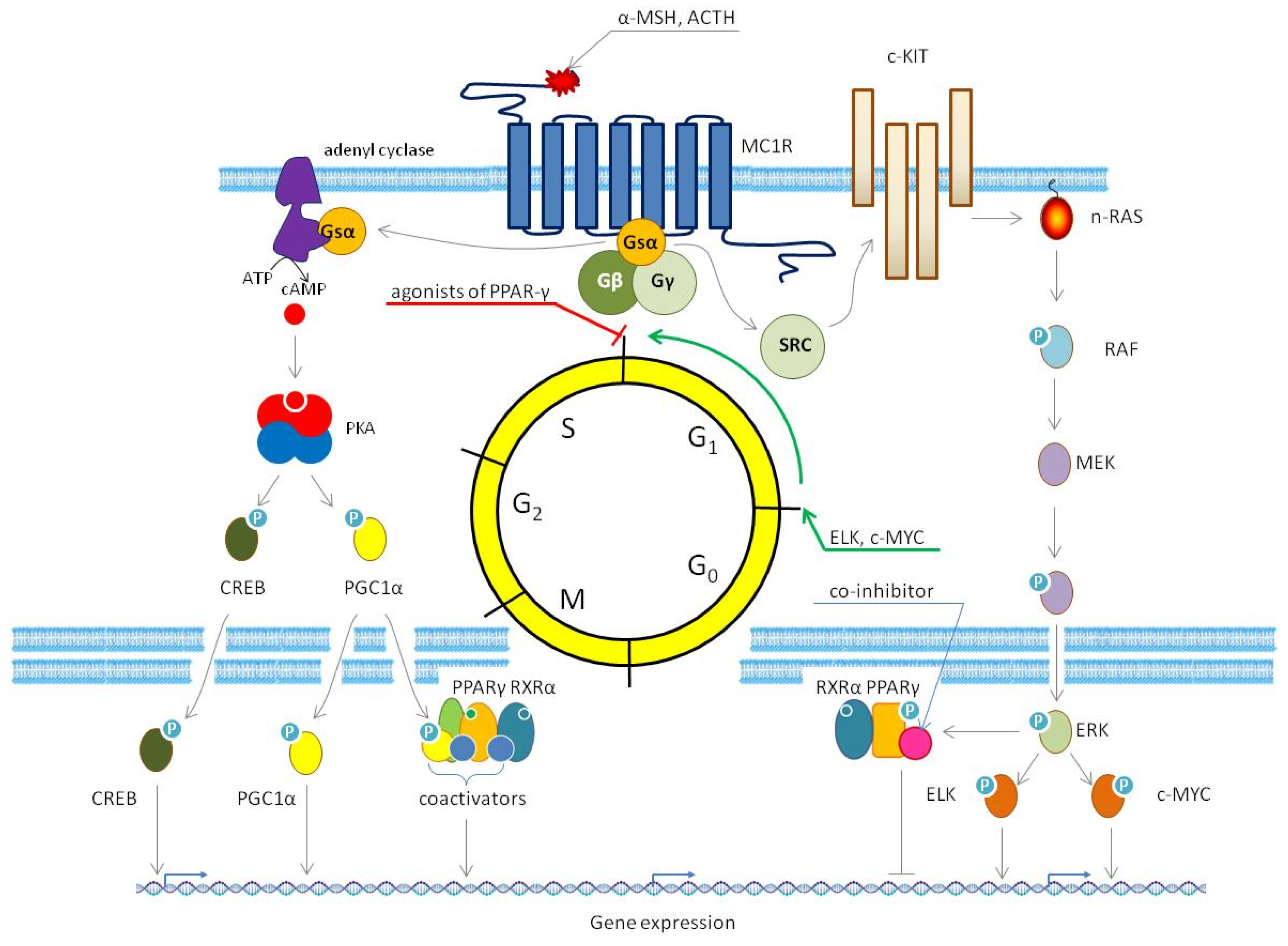

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Function of PPAR-γ | Role of PPAR-γ and Physiological Effect | References |
|---|---|---|---|
| The canonical PPAR-γ signaling pathway | PPAR-γ forms a heterodimer with retinoid X receptor α (RXR-α). Upon binding a ligand, this complex recruits either coactivators or corepressors, depending on the ligand type and cellular context. Then, PPAR-γ-RXRα complexes bind to PPAR-responsive elements (PPREs) in DNA, altering gene expression by modifying chromatin structure and recruiting the transcriptional machinery. | In this pathway, PPAR-γ functions as a nuclear receptor and transcription factor. As a central regulator, PPAR-γ orchestrates fatty acid storage, transport, and metabolism, as well as glucose metabolism, by modulating gene expression in adipocytes, hepatocytes, and other types of cells. | [28,31] |
| The caspase pathway | After cleavage by caspase-1 (CASP1), PPAR-γ translocates to mitochondria, where it binds to and inhibits medium-chain acyl-CoA dehydrogenase (MCAD). | In this pathway, PPAR-γ acts as an endogenous protein inhibitor. Its inhibitory activity promotes lipid droplet accumulation by reducing the oxidation rate of fatty acids via suppression of medium-chain acyl-CoA dehydrogenase (MCAD). | [32] |
| The nuclear factor κB (NF-κB) signaling pathway | PPAR-γ ubiquitinates the p65 subunit of NF-κB, targeting it for proteasomal degradation. Additionally, PPARγ upregulates the gene encoding the inhibitory subunit IκBα, further suppressing NF-κB activity. | In this pathway, PPAR-γ exhibits a dual regulatory role in modulating NF-κB signaling. Firstly, it acts as an E3 ubiquitin ligase, directly ubiquitinating the p65 subunit of NF-κB and targeting it for proteasomal degradation. Secondly, PPAR-γ functions as a transcription factor, inducing IκBα gene expression to enhance NF-κB inhibition. By suppressing the NF-κB pathway through these mechanisms, PPAR-γ attenuates inflammatory responses in affected cells. | [33,34] |
| The activator protein 1 (AP1) signaling pathway | PPAR-γ binds to Jun-Fos heterodimers of the transcription factor AP1, preventing their interaction with DNA. Additionally, PPAR-γ sequesters the transcriptional coactivator of AP1, CREB-binding protein (CBP), further suppressing AP1-dependent gene expression. | In this pathway, PPAR-γ exhibits a dual regulatory role in modulating AP1 activity. Firstly, it acts as a transcriptional repressor of Jun-Fos heterodimers of the transcription factor AP1, blocking their ability to bind DNA and activate target genes. Secondly, PPAR-γ competes with AP1 for shared transcriptional coactivators (e.g., CBP/p300), disrupting the capacity of AP1 to form functional transcriptional complexes. The impact of transrepression depends on the context. For instance, it reduces the expression of pro-inflammatory cytokines (e.g., TNF-α, IL6, and IFN-γ) in immune cells. | [35,36,37] |
| The signal transducer and activator of transcription (STAT) signaling pathway | The heteromers of PPAR-γ STAT1 induce the gene of the scavenger receptor complex of differentiation 36 (CD36). In turn, the heteromers of PPAR-γ and STAT6 upregulate the expression of the fatty acid transporter, fatty acid-binding protein 4 (FABP4). | In this pathway, PPAR-γ acts as a transcriptional coactivator of STAT1 and STAT6, driving changes in gene expression that promote lipid accumulation. By inducing CD36 and FABP4, the heteromers of PPAR-γ with STAT proteins enhance lipid uptake and storage. These alterations result in the accumulation of lipid droplets within affected cells. | [38,39,40] |
| The MAPK pathway | Mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinases (ERKs) and c-Jun N-terminal kinase (JNK), directly phosphorylate PPAR-γ, modulating its transcriptional activity. | In this pathway, PPAR-γ serves as an effector protein of the named MAPKs. Phosphorylation by these kinases reduces the transcriptional activity of PPAR-γ, leading to suppression of its target genes (e.g., the genes controlling lipid metabolism). | [41,42] |
| Therapeutic Agent | Trial ID | Phase | Status | N | Brief Description | Objectives and Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| Anti-PD-L1 immunotherapy (PD-L1—programmed death ligand 1) alone or in combination with either metformin or rosiglitazone (RSG) | NCT04114136 | II | A | 72 | Controlled, randomized, open clinical trial, with parallel groups, without using placebo, three treatment arms | This study aimed to examine whether metformin and RSG will reduce oxygen consumption by tumors, creating a less hypoxic environment for T cells, with pharmacologic remodeling of the tumor microenvironment (TME) to restore the effector function of T cells, acting synergistically with anti-PD-L1 monoclonal antibodies to produce a higher response rate than conventional anti-PD-L1 immunotherapy. | not published |
| Bexarotene in combination with RSG | I | C | 45 | Open-label pilot study | This clinical trial investigated potential synergistic effects of combining bexarotene and RSG in patients with refractory solid tumors. Although the combination demonstrated safety and feasibility in heavily pretreated patients, it failed to induce objective tumor responses. | [55] | |
| Temsirolimus in combination with pioglitazone (PGZ), etoricoxib, and metronomic low-dose trofosfamide versus DTIC (dacarbazine) | EUCTR2011- 002611-29-DE/NCT01614301 | II | C | 48 | Controlled, randomized, open clinical trial, with parallel groups and two treatment arms, without using placebo | This study aimed to evaluate overall survival, objective response rate, time to progression, time to partial response, quality of life, tolerability, and safety of a proposed therapeutic regimen in patients with chemorefractory malignancies. The study found that the proposed therapeutic regimen was active and well tolerated by patients with chemorefractory malignancies. | [56] |
| Bexarotene in combination with efatutazone (EFZ) | NCT01504490 | I | T | 9 | Controlled, randomized, open clinical trial, with parallel groups and unspecified number of treatment arms without using placebo | This study aimed to evaluate the safety and efficacy of a novel drug combination, EFZ and bexarotene, in patients with advanced cancers. The trial was designed to explore the potential synergistic effects of these agents in modulating the tumor microenvironment and improving therapeutic outcomes. However, the study was prematurely terminated following the withdrawal of EFZ from the market, preventing further evaluation of its clinical potential. | not published |
| EFZ | NCT00408434 | I | C | 32 | Open-label dose escalation study | This phase I study aimed to evaluate the safety, tolerability, and preliminary efficacy of EFZ, a novel oral PPAR-γ agonist, in patients with advanced or metastatic solid tumors. The results demonstrated that EFZ has an acceptable tolerability with evidence of disease control in patients with advanced malignancies. | [57] |
| EFZ | NCT00881569 | I | T | 2 | Long-term follow-up open-label safety extension study in non-randomized, observational phase | This study was designed to allow participants who completed a prior clinical trial of EFZ without disease progression or unacceptable toxicity to continue receiving the study drug. The study was terminated after both enrolled patients experienced disease progression, indicating loss of therapeutic benefit. | not published |
| ω3 PUFAs | NCT01032343 | N/A | C | 79 | Double-blind randomized, placebo-controlled nutritional study with two treatment arms | This study investigated whether dietary ω3 polyunsaturated fatty acids (PUFAs), particularly eicosapentaenoic acid (EPA), could mitigate UV-induced cutaneous immunosuppression in healthy human subjects without prior skin cancer diagnoses. The results showed that the dietary supplement used in this study shifted eicosanoid synthesis towards less pro-inflammatory bioactive components, promoting a regulatory milieu under basal conditions and in response to the inflammatory insult. | [58] |
| Resveratol | NCT00098969 | I | A | 40 | Open-label dose escalation study | This clinical trial aimed to assess the safety, tolerability, and optimal dosing of resveratrol in healthy individuals, with secondary exploration of its chemopreventive potential. The results demonstrated that resveratol exhibits a potential chemopreventive effect, by reducing the plasma levels of circulating insulin-like growth factor 1 (IGF1) and insulin growth factor-binding protein 3 (IGFBP3). | [59] |
| Curcumin | NCT01201694 | I | C | 28 | Open-label, non-randomized, dose-escalation study with single group assignment and unspecified number of treatment arms without using placebo | This clinical trial aimed to evaluate the safety, tolerability, and maximum tolerated dose of surface-controlled water-dispersible curcumin in patients with advanced solid tumors. | not published |
| Curcumin | NCT02138955 | IB | C | 32 | Open-label, non-randomized, dose-escalation study with single group assignment and seven treatment arms without using placebo | This clinical trial aimed to evaluate the safety, tolerability, and maximum tolerated dose of liposomal curcumin in patients with advanced or metastatic solid tumors. The results showed that liposomal curcumin was safe and well tolerated by patients with no dose-limiting toxicities observed, supporting its potential for therapeutic use. | [60] |
| Curcumin in combination with cilazapril or losartan, aliskerin, propranolol, aspirin, and metformin | ACTRN12619001078145 | II | C | 50 | Open-label, non-randomized, non-blinded, single treatment arm without using placebo | This clinical trial aimed to evaluate the tolerability and preliminary efficacy of a novel therapeutic regimen targeting the renin–angiotensin system and its downstream signaling pathways in patients with advanced solid tumors. | not published |
| Ligand | Specificity | Mode of Action | PPAR-y Independent Effects | Limitations |
|---|---|---|---|---|
| CGZ | CGZ is a potent and specific PPAR-γ agonist, with a selectivity of over 33-fold compared to other PPARs [128]. | CGZ primarily exerts its effects through the activation of PPAR-γ, resulting in enhanced adipogenesis and fat storage, improved insulin sensitivity, and altered hormone production in adipose tissue, as well as potential anticancer properties. |
|
|
| PGZ | PGZ binds with high affinity to PPAR-γ, a property crucial for its insulin-sensitizing effects [134]. Additionally, it exhibits weak agonistic activity toward PPAR-α [135]. | PGZ primarily acts as a selective agonist of PPAR-γ, with secondary effects on PPAR-α. It improves insulin sensitivity, reduces hepatic glucose production, and enhances adipokine secretion. PGZ effectively lowers blood glucose levels and improves lipid profiles in patients with T2D. |
| |
| RSZ | RRSZ exhibits high specificity for PPAR-γ compared to other PPAR subtypes, showing no significant activity at PPAR-α or PPAR-β/δ [145,146]. | RSZ acts as a potent PPAR-γ agonist, enhancing insulin sensitivity and glucose metabolism by modulating the transcription of insulin-responsive genes involved in glucose production, transport, and utilization [145,146]. |
|
|
| TGZ | Compared to RSZ and PGZ, TGZ exhibits a lower affinity for PPAR-γ. This difference is reflected in the higher clinical dosages required for TGZ (400–800 mg/day) compared to RSZ (4–8 mg/day) and PGZ (15–45 mg/day). Moreover, TGZ displays weaker binding activity to PPAR-α [156]. | TGZ primarily acts as a selective PPAR-γ agonist, inducing genes involved in glucose and lipid metabolism, thereby enhancing insulin sensitivity in muscle and adipose tissue while reducing hepatic gluconeogenesis [156]. However, the metabolic biotransformation of TGZ into reactive metabolites causes idiosyncratic hepatotoxicity, which led to withdrawal of TGZ from the market [156,157]. |
|
|
| EFZ | EFZ demonstrates a significantly higher affinity for PPAR-γ compared to other PPAR subtypes, establishing it as a highly selective agonist. Furthermore, EFZ is considerably more potent than RSZ and TGZ in activating PPAR-γ-mediated transcription. | EFZ selectively activates PPAR-γ, distinguishing itself from other TZDs by downregulating the phosphorylation of PKB (a key enzyme in the prosurvival pathway) without affecting the phosphorylation of ERK. [57,100,165]. |
|
|
| MM902 | MM902 specifically and irreversibly interacts with PPAR-γ [117]. | MM902 functions as a selective and irreversible antagonist of PPAR-γ, inhibiting its activity and thereby affecting cellular processes related to tumorigenesis and metabolism [117]. | MM902 demonstrates weak interactions with a mutant form of BRAF (V600E) and IKK-α, both potentially beneficial for melanoma patients [117] |
|
| T0070907 | T0070907 exhibits an 800-fold selectivity to PPAR-γ over PPAR-α and PPAR-β/δ. | T0070907 operates as a selective and irreversible antagonist of PPARγ, with additional effects that extend beyond PPARγ inhibition. Moreover, in vitro studies found that T0070907 decreased the phosphorylation of PPAR-γ [123]. |
| |
| GW9662 | GW9662 is 100–1000 times more specific to PPAR-γ than PPAR-α and PPAR-β/δ [168,169]. | GW9662 acts as a covalent antagonist of PPAR-γ, irreversibly modifying its ligand-binding domain [120]. |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobolev, V.; Tchepourina, E.; Soboleva, A.; Denisova, E.; Korsunskaya, I.; Mezentsev, A. PPAR-γ in Melanoma and Immune Cells: Insights into Disease Pathogenesis and Therapeutic Implications. Cells 2025, 14, 534. https://doi.org/10.3390/cells14070534
Sobolev V, Tchepourina E, Soboleva A, Denisova E, Korsunskaya I, Mezentsev A. PPAR-γ in Melanoma and Immune Cells: Insights into Disease Pathogenesis and Therapeutic Implications. Cells. 2025; 14(7):534. https://doi.org/10.3390/cells14070534
Chicago/Turabian StyleSobolev, Vladimir, Ekaterina Tchepourina, Anna Soboleva, Elena Denisova, Irina Korsunskaya, and Alexandre Mezentsev. 2025. "PPAR-γ in Melanoma and Immune Cells: Insights into Disease Pathogenesis and Therapeutic Implications" Cells 14, no. 7: 534. https://doi.org/10.3390/cells14070534
APA StyleSobolev, V., Tchepourina, E., Soboleva, A., Denisova, E., Korsunskaya, I., & Mezentsev, A. (2025). PPAR-γ in Melanoma and Immune Cells: Insights into Disease Pathogenesis and Therapeutic Implications. Cells, 14(7), 534. https://doi.org/10.3390/cells14070534