



SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights
 , , , ,
, , , ,  , , ,
, , ,
Abstract
:1. Introduction
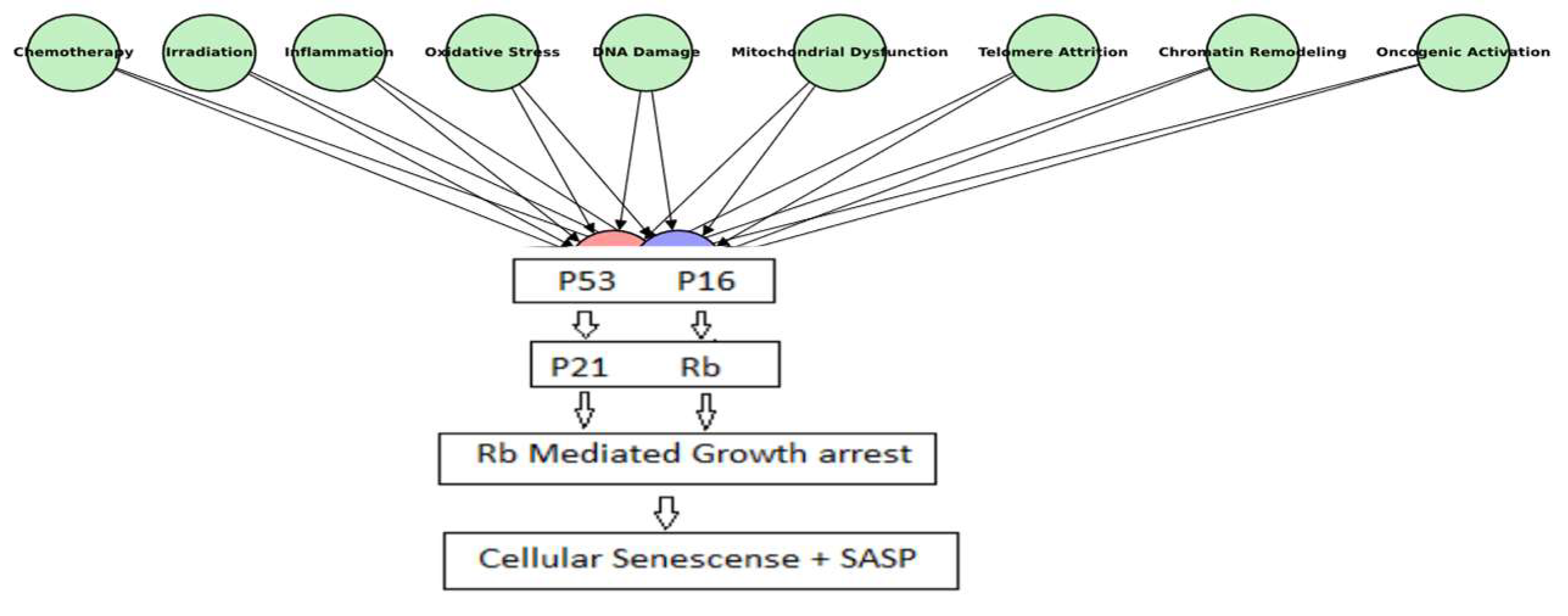
1.1. Cellular Senescence: A Physiological Safeguard and a Pathological Driver
1.2. SASP as the Mediator of Senescence-Induced Effects
1.3. Cellular Rejuvenation and Tissue Homeostasis: The Therapeutic Potential of SASP Modulation
2. SASP: The Molecular Nexus of Cellular Senescence
2.1. Molecular Composition of SASP: Beyond the Core Components
2.2. Temporal Dynamics of SASP: A Shifting Molecular Landscape
2.3. Tissue-Specific Variability: SASP Across Different Microenvironments
2.4. SASP Signaling Pathways
2.5. SASP and the Immune System: Friend or Foe?
3. Cellular Rejuvenation Through SASP Modulation
4. SASP and Its Impact on Tissue Homeostasis
5. Emerging Therapeutic Strategies for SASP Modulation
6. Two Paths to Cellular Rejuvenation: Senescence Clearance vs. SASP Modulation
7. Challenges and Future Directions
- Identifying tissue-specific biomarkers to track SASP dynamics in health and disease;
- Developing next-generation senolytics and senomorphics with greater specificity and improved safety profiles;
- Refining organoid models to better mimic human tissue responses to SASP-targeting interventions;
- Implementing long-term studies to assess the safety and efficacy of SASP modulation therapies.
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, L. The serial cultivation of human diploid cell strains. Nephrol. Dial. Transplant. 1996, 11, 1822–1824. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Dong, Z.; Luo, Y.; Yuan, Z.; Tian, Y.; Jin, T.; Xu, F. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol. Cancer 2024, 23, 181. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Nakao, M.; Tanaka, H.; Koga, T. Cellular senescence variation by metabolic and epigenomic remodeling. Trends Cell Biol. 2020, 30, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.U.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.V.; Deryabin, P.I.; Nikolsky, N.N. “Social life” of senescent cells: What is SASP and why study it? Acta Naturae 2018, 10, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Rodier, F. Dynamic and scalable assessment of the senescence-associated secretory phenotype (SASP). In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2024; Volume 181, pp. 181–195. [Google Scholar]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.G. Cellular senescence and inflammaging in the skin microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef]
- Irvine, K.M.; Skoien, R.; Bokil, N.J.; Melino, M.; Thomas, G.P.; Loo, D.; Gabrielli, B.; Hill, M.M.; Sweet, M.J.; Clouston, A.D.; et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J. Gastroenterol. WJG 2014, 20, 17851. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Wei, W.; Ji, S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell. Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Amaya-Montoya, M.; Pérez-Londoño, A.; Guatibonza-García, V.; Vargas-Villanueva, A.; Mendivil, C.O. Cellular senescence as a therapeutic target for age-related diseases: A review. Adv. Ther. 2020, 37, 1407–1424. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Loghlen, A.; Banito, A.; Raguz, S.; Gil, J. Control of senescence by CXCR2 and its ligands. Cell Cycle 2008, 7, 2956–2959. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Sullivan, J.; Hanley, S.; Price, J.; Tariq, M.A.; McIlvenna, L.C.; Whitham, M.; Sharma-Oates, A.; Harrison, P.; Lord, J.M.; et al. Impact of Senescent Cell-Derived Extracellular Vesicles on Innate Immune Cell Function. Adv. Biol. 2024, 8, 2400265. [Google Scholar] [CrossRef]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef]
- Boccardi, V.; Orr, M.E.; Polidori, M.C.; Ruggiero, C.; Mecocci, P. Focus on senescence: Clinical significance and practical applications. J. Intern. Med. 2024, 295, 599–619. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.U.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Otgonbaatar, A.; Ramamoorthy, A.; Chua, E.H.; Harmston, N.; Gruber, J.; Tolwinski, N.S. Combining stem cell rejuvenation and senescence targeting to synergistically extend lifespan. Aging 2022, 14, 8270. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes. Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Wang, B.; Han, J.; Elisseeff, J.H.; Demaria, M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 2024, 25, 958–978. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes. Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Zhang, X.U.; Kumar, A.; Atkinson, E.J.; Zhu, Y.I.; Jachim, S.; Mazula, D.L.; Brown, A.K.; Berning, M.; Aversa, Z.; et al. The senescence-associated secretome as an indicator of age and medical risk. JCI Insight 2020, 5, e133668. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Weinberger, B. T cells, aging and senescence. Exp. Gerontol. 2020, 134, 110887. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Raulet, D.H. Immunosurveillance of senescent cancer cells by natural killer cells. Oncoimmunology 2014, 3, e27616. [Google Scholar] [CrossRef]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Giroud, J.; Bouriez, I.; Paulus, H.; Pourtier, A.; Debacq-Chainiaux, F.; Pluquet, O. Exploring the communication of the SASP: Dynamic, interactive, and adaptive effects on the microenvironment. Int. J. Mol. Sci. 2023, 24, 10788. [Google Scholar] [CrossRef]
- Alessio, N.; Acar, M.B.; Squillaro, T.; Aprile, D.; Ayaz-Güner, Ş.; Di Bernardo, G.; Peluso, G.; Özcan, S.; Galderisi, U. Progression of irradiated mesenchymal stromal cells from early to late senescence: Changes in SASP composition and anti-tumour properties. Cell Prolif. 2023, 56, e13401. [Google Scholar] [CrossRef]
- Stagni, V.; Ferri, A.; Cirotti, C.; Barilà, D. ATM kinase-dependent regulation of autophagy: A key player in senescence? Front. Cell Dev. Biol. 2021, 8, 599048. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Czaja, A.J. Cellular senescence and its pathogenic and therapeutic implications in autoimmune hepatitis. Expert. Rev. Gastroenterol. Hepatol. 2024, 18, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in hepatic stellate cell activation and liver fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Cai, X.; Guillot, A.; Liu, H. Cellular senescence in hepatocellular carcinoma: The passenger or the driver? Cells 2022, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Zupan, J. Mesenchymal Stem/Stromal cells and fibroblasts: Their roles in tissue Injury and Regeneration, and age-related degeneration. In Fibroblasts—Advances in Inflammation, Autoimmunity and Cancer; IntechOpen: London, UK, 2021; pp. 1–25. [Google Scholar]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.Y.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Marin, I.; Serrano, M.; Pietrocola, F. Recent insights into the crosstalk between senescent cells and CD8 T lymphocytes. Npj Aging 2023, 9, 8. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescence-associated secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Zhang, B.; Trapp, A.; Kerepesi, C.; Gladyshev, V.N. Emerging rejuvenation strategies—Reducing the biological age. Aging Cell 2022, 21, e13538. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Zhu, B.; Deng, M.; Qiu, J.; Feng, Y.; Ding, N.; Huang, C. Metabolic Reprogramming Induced by Aging Modifies the Tumor Microenvironment. Cells 2024, 13, 1721. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Dasgupta, N.; Arnold, R.; Equey, A.; Gandhi, A.; Adams, P.D. The role of the dynamic epigenetic landscape in senescence: Orchestrating SASP expression. Npj Aging 2024, 10, 48. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Zhao, M. An updated review of the epigenetic mechanism underlying the pathogenesis of age-related macular degeneration. Aging Dis. 2020, 11, 1219. [Google Scholar] [CrossRef]
- Lu, Q.; Quinn, A.M.; Patel, M.P.; Semus, S.F.; Graves, A.P.; Bandyopadhyay, D.; Pope, A.J.; Thrall, S.H. Perspectives on the discovery of small-molecule modulators for epigenetic processes. J. Biomol. Screen. 2012, 17, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting histone deacetylases in diseases: Where are we? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, M.M.; Garbarino, V.R.; Kautz, T.; Palavicini, J.P.; Lopez-Cruzan, M.; Dehkordi, S.K.; Mathews, J.; Zare, H.; Xu, P.; Zhang, B.; et al. Senolytic therapy to modulate the progression of Alzheimer’s Disease (SToMP-AD)–Outcomes from the first clinical trial of senolytic therapy for Alzheimer’s disease. Res. Sq. 2023, 9, 22–29. [Google Scholar]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for cancer therapy: Is all that glitters really gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef]
- Zhu, M.; Meng, P.; Ling, X.; Zhou, L. Advancements in therapeutic drugs targeting of senescence. Ther. Adv. Chronic Dis. 2020, 11, 2040622320964125. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free. Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Ahmad, S.I. (Ed.) Handbook of Mitochondrial Dysfunction; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Reiten, O.K.; Wilvang, M.A.; Mitchell, S.J.; Hu, Z.; Fang, E.F. Preclinical and clinical evidence of NAD+ precursors in health, disease, and ageing. Mech. Ageing Dev. 2021, 199, 111567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, S.W.; Park, H. Targeting senescence-associated secretory phenotype (SASP) to combat aging-related diseases. Exp. Mol. Med. 2016, 48, e269. [Google Scholar]
- Saito, Y.; Yamamoto, S.; Chikenji, T.S. Role of cellular senescence in inflammation and regeneration. Inflamm. Regen. 2024, 44, 28. [Google Scholar] [CrossRef]
- Salminen, A. The role of the immunosuppressive PD-1/PD-L1 checkpoint pathway in the aging process and age-related diseases. J. Mol. Med. 2024, 102, 733–750. [Google Scholar] [CrossRef]
- Wang, M.J.; Zhang, H.L.; Chen, F.; Guo, X.J.; Liu, Q.G.; Hou, J. The double-edged effects of IL-6 in liver regeneration, aging, inflammation, and diseases. Exp. Hematol. Oncol. 2024, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Zheng, L.; He, S.; Wang, H.; Li, J.; Liu, Y.; Liu, S. Targeting cellular senescence in aging and age-related diseases: Challenges, considerations, and the emerging role of senolytic and senomorphic therapies. Aging Dis. 2024, 15, 2554. [Google Scholar]
- Elder, S.S.; Emmerson, E. Senescent cells and macrophages: Key players for regeneration? Open Biol. 2020, 10, 200309. [Google Scholar] [CrossRef]
- Mavrogonatou, E.; Papadopoulou, A.; Pratsinis, H.; Kletsas, D. Senescence-associated alterations in the extracellular matrix: Deciphering their role in the regulation of cellular function. American J. Physiol.-Cell Physiol. 2023, 325, C633–C647. [Google Scholar] [CrossRef]
- Papismadov, N.; Solomonov, I.; Sagi, I.; Krizhanovsky, V. The ECM path of senescence in aging: Components and modifiers. FEBS J. 2020, 287, 2636–2646. [Google Scholar]
- Usman, K.; Hsieh, A.; Hackett, T.L. The role of miRNAs in extracellular matrix repair and chronic fibrotic lung diseases. Cells 2021, 10, 1706. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Lofaro, F.D.; Bonacorsi, S.; Mazzilli, A.; Garcia-Fernandez, M.; Quaglino, D. The role of fibroblasts in skin homeostasis and repair. Biomedicines 2024, 12, 1586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A: Biomed. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Coppé, J.P.; Lam, E.W. Cellular senescence: The sought or the unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Velarde, M.C.; Demaria, M. Targeting senescent cells: Possible implications for delaying skin aging: A mini-review. Gerontology 2016, 62, 513–518. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, S.; Qu, J.; Belmonte, J.C.; Liu, G.H. Rejuvenation of tissue stem cells by intrinsic and extrinsic factors. Stem Cells Transl. Med. 2022, 11, 231–238. [Google Scholar] [CrossRef]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef]
- Zhao, S.; Qiao, Z.; Pfeifer, R.; Pape, H.C.; Mao, K.; Tang, H.; Meng, B.; Chen, S.; Liu, H. Modulation of fracture healing by senescence-associated secretory phenotype (SASP): A narrative review of the current literature. Eur. J. Med. Res. 2024, 29, 38. [Google Scholar] [CrossRef]
- Yue, Z.; Nie, L.; Zhao, P.; Ji, N.; Liao, G.; Wang, Q. Senescence-associated secretory phenotype and its impact on oral immune homeostasis. Front. Immunol. 2022, 13, 1019313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, J.; Yu, S.; Zheng, Y.; Duan, M.; Zhao, L.; Wang, Y.; Yang, Z.; Jiang, X. Cellular senescence and metabolic reprogramming: Unraveling the intricate crosstalk in the immunosuppressive tumor microenvironment. Cancer Commun. 2024, 44, 929–966. [Google Scholar] [CrossRef] [PubMed]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The SASPin the challenging future of cancer therapy and age-related diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Abubakar, M.; Hameed, Y.; Kiani, M.N.; Aftab, A. Common features between aging and cancer: A narrative review. Aging Adv. 2024, 1, 118–134. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κ B activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Cech, M. Therapeutic Potential of Senotherapeutics. Bachelor’s Thesis, Faculty of Science, University of Hradec Králové, Hradec Králové, Czech Republic, 2024; 80p. [Google Scholar]
- Shin, Y.J.; Kwon, K.S.; Suh, Y.; Lee, K.P. The role of non-coding RNAs in muscle aging: Regulatory mechanisms and therapeutic potential. Front. Mol. Biosci. 2024, 10, 1308274. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Baniahmad, A.; Branicki, W.; Taheri, M.; Eghbali, A. Emerging role of non-coding RNAs in senescence. Front. Cell Dev. Biol. 2022, 10, 869011. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.S.; Burton Sojo, G.; Sun, H.; Friedland, B.N.; McNamara, M.E.; Schmidt, M.O.; Wellstein, A. The Role of Aging and Senescence in Immune Checkpoint Inhibitor Response and Toxicity. Int. J. Mol. Sci. 2024, 25, 7013. [Google Scholar] [CrossRef]
- Huang, M.; Wang, Y.; Fang, L.; Liu, C.; Feng, F.; Liu, L.; Sun, C. T cell senescence: A new perspective on immunotherapy in lung cancer. Front. Immunol. 2024, 15, 1338680. [Google Scholar] [CrossRef]
- Giannoula, Y.; Kroemer, G.; Pietrocola, F. Cellular senescence and the host immune system in aging and age-related disorders. Biomed. J. 2023, 46, 100581. [Google Scholar] [CrossRef]
- Hou, Y.; Chen, M.; Bian, Y.; Hu, Y.; Chuan, J.; Zhong, L.; Zhu, Y.; Tong, R. Insights into vaccines for elderly individuals: From the impacts of immunosenescence to delivery strategies. Npj Vaccines 2024, 9, 77. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, M.; Ren, Y.; Xu, H.; Weng, S.; Ning, W.; Ge, X.; Liu, L.; Guo, C.; Duo, M.; et al. Recent advances and applications of CRISPR-Cas9 in cancer immunotherapy. Mol. Cancer 2023, 22, 35. [Google Scholar] [CrossRef] [PubMed]
- Herbstein, F.; Sapochnik, M.; Attorresi, A.; Pollak, C.; Senin, S.; Gonilski-Pacin, D.; Ciancio del Giudice, N.; Fiz, M.; Elguero, B.; Fuertes, M.; et al. The SASP factor IL-6 sustains cell-autonomous senescent cells via a cGAS-STING-NFκB intracrine senescent noncanonical pathway. Aging Cell 2024, 23, e14258. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.G.; Barajas, M.; Razquin, N.; Berraondo, P.; Rodrigo, M.; Wu, C.; Qian, C.; Fortes, P.; Prieto, J. In vitro and in vivo comparative study of chimeric liver-specific promoters. Mol. Ther. 2003, 7, 375–385. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.E. Tissue homeostasis and non-homeostasis: From cell life cycles to organ states. Annu. Rev. Cell Dev. Biol. 2022, 38, 395–418. [Google Scholar] [CrossRef]
- Watson, N.; Kuppuswamy, S.; Ledford, W.L.; Sukumari-Ramesh, S. The role of HDAC3 in inflammation: Mechanisms and therapeutic implications. Front. Immunol. 2024, 15, 1419685. [Google Scholar] [CrossRef]
- Jin, S.; Wang, Y.; Wu, X.; Li, Z.; Zhu, L.; Niu, Y.; Zhou, Y.; Liu, Y. Young exosome bio-nanoparticles restore aging-impaired tendon stem/progenitor cell function and reparative capacity. Adv. Mater. 2023, 35, 2211602. [Google Scholar] [CrossRef]
- Rossi, M.; Abdelmohsen, K. The emergence of senescent surface biomarkers as senotherapeutic targets. Cells 2021, 10, 1740. [Google Scholar] [CrossRef]
- Ray, K. Clearance of nanomaterials in the liver. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 560. [Google Scholar] [CrossRef]
- Bennett, G. Senotherapy: A Potential Pharmacological Strategy for Prolonging Human Lifespan and Healthspan. In SpringerBriefs in Modern Perspectives on Disability Research; Kindle Edition; Springer: Cham, Switzerland, 2023. [Google Scholar]
- Aquino-Martinez, R.; Eckhardt, B.A.; Rowsey, J.L.; Fraser, D.G.; Khosla, S.; Farr, J.N.; Monroe, D.G. Senescent cells exacerbate chronic inflammation and contribute to periodontal disease progression in old mice. J. Periodontol. 2021, 92, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J.P. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 2015, 4, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Fujieda, K.; Senju, S.; Ikeda, T.; Oshiumi, H.; Nishimura, Y. Immune-suppressive effects of interleukin-6 on T-cell-mediated anti-tumor immunity. Cancer Sci. 2018, 109, 523–530. [Google Scholar] [CrossRef]
- Wan, M.; Gray-Gaillard, E.F.; Elisseeff, J.H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Rim, C.; You, M.J.; Nahm, M.; Kwon, M.S. Emerging role of senescent microglia in brain aging-related neurodegenerative diseases. Transl. Neurodegener. 2024, 13, 10. [Google Scholar] [CrossRef]
- Lau, L.; David, G. Pro-and anti-tumorigenic functions of the senescence-associated secretory phenotype. Expert. Opin. Ther. Targets 2019, 23, 1041–1045. [Google Scholar] [CrossRef]
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The heterogeneity of cellular senescence: Insights at the single-cell level. Trends Cell Biol. 2023, 33, 9–17. [Google Scholar] [CrossRef]
- Theodorakis, N.; Feretzakis, G.; Tzelves, L.; Paxinou, E.; Hitas, C.; Vamvakou, G.; Verykios, V.S.; Nikolaou, M. Integrating Machine Learning with Multi-Omics Technologies in Geroscience: Towards Personalized Medicine. J. Pers. Med. 2024, 14, 931. [Google Scholar] [CrossRef] [PubMed]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Olshansky, S.J.; Perry, D.; Miller, R.A.; Butler, R.N. Pursuing the longevity dividend: Scientific goals for an aging world. Ann. N. Y. Acad. Sci. 2007, 1114, 11–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Feature | Early SASP (Pro-Repair) | Late SASP (Pro-Degradation) |
|---|---|---|
| Key factors | Growth factors, anti-inflammatory cytokines, matrix remodeling proteins | Pro-inflammatory cytokines (IL-6,8), chemokines, matrix metalloproteins (MMPs) |
| Function | Facilitates tissue repair and regeneration | Promote tissue degradation and chronic inflammation |
| Temporal dynamics | Occurs in the initial stages of senescence, transient and resolves upon completion of repair | Develops during prolonged senescence, is persistent, and can contribute to age-related pathologies |
| Impact on environment | Supports regenerative processes and maintains tissue homeostasis | Disrupts tissue homeostasis, promotes inflammatory factors; may lead to tumor progression or fibrosis depending on the context |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, S.; Alqahtani, T.; Venkatesan, K.; Sivadasan, D.; Ahmed, R.; Sirag, N.; Elfadil, H.; Abdullah Mohamed, H.; T.A., H.; Elsayed Ahmed, R.; et al. SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights. Cells 2025, 14, 608. https://doi.org/10.3390/cells14080608
Alqahtani S, Alqahtani T, Venkatesan K, Sivadasan D, Ahmed R, Sirag N, Elfadil H, Abdullah Mohamed H, T.A. H, Elsayed Ahmed R, et al. SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights. Cells. 2025; 14(8):608. https://doi.org/10.3390/cells14080608
Chicago/Turabian StyleAlqahtani, Saud, Taha Alqahtani, Krishnaraju Venkatesan, Durgaramani Sivadasan, Rehab Ahmed, Nizar Sirag, Hassabelrasoul Elfadil, Hanem Abdullah Mohamed, Haseena T.A., Rasha Elsayed Ahmed, and et al. 2025. "SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights" Cells 14, no. 8: 608. https://doi.org/10.3390/cells14080608
APA StyleAlqahtani, S., Alqahtani, T., Venkatesan, K., Sivadasan, D., Ahmed, R., Sirag, N., Elfadil, H., Abdullah Mohamed, H., T.A., H., Elsayed Ahmed, R., Muralidharan, P., & Paulsamy, P. (2025). SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights. Cells, 14(8), 608. https://doi.org/10.3390/cells14080608